
Abstract
Introduction
This randomized, double-blind, parallel-group study investigated the safety, tolerability, pharmacokinetics (PK), and cognitive outcomes of BI 409306—a selective phosphodiesterase 9A (PDE9A) inhibitor—in patients with schizophrenia.
Methods
Patients with mild-to-moderate schizophrenia were randomized (1:1:1:1) to receive BI 409306 at 25, 50, or 100 mg or placebo once daily over 14 days. The primary endpoints were safety and tolerability; the secondary endpoints were PK and cognitive outcomes.
Results
Of the 40 randomized patients, 38 (95%) completed the study. Patients were predominantly male (87.5%; mean age, 40.2 years). After a single dose, C max was reached within 30–45 min. The geometric mean (gMean) C max and AUC0-∞ ranged from 138 to 998 nmol/L and 217 to 2020 nmol∙h/L, respectively. Elimination was rapid (gMean t 1/2 range 1.10–1.85 h). After multiple doses, C max,ss was reached within 1 h; elimination was similar to that observed after a single dose. Total exposure at steady state and after a single dose were similar (accumulation ratio range: AUC, 0.758–1.13 and Cmax, 0.768–1.40). No deaths, adverse events (AEs) leading to discontinuation, or serious AEs were observed. Treatment-emergent AEs were mild, with no apparent dose-related trends. There was no worsening of schizophrenia symptoms (Positive and Negative Syndrome Scale) and no trends in suicidality (Columbia Suicide Severity Rating Scale). The Hopkins Verbal Learning Test–Revised (HVLT-R) and Brief Visuospatial Memory Test–Revised (BVMT-R) showed no effect on cognitive function.
Conclusion
Administration of BI 409306 in patients with mild-to-moderate schizophrenia resulted in satisfactory safety and tolerability. BI 409306, PK was characterized by rapid absorption, monophasic to biphasic elimination, and minor accumulation with multiple dosing.
Trial Registration
ClinicalTrials.gov identifier NCT01892384.
Funding
Boehringer Ingelheim Pharma GmbH & Co. KG.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Impaired N-methyl-d-aspartic acid (NMDA) receptor signaling and reduced levels of cyclic guanosine monophosphate (cGMP) have been demonstrated in animal models of schizophrenia [1, 2], and increasing evidence suggests that glutamatergic dysfunction may play a key role in mediating risk for conversion to psychosis [3,4,5]. Phosphodiesterase 9A (PDE9A), which has the highest affinity of all PDEs, hydrolyzes cGMP to regulate its intracellular concentration within glutamatergic neurons [6]. Consequently, PDE9A inhibition may augment synaptic plasticity and memory function through increased cGMP availability and, thus, improved NMDA receptor signaling [6, 7]. PDE9A is highly expressed in the neocortex and hippocampus; therefore, it is likely to be a significant determinant of intracellular basal cGMP levels in these brain regions [6, 8].
BI 409306 is a potent and selective PDE9A inhibitor. Application of BI 409306 to rat hippocampal slices resulted in significant enhancement of long-term potentiation (LTP) [9]. Furthermore, BI 409306 induced a dose-dependent increase in cGMP in rat cerebrospinal fluid (CSF) and prefrontal cortex and improved memory performance [10]. In healthy male volunteers, BI 409306 was rapidly absorbed and eliminated and showed a good safety and tolerability profile [11,12,13]. In these studies, the most frequent adverse events (AEs) were visual symptoms such as photopsia (flashing light), photophobia (increased sensitivity to light), chromatopsia (change in color perception), and blurred vision. These AEs occurred shortly after administration and were resolved within 1–2 h. In a proof-of-mechanism study conducted in healthy male volunteers, BI 409306 administered orally as a single dose crossed the blood–brain barrier and triggered a dose- and concentration-dependent increase in cGMP in the CSF [14].
The objective of this study was to investigate the safety and tolerability of BI 409306, a novel PDE9 inhibitor, in patients with mild-to-moderate schizophrenia. The associated pharmacokinetics (PK) and cognitive outcomes were also explored. BI 409306 was administered orally at a dosage of 25, 50, or 100 mg for 14 days.
Methods
Study Design
This was a randomized, double-blind, placebo-controlled, parallel-group, phase I study in patients with mild-to-moderate schizophrenia. The study was conducted from 8 July 2013 to 5 December 2013 at the Community Clinical Research, Inc., Austin, TX. The duration of the study from screening through completion of the end-of-treatment visit was up to 8 weeks. The end-of-treatment visit was planned 7–14 days after the administration of the last dose of the study drug (Fig. 1). Patients were randomized (1:1:1:1) on day 1 to receive BI 409306 at 25, 50, or 100 mg or placebo, administered orally once daily (qd) for 14 days. Sequentially numbered blinded medication kits were assigned to the patients at the study site. All the patients provided written informed consent. All procedures followed in the study (ClinicalTrials.gov: NCT01892384) were in accordance with the protocol, ethical standards laid down in the Helsinki Declaration of 1964 (as revised in 2013), the International Conference on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Harmonised Tripartite Guidelines for Good Clinical Practice, and relevant Boehringer Ingelheim standard operating procedures.

Study design
Patients
Patients included in the study were 18–55 years of age, with an established diagnosis of schizophrenia as defined by the Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV criteria, with the following clinical features: clinically stable and in the residual (non-acute) phase of illness or maintained on current antipsychotic medications and dose for ≥ 8 weeks; and having no more than a “moderate” severity rating on hallucinations and delusions, positive formal thought disorder and negative symptoms, and minimal extrapyramidal and depressive symptoms. All patients were genotyped for cytochrome P450 2C19 (CYP2C19).
Patients were excluded if they were treated with > 1 antipsychotic agent, not stabilized on antipsychotic treatment, or had electroconvulsive therapy within the last 30 days, or if the severity of their cognitive impairment would affect the study outcome in the clinical judgment of the investigator. In addition, patients with any of the following were excluded from the study: suicidal behavior within the past 2 years or type 4 or 5 suicidal ideation on the Columbia Suicide Severity Rating Scale (C-SSRS) within the past 3 months; findings from medical examination or laboratory values deviating from normal limits and of clinical relevance; planned elective surgery; a history or diagnosis of symptomatic and unstable/uncontrolled gastrointestinal, hepatic, renal, respiratory, cardiovascular, metabolic, immunological, hematological, or hormonal disorders; a history or new diagnosis of human immunodeficiency virus infection or malignancy; or a history of neurologic or psychiatric condition that might interfere with the interpretation of data. Pregnant or breastfeeding women were also excluded.
Study Assessments
Safety Endpoints
The primary endpoints for safety and tolerability were evaluated based on AEs, vital signs, 12-lead electrocardiogram (ECG), clinical laboratory tests, physical examination, Clinical Global Impression–Improvement (CGI-I) scale, disease severity on the Positive and Negative Syndrome Scale (PANSS), suicidality assessment by the C-SSRS, and visual acuity and color vision assessed by Snellen chart and Ishihara plates, respectively.
Pharmacokinetic Endpoints
The following PK parameters were assessed as secondary endpoints: area under the plasma concentration–time curve of BI 409306 from zero extrapolated to infinity after a single dose (AUC0−∞); area under the plasma concentration–time curve of BI 409306 in plasma at steady state over a uniform dosing interval tau (AUCtau,ss); maximum measured concentration of BI 409306 in plasma following a single dose (C max) and at steady state (C max,ss); time from dosing to maximum measured concentration of BI 409306 in plasma following a single dose (t max) and at steady state (t max,ss); and terminal elimination half-life of BI 409306 (t 1/2).
Cognitive Outcomes
Additional secondary endpoints included measurement of the total score on the Hopkins Verbal Learning Test–Revised (HVLT-R) and total score on the Brief Visuospatial Memory Test–Revised (BVMT-R).
Statistical Analyses
Dose proportionality was explored after a single dose (day 1, C max and AUC0−∞) and after multiple dosing (day 14, C max,ss and AUCtau,ss) using a linear regression model with log-transformed dose as an independent variable applied to log-transformed data. Two-sided 95% confidence intervals (CIs) for the slope were computed from this model. For PK endpoints AUC0−∞ and AUCtau,ss, a linear model with patient and order as fixed effects was applied to log-transformed data to assess linearity for each dose separately. Attainment of steady state was assessed using a linear model with patient as a fixed effect, time as a repeated effect, and log-transformed pre-dose concentration from days 1 to 14 as the dependent variable for each dose separately.
Changes in HVLT-R and BVMT-R total scores were compared between doses of BI 409306 and placebo using an analysis of covariance model with treatment as a fixed effect and baseline as a covariate. Safety analyses were performed for all patients who received ≥ 1 dose of the study drug; the results are summarized descriptively.
Results
Patient Characteristics
Of the 50 patients enrolled, 40 were randomized to receive either BI 409306 or placebo; 38 patients completed the study and two withdrew consent (Fig. 2). There were four important protocol deviations: one patient in the 25-mg group received a prohibited medication (lorazepam) on day 7; one patient in the 50-mg group tested positive for cocaine on day −1, but had initiated treatment before the drug screen results were available; another patient had a prior suicide attempt; and one patient in the placebo group declined to provide informed consent for the optional pharmacogenetic sample collection for DNA banking. However, none of these patients were excluded from the study analysis. Patients were predominantly male (87.5%) and African American (52.5%) or white (47.5%), with a mean (standard deviation [SD]) age of 40.2 (9.0) years. Demographics and baseline characteristics were well balanced across all treatment groups, with the exception of cognition (HVLT-R and BVMT-R; Table 1). As BI 409306 oxidative metabolism is dependent on CYP2C19, CYP2C19 phenotype was predicted for each patient based on genotype. Of the 40 randomized patients, 16 (40.0%) were predicted to be ultra-rapid metabolizers (UM) for CYP2C19; 11 (27.5%), extensive metabolizers (EM); and 8 (20.0%), intermediate metabolizers (IM). No patients were predicted to be poor metabolizers (PM) for CYP2C19. For five patients, the predicted CYP2C19 phenotype could not be determined based on genotype.

Patient disposition
Safety
Table 2 summarizes the AEs. BI 409306 was well tolerated; treatment-emergent AEs were reported in 23 (57.5%) patients. All AEs were mild in intensity, and no apparent dose-related trends were observed for any AE. Eleven (27.5%) patients reported a total of 13 eye-related AEs, including blurred vision (n = 8), photopsia (flashing lights; n = 2), visual impairment (n = 2), and visual field defect (n = 1). The number of patients reporting eye-related AEs was similar across all BI 409306 dose groups and the placebo group. All eye-related AEs were non-serious and mild in intensity, and resolved spontaneously without medical treatment. All but three events (blurred vision in one patient each in the 25-mg and 50-mg groups, and visual field defect in one patient in the 100-mg group) were assessed as related to treatment. There were no deaths, serious AEs or discontinuation due to AEs. The incidence of treatment-emergent AEs was similar between BI 409306 and placebo. No clinically relevant or dose-related changes were observed in laboratory parameters, vital signs, or ECGs in patients receiving BI 409306 or placebo.
CGI-I indicated no change or minimal improvement in the patients’ overall disease status. The mean change from baseline in total PANSS scores for the 25-, 50-, and 100-mg BI 409306 and placebo groups were −0.1, −0.2, −1.4, and −0.9, respectively, at day 7, and 2.3, 0.0, −3.6, and 0.0, respectively, at day 13, suggesting no deterioration in disease symptoms or clinical status. Mild suicidal ideation after treatment initiation (C-SSRS score = 1) was reported in one patient each in the 50-mg BI 409306 and placebo groups. Changes in visual acuity were reported intermittently for a few patients across all treatment groups. Color discrimination was reported as normal at maximum time points in all the patients. Visual examination of box-plots of C max,ss of BI 409306 or its major metabolites did not suggest any association between higher exposure and worsening of visual acuity or color vision. Administration of BI 409306 had a negligible effect on the mean pupil diameter relative to placebo.
Pharmacokinetics
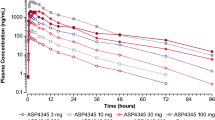
PK profiles of BI 409306 following single- and multiple-dose administration were characterized by a rapid increase in plasma concentration and rapid elimination. Table 3 shows the PK endpoints after single- and multiple-dose administration of BI 409306. After single-dose administration, BI 409306 was rapidly absorbed, reaching C max within 30–45 min (Fig. 3). Following absorption, BI 409306 was rapidly eliminated, with geometric mean (gMean) t 1/2 ranging from 1.10 to 1.85 h. After multiple-dose administration, BI 409306 was rapidly absorbed, with C max,ss attained within the first hour after dosing. Total exposure at steady state was similar to that following a single dose, with an accumulation ratio close to 1 (ranges: AUC 0.758–1.13; C max 0.768–1.40), indicating minor to no accumulation with multiple dosing.

Geometric mean plasma concentration–time profiles after single and multiple doses of BI 409306 (25, 50, and 100 mg)
Dose-normalized gMean C max, C max,ss, AUC0−∞ and AUCtau,ss values were similar for the 50- and 100-mg dose groups following single- and multiple-dose administration, suggesting dose proportionality. However, dose-normalized parameters for the 25-mg dose group were lower than expected, with approximately threefold lower C max,ss,norm and AUCtau,ss, norm compared with the higher dose groups. Linearity with respect to multiple dose administration could not be established due to high inter-individual variability in the linearity index within each dose group.
Inter-individual variability in BI 409306 PK parameters was moderate to high. The geometric coefficient of variation (gCV) after a single dose ranged from 98.4% to 108% for AUC0−∞ and from 73.3% to 106% for C max; gCV at steady state ranged from 86.8% to 112% for AUCtau,ss and from 74.4% to 90.9% for C max,ss. Exposure was highest in patients with the CYP2C19 IM predicted phenotype, followed by EM and UM patients. However, inter-individual variability was lower in the IM patients than in the EM and UM patients, with gCV% of 48.4%, 152%, and 154%, respectively.
Cognitive Outcomes
No significant treatment differences were observed with 25-, 50-, or 100-mg BI 409306 in HVLT-R total scores (p = 0.0867, p = 0.2011, and p = 0.4103, respectively) or BVMT-R total scores (p = 0.5791, p = 0.605, and p = 0.7171, respectively) compared with placebo.
Discussion
In this study, BI 409306 at a dosage of 25, 50, or 100 mg per day for 14 days was well tolerated in patients with mild-to-moderate schizophrenia. The PK of BI 409306 following single- and multiple-dose administration was characterized by very rapid absorption followed by rapid monophasic to biphasic elimination. No evidence of deterioration in schizophrenia symptoms, suicidality, or overall clinical status was observed in this study.
The safety results for BI 409306 in this study are comparable to those from studies conducted in healthy male volunteers. In previous studies, AEs were mild to moderate in intensity and of short duration [11, 14]. The most frequently reported AEs were visual AEs. Consistent with the present study, there was no apparent dose-dependent increase in AEs [14]. In young and elderly healthy subjects administered 25, 50, or 100 mg BI 409306 or placebo, a majority of the AEs were mild in intensity, with a dose-dependent increase in subjects reporting eye disorders such as photophobia (increased sensitivity to light), chromatopsia (change in color perception), visual impairment, blurred vision, abnormal sensation in the eye, asthenopia, and eye pain [12]. Similarly, in healthy male Chinese and Japanese subjects, most AEs were mild; the most frequently reported AEs in subjects receiving BI 409306 were eye disorders (29.8%), which were resolved within 1–2 h [13]. Visual side effects have been proposed to be associated with PDE9A, which regulates the level of cGMP by hydrolyzing it and modulates inhibitory processes in the cone pathway of the retina [15]. Given the results in healthy subjects, eye-related AEs were of interest in this study, and indeed, these were the most frequently reported AEs. Unlike previous studies, however, eye-related AEs in this study were reported with similar frequency in all treatment groups. Overall, changes in visual acuity were reported intermittently and with similar severity and frequency by patients across treatment groups, and were typically mild and transient. While some changes in visual acuity in BI 409306-treated patients occurred around the anticipated time of peak plasma concentrations of BI 409306, a consistent relationship between changes in visual acuity and BI 409306 C max was not observed. Interestingly, the frequency of total AEs in this study is comparable to that of a study in which the selective PDE9A inhibitor PF-04447943 was used in patients with mild-to-moderate probable Alzheimer’s disease [16].
The PK of BI 409306 in this study is in line with that observed in previous studies in healthy subjects, which is characterized by very rapid absorption and elimination, with minor accumulation. In the current study, exposure was dose-proportional from 50 to 100 mg and was comparable to that observed in a previous study [12]. However, exposure in the 25-mg dose group was lower than expected, resulting in a lack of dose proportionality over the 25- to 100-mg dose range. Linearity with respect to multiple-dose administration could not be established due to the high inter-subject variability in the linearity index within each dose group. This variability could be partly due to differences in CYP2C19 predicted phenotype, as exposure in IM was highest, followed by exposure in EM and UM.
A previous study reported that BI 409306 crossed the blood–brain barrier, with the maximum concentration in CSF reaching 28.31% of the maximum plasma concentration [14]. BI 409306 had no effect on the chosen cognitive tests over the 14-day treatment period in patients with schizophrenia; however, that study was not of sufficient power or duration to adequately assess the overall effect on cognition [17]. In a phase II placebo-controlled study, PF-04447943 reached maximal concentrations of approximately five times the IC50 [16]. Moreover, the estimated median exposure remained above the IC50 for the entire dosing duration. Treatment with PF-04447943 over a 12-week period was not more effective than placebo in terms of cognition, behavior, or clinician-rated global change.
A limitation of this study is that the population was drawn from a single center. Furthermore, the patients who participated in this study may not represent the larger population with chronic schizophrenia, many of whom are on multiple medications or on medications that were excluded. Nevertheless, the study results provide guidance for design of future trials with BI 409306.
Conclusion
Overall, BI 409306 showed satisfactory safety and tolerability at 25-, 50-, or 100-mg qd doses for 14 days in patients with mild-to-moderate schizophrenia. The PK profile of BI 409306 was characterized by rapid absorption, rapid monophasic to biphasic elimination, and minor accumulation with multiple dosing. There were no apparent effects on cognitive function over 14 days of treatment with BI 409306.
References
Neill JC, Barnes S, Cook S, et al. Animal models of cognitive dysfunction and negative symptoms of schizophrenia: focus on NMDA receptor antagonism. Pharmacol Ther. 2010;128:419–32.
Nabeshima T, Mouri A, Murai R, Noda Y. Animal model of schizophrenia: dysfunction of NMDA receptor-signaling in mice following withdrawal from repeated administration of phencyclidine. Ann N Y Acad Sci. 2006;1086:160–8.
Salisbury DF, Shenton ME, Griggs CB, Bonner-Jackson A, McCarley RW. Mismatch negativity in chronic schizophrenia and first-episode schizophrenia. Arch Gen Psychiatry. 2002;59:686–94.
Schobel SA, Chaudhury NH, Khan UA, et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron. 2013;78:81–93.
Medoff DR, Holcomb HH, Lahti AC, Tamminga CA. Probing the human hippocampus using rCBF: contrasts in schizophrenia. Hippocampus. 2001;11:543–50.
Dorner-Ciossek C, Baum-Kroker KS, Rosenbrock H. CNS functions and diseases-Chapter 15: role of PDE9 in cognition. In: Zhang H, Xu Y, O’Donnell JM, editors. Advances in neurobiology, vol. 17. Basel: Springer International Publishing; 2017.
Reneerkens OA, Rutten K, Steinbusch HW, et al. Selective phosphodiesterase inhibitors: a promising target for cognition enhancement. Psychopharmacology. 2009;202:419–43.
Schmidt CJ. Phosphodiesterase inhibitors as potential cognition enhancing agents. Curr Top Med Chem. 2010;10:222–30.
Dorner-Ciossek C, Giovannini R, Rosenbrock H. BI 409306, a novel phosphodiesterase 9A inhibitor, part I: potency, selectivity and in vitro functional characterization on synaptic plasticity. International Congress on Schizophrenia Research. Schizophrenia Bull. 2015; p. S31.
Rosenbrock H, Marti A, Koros E, et al. BI 409306, a novel phosphodiesterase 9A inhibitor, part II: in vivo characterization regarding target engagement and cognition tasks in rodents. International Congress on Schizophrenia Research. Schizophrenia Bull. 2015; p. S36.
Moschetti V, Boland K, Feifel U, Hoch A, Zimdahl-Gelling H, Sand M. First-in-human study assessing safety, tolerability and pharmacokinetics of BI 409306, a selective phosphodiesterase 9A inhibitor, in healthy males. Br J Clin Pharmacol. 2016;82:1315–24. https://doi.org/10.1111/bcp.13060.
Moschetti V, Boland K, Hoch A, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple rising doses of BI 409306 film-coated tablets given orally once or twice daily for 14 days in young and elderly healthy volunteers. Poster presented at: Alzheimer’s Association International Conference, 18–23 Jul 2015, Washington, DC, USA.
Wunderlich G, Kim JM, Yum S.-Y.A., et al. Safety, tolerability, and pharmacokinetics of BI 409306: a randomized, placebo-controlled, double-blinded phase I study in Chinese and Japanese healthy male volunteers. Poster presented at: Alzheimer’s Association International Conference, 18–23 Jul 2015, Washington, DC, USA.
Boland K, Moschetti V, Dansirikul C, et al. A phase I, randomized, proof-of-clinical-mechanism study assessing the pharmacokinetics and pharmacodynamics of the oral PDE9A inhibitor BI 409306 in healthy male volunteers. Hum Psychopharmacol. 2017;. https://doi.org/10.1002/hup.2569.
Dhingra A, Tummala SR, Lyubarsky A, et al. PDE9A is expressed in the inner retina and contributes to the normal shape of the photopic ERG waveform. Front Mol Neurosci. 2014;7:60. https://doi.org/10.3389/fnmol.2014.00060.
Schwam EM, Nicholas T, Chew R, et al. A multicenter, double-blind, placebo-controlled trial of the PDE9A inhibitor, PF-04447943, in Alzheimer’s disease. Curr Alzheimer Res. 2014;11:413–21.
Brown A, Daniels K, Zhang S, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of BI 409306 film-coated tablets given orally qd for 14 days in patients with schizophrenia. International Congress on Schizophrenia Research. Schizophrenia Bull. 2015; p. S04.
Acknowledgements
The study and the article processing charges were funded by Boehringer Ingelheim Pharma GmbH & Co. KG (Ingelheim am Rhein, Germany). All authors had full access to all the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis. The authors would like to thank Diana Brewer of Community Clinical Research, Inc., Austin, TX, USA, for her significant assistance in the operational conduct of this study, and Dr. Lauren Liss for her help in drafting the Introduction section. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole and have given final approval of the version to be published. Writing, editorial support and formatting assistance was provided by Suchita Nath-Sain, PhD, of Cactus Communications, which was contracted and funded by Boehringer Ingelheim for these services. Boehringer Ingelheim was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations. The results presented in this paper were published in part at the 15th International Congress on Schizophrenia Research, 28 March to 1 April 2015, Colorado Springs, CO, USA, and the 70th Annual Scientific Meeting of the Society of Biological Psychiatry, 14–16 May 2015, Toronto, Ontario, Canada.
Disclosures
David Brown received a fee from Boehringer Ingelheim for consulting services on the clinical trial protocol. Kristen Daniels is employed by Boehringer Ingelheim. Michael Sand is employed by Boehringer Ingelheim. Solen Pichereau was an employee of Boehringer Ingelheim at the time that the study was conducted but is now employed by Roche (Basel, Switzerland). Solen Pichereau has no conflicts to declare. The authors received no direct compensation related to the development of the manuscript. The journal’s article processing charges will be funded by the sponsor.
Compliance with Ethics Guidelines
All procedures followed in this study (ClinicalTrials.gov: NCT01892384) were in accordance with the protocol, ethical standards laid down in the Helsinki Declaration of 1964 (as revised in 2013), the International Conference on Harmonisation Harmonised Tripartite Guidelines for Good Clinical Practice and relevant Boehringer Ingelheim standard operating procedures. Informed consent was obtained from all patients for inclusion in the study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/62DCF0600F4FDF01.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Brown, D., Daniels, K., Pichereau, S. et al. A Phase IC Study Evaluating the Safety, Tolerability, Pharmacokinetics, and Cognitive Outcomes of BI 409306 in Patients with Mild-to-Moderate Schizophrenia. Neurol Ther 7, 129–139 (2018). https://doi.org/10.1007/s40120-017-0085-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-017-0085-5