
Abstract
Global oxycodone consumption has increased sharply during the last two decades, and, in 2008, oxycodone consumption surpassed that of morphine. As oxycodone was synthesized in 1916 and taken to clinical use a year later, it has not undergone the same approval process required by today’s standards. Most of the basic oxycodone pharmacokinetic (PK) data are from the 1990s and from academic research; however, a lot of additional data have been published over the last 10 years. In this review, we describe the latest oxycodone data on special populations, including neonates, children, pregnant and lactating women, and the elderly. A lot of important drug interaction data have been published that must also be taken into account when oxycodone is used concomitantly with cytochrome P450 (CYP) 3A inducers and inhibitors and/or CYP2D6 inhibitors. In addition, we gathered data on abuse-deterrent oxycodone formulations, and the PK of alternate administration routes, i.e. transmucosal and epidural, are also described.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
There is extensive between-subject variation in the pharmacokinetics (PK) of oxycodone in preterm neonates and newborns, and their hepatic clearance (CL) is low and volume of distribution large. In elderly individuals, oxycodone CL is slightly lower and volume of distribution is slightly smaller compared with young adults. |
In pregnant or labouring women, oxycodone passes freely through the placenta; oxycodone concentrations are similar in maternal and fetal plasma. Low concentrations of oxycodone are found in mothers’ milk. |
Interactions with cytochrome P450 (CYP) 3A inhibitors and inducers with/without CYP2D6 inhibitors can substantially affect the PK and pharmacodynamics (PD) of oxycodone. These interactions must be taken into account in patients using, starting, or discontinuing CYP3A inducer(s) or inhibitor(s) with/without CYP2D6 inhibitor(s) during oxycodone treatment. |
Transmucosal administration is a feasible option in acute pain management for those patients with no intravenous line in place, while epidural administration is an option in those patients with an epidural catheter in place. However, further data are needed on the safety and efficacy of intrathecal oxycodone. |
1 Introduction
There has been a sharp increase in global oxycodone consumption since 1995, when a controlled-release (CR) tablet formulation was approved and launched for use in the following year. Twelve years later, in 2008, oxycodone consumption surpassed that of morphine. Much of the oxycodone consumption is from the US (73% of the world total), while other major consumer countries include Canada, Germany, Australia, France, China, the UK and Italy. For decades, oxycodone consumption per capita was similar in the US, Australia and Finland, but, during the last 15–20 years, use in the US and Australia has surpassed that in Finland by severalfold. In 2012, oxycodone consumption in Australia and the US was almost threefold and eightfold, respectively, higher than in Finland (Fig. 1) [1].

a Global consumption of total opioids, oxycodone, and morphine for the years 1992, 2002 and 2012. In 2012, oxycodone covered half of the total opioid use. b Oxycodone use per capita in the US, Australia and Finland for the years 1992, 2002 and 2012
Oxycodone was synthesized in 1916 and introduced for clinical use in 1917 in Germany [2]. It became available for use in the US in 1939. In Finland, oxycodone has been the most commonly used opioid analgesic since the 1960s, and, during the last 10 years, oxycodone has accounted for 70–80% of total opioid consumption in Finland [3]. Like other old drugs, oxycodone has not undergone the same approval process required by today’s standards. It was formally approved in Finland in 1973 and in the US in 1991.
To receive marketing authorisation for a medicine, applicants should provide a dossier containing data on the efficacy, safety and quality of the product. The efficacy data should contain pharmacokinetic (PK) data in healthy volunteers and patients with regard to absorption, distribution, metabolism, elimination, bioavailability and interactions. Data on adverse reactions are also required, and whether there is any reason to suspect that the adverse reaction is caused by the altered PK of the substance based on genetic PK variations. Data should also be provided for special patient groups [4]. Oxycodone has a long history of clinical use but without appropriate knowledge on its PK, with most of the oxycodone PK data originating from the 1990s.
The basic PK of oxycodone have been previously described (see, for example, Lalovic et al. [5] and Pöyhiä et al. [6]). In this review, our focus is on special populations, including children, pregnant and lactating women, the elderly, and those with liver/renal failure. We also review drug interactions with the principal cytochrome P450 (CYP) enzymes involved in oxycodone metabolism. In addition, we have gathered PK data on abuse-deterrent oxycodone formulations. For these purposes, we conducted a MEDLINE search to identify literature published in 2006 and beyond that was relevant to the PK of oxycodone.
2 Basic Pharmacokinetics (PK)
Oxycodone, 4,5α-epoxy-14-hydroxy-3-methoxy-17-methylmorphinan-6-one hydrochloride, is derived from the opioid alkaloid thebaine. It is a full opioid agonist, has two planar rings, two aliphatic rings and four chiral centres, and its molecular weight is 351.83 g/mol. It is hygroscopic, freely soluble in water and slightly soluble in ethanol [7].
Oxycodone is relatively selective for the µ-opioid receptor, although it can bind to other opioid receptors at higher doses. Opioid receptors are G-protein-coupled receptors, where the link with effector proteins is relayed via a G-protein. Oxycodone binding of the opioid receptor stimulates the exchange of guanosine-5′-triphosphate (GTP) for guanosine-5′-diphosphate (GDP) on the G-protein complex. This inhibits adenylate cyclase and prevents cyclic adenosine-monophosphate (cAMP) production. As a consequence, the release of nociceptive neurotransmitters, substance P, γ-aminobutyric acid (GABA), dopamine, acetylcholine, and noradrenaline are inhibited. Oxycodone also inhibits the release of vasopressin, somatostatin, insulin, and glucagon. Oxycodone closes N-type voltage-gated calcium channels and opens G-protein-coupled, inwardly rectifying potassium channels. Subsequently, neuronal excitability is reduced and hyperpolarisation occurs [8].
Oxycodone is extensively metabolised in the liver via CYP3A4/5 to noroxycodone (45%) and via CYP2D6 to oxymorphone (19%). Noroxycodone exhibits only weak antinociceptive potency; its affinity for µ-opioid receptors is one-third and potency five- to tenfold less than oxycodone. On the contrary, oxymorphone has a 10- to 45-fold higher affinity for µ-opioid receptors and is 8- to 30-fold more potent compared with oxycodone. Oxymorphone does not readily cross the blood–brain barrier (BBB) and, thus, in humans, the approximate oral equianalgesic dose ratio of oxymorphone:oxycodone is 1:2. Both noroxycodone and oxymorphone are further metabolised to noroxymorphone, which has a two- to fourfold higher affinity for µ-opioid receptors and is twofold more potent than oxycodone. However, noroxymorphone does not cross the BBB and is unlikely to contribute to the analgesic action of oxycodone. In general, the contribution of active metabolites to analgesia following oxycodone administration is thought to be clinically insignificant (Fig. 2) [5, 9].

Metabolism of oxycodone. Oxycodone is metabolised via CYP3A4 and CYP2D6 to noroxycodone, oxymorphone, and noroxymorphone. Oxycodol, noroxycodol and oxymorphol are reductive metabolites of oxycodone, noroxycodone and oxymorphone, with each of them having two stereoisomers (α and β). CYP cytochrome P450
The PK of oxycodone and its main metabolites in healthy subjects have been described in several reviews. Following intravenous administration, the median steady-state volume of distribution (Vdss) for oxycodone was 2.6 L/kg (range 1.8–3.7), the total plasma clearance (CL) was 0.82 L/min (range 0.4–1.3), elimination half-life (t½) was 3.0 h (range 1.8–9.7) and the noroxycodone:oxycodone (NOR:OXY) ratio for area under the concentration–time curve (AUC) was 0.3 (range 0.06–0.83) [10].
Following intramuscular administration of oxycodone 0.14 mg/kg, the mean maximum plasma concentration (Cmax) was 34 ng/mL (standard deviation [SD] 10), median time to Cmax (Tmax) was 1.0 h (range 0.5–1.5), CL was 0.78 L/min (SD 0.2), t½ was 4.9 h (SD 0.8) and NOR:OXY ratio was 0.45 (SD 0.23) [11].
After oxycodone 0.28 mg/kg oral liquid, the mean Cmax was 38 ng/mL (SD 14), median Tmax was 1 h (range 0.5–1), t½ was 5.1 h (SD 1.7), bioavailability relative to intramuscular administration was 0.6 (SD 0.2) and the NOR:OXY ratio was 0.64 (SD 0.24) [11]. After oral administration of an oxycodone 0.15 mg/kg immediate-release (IR) capsule, the mean Cmax was 26 ng/mL, median Tmax was 1.5 h (range 1–5), t½ was 3.8 h (SD 0.9), bioavailability relative to intravenous administration was 0.69 (SD 0.1) and the NOR:OXY ratio was 0.97 (SD 0.39) [12].
Oxycodone bioavailability with CR and IR tablets is similar to the IR solution [13, 14]; however, after administration of oxycodone IR tablets, the Cmax was twofold higher than with CR tablets, Tmax was earlier (1.3 and 2.6 h) and t½ was shorter with IR tablets than CR tablets (3 vs. 4–5 h, respectively) (Table 1) [14].
Oxycodone is 38–45% protein-bound, mainly to albumin, and oxycodone and its metabolites are excreted primarily via the kidney: oxycodone 9–11%, noroxycodone 23%, oxymorphone 10%, noroxymorphone 14%, reduced metabolites ≤ 18%. Oxycodone crosses the placenta freely and is excreted in human breast milk [5, 6, 15].
3 Cytochrome P450 2D6 Polymorphism and Oxycodone
The genetic polymorphism of CYP2D6 influences the PK of oxycodone [16]. Based on functional alleles, the population can be classified into four CYP2D6 phenotypes: poor metabolisers (PMs) with no functional alleles, intermediate metabolisers (IMs) with decreased functional alleles, normal metabolisers (NMs) with normal functional alleles, and ultrarapid metabolisers (UM) with increased functional alleles. Data on whether CYP2D6 phenotypes affect analgesic efficacy and adverse effects of oxycodone are inconsistent. In experimental settings in healthy subjects taking two oxycodone 10 mg capsules orally, the plasma oxymorphone:oxycodone ratio was lower in PMs compared with more rapid metabolisers (0.004 vs. 0.01, respectively). However, the variation in plasma concentrations at 1 h after ingestion was extensive (between 3.0 and 104 ng/mL in PMs and 0.1 and 72.7 ng/mL in others), indicating that there has also been significant variation in the absorption of oxycodone. In both groups, oxycodone induced a significant analgesic effect compared with placebo for all the pain variables tested. Analgesic efficacy was marginally less in PMs compared with others; the electrical stimulation pain detection threshold was increased compared with baseline (9% vs. 20%) as well as the tolerance thresholds (15% vs. 26%), the cold pressor test (14% vs. 26%), the pain summation threshold to repetitive electrical stimulation (20% vs. 26%), and the discomfort rating of the cold pressor test (17% vs. 23%) in PMs versus others, respectively [17]. Supporting data were presented by Samer et al. [18, 19], who reported that pain tolerance to electrical stimulation and cold correlated with CYP2D6 activity. However, the main limitation of that study was its small sample size, and only 10 subjects were tested: one PM, one IM, six NMs and two UMs [18, 19].
In clinical studies, no meaningful differences between the phenotypes have been found. In postoperative pain treatment in patients undergoing thyroidectomy, mastectomy or hysterectomy, there was no difference in intravenous oxycodone analgesia between PMs (n = 24) and other phenotypes (n = 246). The mean oxycodone consumption during the first 24 postoperative hours was 15 and 13 mg, respectively, in the two groups [20]. In patients having major abdominal surgery, supporting data were presented in eight PMs, 38 IMs, 70 NMs, and five UMs. In the early phase of the recovery, oxycodone consumption was marginally higher in PMs; however, after the first 12 postoperative hours, there was no difference in oxycodone consumption or pain scores between the CYP2D6 phenotypes [21].
4 Central Nervous System Penetration
The main site of analgesic action of oxycodone is in the central nervous system (CNS), the brain and the spinal cord. Oxycodone may also have some peripheral action that can contribute to analgesia, particularly in inflammatory and visceral pain conditions [22]. Pharmacokinetic/pharmacodynamic (PK/PD) models evaluating oxycodone analgesia in experimental visceral pain indicate a peripheral effect and show a direct linear drug–effect correlation between the plasma oxycodone concentration and visceral pain tolerance [23, 24].
To have CNS effects, oxycodone should penetrate the BBB. Experimental data indicate that oxycodone is actively transferred through the BBB. The concentration of unbound oxycodone is three- to sixfold higher in the brain than in the plasma [25,26,27]. Interestingly, the oxycodone brain:plasma ratio seems to be dose-dependent. In a recent study in mice, plasma and brain concentrations were measured after 6-day oxycodone infusions. The brain:plasma ratios were 3.1 in the low-dose group (20 mg/kg/day), 1.5 in the medium-dose group (45 mg/kg/day) and 1.0 in the high-dose group (120 mg/kg/day) [28].
A proton-coupled antiporter for organic cations in the endothelial cells has been proposed as the active influx transporter for oxycodone at the BBB [29,30,31]. Perivascular cells and pericytes mediate BBB properties but do not seem to be involved in oxycodone BBB distribution in rats [32]. Neurodegenerative disease is supposed to alter BBB function and drug delivery into the CNS; however, oxycodone transport at the BBB seems to not be affected in experimental models of neurodegenerative disease [33].
Permeability glycoprotein (P-gp) is one of the efflux proteins at the BBB. Data on whether oxycodone is a substrate of P-gp are conflicting. Earlier data indicated that coadministration with a P-gp inhibitor did not affect transport of oxycodone into the brain [34]. More recently, it has been shown that oxycodone may stimulate the P-gp activity in a concentration-dependent manner and that prolonged use of oxycodone may induce overexpression of P-gp [35].
Active transport across the BBB is supported by experimental data showing that the time to reach 50% equilibrium to the deep brain compartment for oxycodone is 7 min [27]. PD data support active transport. The onset of oxycodone analgesic action is relatively fast, i.e. 15 min after intravenous administration in rats [36] and 28 min in humans [37], both of which are faster than in morphine.
In experimental studies ethanol, (nor)ketamine and pregabalin have attenuated oxycodone tolerance without affecting oxycodone brain concentrations [28, 38,39,40]. In humans, cerebrospinal fluid (CSF) concentrations are often used as a surrogate parameter of CNS exposure of compounds. After intravenous oxycodone 0.1 mg/kg, Cmax, Tmax and t½ in CSF were 31 ng/mL, 1.1 h and 4.0 h, respectively. After intravenous administration, the AUCCSF; AUCplasma ratio of oxycodone was 1.2; however, after epidural administration, oxycodone exposure was significantly higher, Cmax and AUC in the CSF were 320- and 120-fold higher, respectively, compared with intravenous administration, and the AUCCSF:AUCplasma ratio of oxycodone after epidural administration was 110 [41]. A recent physiology-based PK model was able to predict the CSF concentration–time profile of oxycodone within a 1.6-fold error [42].
Oxycodone is metabolised via CYP2D6 into oxymorphone, which has a 10- to 45-fold higher affinity and 8- to 30-fold higher activation of the μ-opioid receptor compared with oxycodone [9]. Oxymorphone is suggested to have an active influx into the CNS as the oxymorphone concentrations in the brain are twofold higher than in the blood [43]. However, it is unlikely that oxymorphone accounts for the analgesic effect of oxycodone after peripheral administration because the plasma concentrations of oxymorphone produced by metabolism are negligible and the influx CL for oxymorphone is low compared with oxycodone [9].
Noroxycodone is the main primary metabolite of oxycodone metabolised via CYP3A4/5 and is further metabolised to the secondary metabolite noroxymorphone via CYP2D6. Noroxymorphone has a higher affinity to the µ-opioid receptor than oxycodone and is a more potent G-protein activator than oxycodone [9]. Experimental data show that after systemic administration, noroxymorphone does not have any analgesic efficacy, but after intrathecal administration it may be more potent than oxycodone [44]. Human and experimental data show that noroxymorphone is present at relatively high concentrations in circulation but does not appear to cross the BBB to any significant extent [41]. In conclusion, noroxymorphone is assumed to not contribute to the analgesic action of oxycodone after systemic administration.
Recent data suggest that CYPs are present and active in the brain, and that brain CYP2D6 may contribute to the metabolism of oxycodone. Moreover, smoking and ethanol consumption may increase CYP2D6 expression in the brain [45], which may have some impact on the PK and PD of oxycodone.
After intrathecal administration, the analgesic efficacy of oxymorphone is 40-fold higher and the intrathecal oxymorphone dose for 50% maximum potential effect (MPE) is significantly lower at 0.63 μg/kg, compared with oxycodone at 20 μg/kg [46]. Interestingly, inhibition of CYP2D6 with intracerebroventricular propranolol increases brain oxycodone concentrations and analgesia, whereas induction of CYP2D6 with nicotine increases brain oxymorphone concentrations but decreases analgesia [47]. Further studies may more precisely elucidate the role of brain CYPs on the PK and PD of oxycodone and its metabolites.
5 Oxycodone in Renal and Hepatic Insufficiency
Renal function has some effects on the PK of oxycodone as small amounts of oxycodone (approximately 10%) and the majority of metabolites are excreted via kidneys. Moreover, uraemia affects body composition by decreasing the lean tissue mass index and increasing the fat tissue index, which also have an impact on the PK [48]. Since publication of the study by Kirvelä et al. [49], only a few studies have evaluated the PK of oxycodone in renal insufficiency. In their study, Kirvelä et al. show that elimination of oxycodone is slightly prolonged in patients with renal deterioration compared with healthy controls (t½ median 3.9 vs. 2.3 h), due to increased Vd (4.0 vs. 2.4 L/kg) and reduced CL (0.8 vs. 1.1 L/min), and, most importantly, that the interindividual variation is extensive in those with renal insufficiency (t½ 1.8–25.7 vs. 1.3–4.0 h) [49].
An abuse-deterrent formulation of extended-release (ER) oxycodone (Remoxy®; Pain Therapeutics, Inc., Austin, TX, USA) was evaluated in patients with normal or mild, moderate and severely impaired renal function. Following oxycodone 20 mg orally, the Cmax (31.6 vs. 17.6 ng/mL) and AUC (494 vs. 211 ng·h/mL) increased up to two- to threefold in patients with declining renal function compared with those with normal renal function [50].
Oxycodone is extensively metabolised, and hepatic function significantly affects the PK. In patients undergoing hepatic transplantation, the median oxycodone Vdss after an intravenous dose of 0.05 mg/kg decreased from 3.1 L/kg (range 2.1–6.0) before surgery to 2.0 L/kg (range 0.9–5.9) after surgery, CL increased from 0.26 L/min (range 0.15–0.73) to 1.1 L/min (range 0.7–4.0), and t½ decreased from 14 h (range 5–24), similar to that reported in healthy subjects [3.4 h (range 2.6–5.1)]. AUC values were 3.7-fold higher before transplantation than after, and significantly more unchanged oxycodone was excreted in urine in end-stage cirrhosis [51]. It was later shown that even mild hepatic impairment significantly affects the PK of oxycodone. After administration of a CR oxycodone 20 mg tablet, Cmax was higher (25 vs. 17 ng/mL), t½ was longer (7.7 vs. 5.4 h) and AUC was also higher (387 vs. 199 ng·h/mL) in patients with hepatic impairment compared with healthy controls [52]. Consistent with this, in patients with mild to moderate liver insufficiency administered a tamper-resistant formulation of ER oxycodone (Remoxy®) 10 mg orally, Cmax increased from 7.6 to 13.1 ng/mL and AUC increased from 106 to 218 ng·h/mL when hepatic function declined from normal to moderate insufficiency [50].
In both renal and hepatic insufficiency, low elimination of oxycodone must be taken into consideration and close monitoring of patients is necessary when repeated doses are used. In patients with mild or moderate renal impairment, the initial dose should be 50–75%, and 50% of the standard starting dose in patients with severe kidney failure; a prolonged dosage interval is advised. Further doses should be adjusted according to the clinical situation. In patients with hepatic impairment, the initial dose should be 50% of the standard starting dose and either the dose interval should be longer or further doses smaller than standard doses. Patients must be closely monitored, and the dose should be adjusted based on clinical response.
In long-term use, drug formulation must also be considered as t½ and release of oxycodone from ER and CR tablets are prolonged. Patients with renal and hepatic function deterioration are more prone to oxycodone accumulation and adverse events in chronic use. However, recent data indicate that the PK parameters for a single dose of CR oxycodone-naloxone tablets in patients with end-stage renal disease are similar to those reported in subjects with normal renal function [53].
6 Sex Differences in the PK of Oxycodone
Women experience more pain and have more chronic pain than men and thus may need analgesics more often [54]. Recent data indicate significant differences between female and male structural and functional brain connectivity in regions involved in pain processing. These differences are already evident in neonates, and painful stimulus induces more widespread response in female newborns compared with male infants [55].
Sex differences in the PK of oxycodone have been sparsely evaluated. An early study with few subjects (n = 7) in each of four study groups indicated higher exposure of oxycodone and less metabolism to oxymorphone in women and the elderly compared with men and younger adults [56]. Most of the recent studies on oxycodone have not found this type of difference between women and men [57,58,59,60]; however, data on chronic oxycodone therapy show that the dose-adjusted Cmax value of oxycodone is lower, metabolism to oxymorphone is less, and the NOR:OXY ratio is higher in women compared with men [61, 62]. These findings support the earlier data by Kaiko et al. [56] and could reflect the fact that CYP3A4 expression is greater and/or phenotype is more active in women than men [63].
7 Oxycodone During Pregnancy and Lactation
Prolonged use of opioids during pregnancy can result in physical dependence and withdrawal syndrome in neonates. Opioid use during labour is also a concern as opioids cross the placenta freely and may produce respiratory and CNS depression in newborns. Low concentrations of oxycodone are detected in breast milk, and thus nursing mothers are advised to avoid oxycodone. The infant’s exposure depends on the mother’s plasma concentration and the amount of milk ingested. If opioid use is considered necessary, oxycodone use should be limited to a few days; a maximum daily dose of 30 mg orally is suggested [64].
Pregnancy and labour induce several physiological and anatomical changes in a female body that affect body composition and gastrointestinal, circulatory, renal and hepatic functions. These changes can have a profound effect on PK [65, 66]. Oxycodone has been evaluated in pregnant females in both experimental and clinical settings. In pregnant ewes, after administration of 0.1 mg/kg intravenous oxycodone, CL varied between 4.6 and 6.2 L·h/kg and Cmax varied between 50 and 74 ng/mL, but Vd varied significantly, between 1.5 and 4.7 L/kg. No accumulation of oxycodone was detected, and the fetal:maternal (F:M) ratio was between 0.8 and 2.0 after intravenous administration and between 0.5 and 1.0 after epidural administration. On the contrary, the active metabolite, oxymorphone, was noted to accumulate into the fetus after intravenous administration (median oxymorphone F:M ratio 2.1) but not after epidural administration (F:M ratio 1.3) [67].
The PK of oxycodone have been studied in early labour pain in full-term women with singleton pregnancy. After intravenous oxycodone 2–5 mg, Cmax ranged between 9 and 41 ng/mL. As Vd was less, i.e. 2.4 L/kg, than that reported in non-pregnant healthy subjects, t½ was relatively short at between 1.8 and 2.8 h. At the time of birth, a median of 7 h after the last dose of oxycodone, fetal exposure was low, the umbilical vein and arterial plasma oxycodone were between 0.1 and 14.8 ng/mL, and the F:M ratio was 0.9–1.5. No fetal adverse effects or accumulation of the metabolites to the fetal side were noted [68].
As oxycodone is a weak base (pKa 8.5) and is moderately protein bound (38–45%), it may accumulate in a mother’s milk (pH 7.2) [69]. Seaton et al. [70] administered oxycodone 30 mg rectally after caesarean section, and thereafter at 30–90 mg on day 1, ≤ 90 mg on day 2 and ≤ 50 mg on day 3. Oxycodone concentration in colostrum varied between 7 and 130 ng/mL at 24 h, 0 and 168 ng/mL at 48 h, and between 0 and 31 ng/mL at 72 h. The median colostrum:mother plasma ratio was 3.2:1 and there was a strong correlation between plasma and milk oxycodone concentrations at 24 h, with moderate correlation thereafter. However, oxycodone was detected in only 1 of 45 child plasma samples (concentration 7.4 ng/mL), which is consistent with the data indicating that during the first postpartum days, mothers produce small amounts of colostrum but not mature milk. Therefore, newborns are only exposed to small amounts of oxycodone. The amount of consumed colostrum/milk during the first days of life is low at < 4 mL/kg on day 1 and < 44 mL/kg on day 3 [71]; these small amounts of milk may not be enough to deliver any clinically significant amount of oxycodone to newborns.
However, if nursing mothers use oxycodone for a prolonged period of time, this is a risk to the health of the infant. In a retrospective analysis of nursing mothers using oxycodone, 20% (28/139) reported signs of CNS depression in their infants. Mothers who reported infant sedation were taking a higher dose of oxycodone (0.4 mg/kg/day) than mothers with asymptomatic infants (0.15 mg/kg/day). In addition, they were using oxycodone for a longer period of time (7 vs. 3 days) and were more likely to experience CNS depression themselves after oxycodone use (88%) than those with asymptomatic infants (58%). Thirty-eight of 39 mothers reported that neonatal symptoms ceased with maternal oxycodone discontinuation [72]. Sixty-seven of these mothers were genotyped for CYP2D6, CYP3A5, P-gp and the µ-opioid receptor encoding gene, OPRM1, polymorphisms; however, none of these were associated with oxycodone-induced CNS depression in newborns [73].
In summary, a few days of maternal use of oral oxycodone < 30 mg/24 h seems to be well tolerated by newborns; however, close monitoring of infants is necessary and if any signs of increased sleepiness, difficulty breastfeeding, breathing difficulties, or limpness are noted, a physician should be contacted immediately [64]. If oxycodone use is needed after the first few postpartum days, a safe approach is to instruct mothers not to undertake nursing while receiving oxycodone, or to do so under close supervision by a physician.
8 Oxycodone in Children
Both acute and chronic pain are common in children and effective pain treatment is needed [74]. Non-pharmacological methods are often highly effective and many painful conditions can be relieved with non-steroidal anti-inflammatory drugs (NSAIDs) with/without acetaminophen [75]. However, there is a need for opioid analgesics when NSAIDs are contraindicated [76] or when the efficacy of NSAIDs/acetaminophen is insufficient.
In acute pain management, oxycodone has been used in paediatric patients for several decades [77, 78], and per-oral oxycodone preparations, both IR and CR formulations, have recently been approved for paediatric patients ≥ 11 years of age [79, 80].
The PK and PD of oxycodone in children are generally similar to that in adults [80,81,82,84]. Older infants have a slightly faster CL per kilogram compared with adults, while neonates and younger infants have slower CL and extensive between-subject variation in PK parameters.
Oxycodone CL increases with age in the first 6 months following birth. An in vitro study by Korjamo et al. suggests that fewer metabolites of oxycodone are formed in infants than in older subjects [84], indicating that metabolism of oxycodone is not fully matured in neonates. This was also found in a PK study in infants aged < 6 months. Both CL and t½ were correlated to age, and excessive interindividual variation in the PK of oxycodone was found in the youngest infants aged ≤ 2 months [85]. In a later study, 79 infants with a gestational age of 23–42 weeks and postnatal age of 0–650 days received intravenous oxycodone 0.1 mg/kg during surgery [86]. Preterm neonates had the highest interindividual variation in PK parameters. In extremely preterm neonates, CL was lowest, Vd was highest, and thus t½ was longer at 6.8–12.5 h. Clear maturation in PK was observed, and, in infants aged 6–24 months, CL per kilogram was higher than in adults and thus t½ was also relatively short at 1.7–2.6 h. The metabolite profile in plasma and urine suggests that CYP3A-mediated N-demethylation is also the main elimination route in infants as noroxycodone was the major metabolite. The data indicated that renal CL is rather constant across age groups, but hepatic CL is significantly decreased in preterm, compared with full-term, newborns and older infants. In that study, similar to adults [5, 49], 12–13% of the dose was recovered as oxycodone in urine [86].
Two earlier studies evaluated the PK of oxycodone in children aged 6–93 months [82, 83]. These two studies indicate that in older infants, PK parameters are fairly similar to those reported in adults [81]. In these studies, transmucosal administration was also evaluated. Data indicate that after buccal and sublingual administration of oxycodone 0.1 mg/kg, the rate and extent of absorption is appropriate for clinical use. After transmucosal administration, Cmax was between 16 and 22 ng/mL, Tmax was 60 min, and bioavailability was 55% [82, 83]. Most children (24/30) had Cmax above the proposed minimum effective concentration (MEC) of oxycodone in children (12 ng/mL) and the analgesic plasma concentrations were sustained for 2.5–3 h. In a feasibility study, this administration route (oral transmucosal dosing) and dose (0.1 mg/kg) were found to be appropriate for children aged 5–14 years who presented at the hospital with acute abdominal pain [76]. These data indicate that oral transmucosal oxycodone is an appropriate dosage form for acute pain management in children with no intravenous access in place. By administering oxycodone orally, painful and frightening skin-penetrating injections can be avoided.
A population PK model combined the data of three previous studies [83, 85, 87] and indicated that CL matures rapidly after birth, and between-subject variability is pronounced in youngest neonates but is decreased in older infants and children. A population PK model was developed to evaluate optimal dosing regimens in order to keep the oxycodone plasma concentration between 10 and 50 ng/mL. Based on the modelling, the authors were able to give preliminary dosing recommendations for neonates of different age groups [86, 88].
Balyan et al. [89] evaluated the impact of genetic polymorphism of CYP2D6 on the PK of oxycodone in 30 children aged 2–17 years receiving oxycodone 0.07–0.1 mg/kg orally in the early postoperative period. Consistent with earlier data, delayed absorption was observed in some subjects, with a Tmax of 12 h indicating that absorption of swallowed oxycodone is erratic early after surgery and anaesthesia. All Cmax values of oxycodone were below 10 ng/mL, Cmax of oxymorphone was below 0.3 ng/mL, and t½ of oxycodone was 2.7 h. Patients with a more active CYP2D6 phenotype (n = 13 or 16) had higher concentrations of oxymorphone in comparison with phenotypes with less activity of CYP2D6 (n = 17 or 14). However, oxymorphone Cmax and oxymorphone:oxycodone ratios were low, between 0.01 and 0.05, in all three phenotypes (PMs, IMs and NMs) [89].
9 Oxycodone in the Elderly
In the elderly, pain is more common than among young adults. In elderly patients, NSAIDs are often contraindicated and the efficacy of acetaminophen is weak, thus opioid analgesics are often appropriate treatment in this population. Body composition undergoes changes and organ functions decline with advancing age, both of which can substantially impact the PK and PD of opioids.
Oxycodone exposure is increased with advancing age, but the exposure to metabolites seems to be similarly low across age groups [14, 90]. However, one study found a higher NOR:OXY ratio in patients 70 years of age or older after intravenous administration, but the number of subjects was too small to draw any firm conclusions [60] and no such difference has been shown after oral oxycodone administration.
After intravenous administration, oxycodone CL is reduced in the elderly (0.5–0.8 L/min vs. 0.8–1.2 L/min in the younger population), Vd is slightly smaller (3.2–3.7 L/kg vs. 3.7 L/kg), and, correspondingly, t½ is prolonged up to 5.0 h compared with 4.0 h and AUC is 38–80% higher than in young adults. After administration of intravenous oxycodone 5 mg, Cmax was 40–43 ng/mL in elderly patients, which is higher compared with young adult patients (29 ng/mL) [14, 60, 91]. Contradictory data have been presented. Villesen et al. [92] administered intravenous oxycodone 0.05 mg/kg to eight patients older than 70 years of age and found similar PK parameters compared with the younger population.
After oral administration of an oxycodone 10 mg IR capsule, Tmax was later in older patients (3.1–3.5 h vs. 1.9 h), Cmax was slightly higher (16–20 vs. 15 ng/mL), CL was reduced to 45–69%, t½ was prolonged (5.2–5.7 vs. 3.7 h) and AUC increased 50–80% compared with young adults [14, 60, 90]. The bioavailability of three enteral formulations of oxycodone (solution, IR capsule and CR tablet) was 55–64% in patients > 75 years of age [14], which is similar to that reported in the younger population [11, 93].
A recent study shows that oxycodone has a more significant ventilatory depressant effect in the elderly than in younger subjects. After administration of oral oxycodone 20 mg coadministered with ethanol, minute ventilation (MV), at a baseline of 8 L/min, decreased to 4.1 L/min (95% confidence interval) in the elderly, compared with 5.4 L/min (95% confidence interval) in young adults. Apnoeic events were more common among the elderly subjects after oxycodone administration, and this effect was augmented significantly in the elderly, but not in younger subjects, at ethanol plasma concentrations of 0.5 and 1.0 g/L [94].
In healthy elderly subjects, the PK and PD of oxycodone are only nominally affected, but in debilitated elderly patients caution is needed in oxycodone dosing. The decline of organ functions varies significantly, interindividual variation in the PK of oxycodone in the elderly appears to be large, and PD responses can be more significant than among young adults. These characteristics necessitate careful administration of oxycodone with low starting doses, longer dosing intervals and close follow-up of patients. In the elderly aged > 70 years, the starting dose should be 50% of the standard dose, and in those aged > 80 years the starting dose should be 30% of the standard dose [14, 60, 90, 92].
10 Oxycodone in Patients Undergoing Laparoscopic versus Open Surgery
Because most patients need opioid analgesics after surgery, CR oxycodone has been investigated as part of proactive analgesia [77]. Jokela et al. [95] coadministered CR oxycodone 15 mg and ibuprofen 800 mg orally 1 h prior to day-case gynaecological laparoscopic surgery; however, oxycodone did not provide better pain relief than placebo. All Cmax values for oxycodone were below 15 ng/mL, significantly lower than the proposed minimum effective analgesic concentration (MEAC) of 50 ng/mL for pain relief after laparoscopic surgery [96]. Furthermore, Tmax was also late at a mean of 5 h (range 2–8) [Table 2] [95].
The dosage of oxycodone was rather conservative in the study by Jokela et al. [95]. The data indicate that, on average, the Cmax for adult subjects after administration of a CR oxycodone 15 mg tablet was 17 ng/mL [14, 97]. This relatively low Cmax could also be explained by posture after ingestion, which affects the absorption of swallowed drugs [95]. After premedication, patients are often in the supine position, which is known to retard absorption [98]. Moreover, preoperative anxiety, anaesthesia and surgery are known to delay gastric emptying, and all these factors may further retard absorption of swallowed analgesics during the perioperative period [99].
Lenz et al. [100] compared intravenous oxycodone with intravenous morphine in patients who underwent laparoscopic hysterectomy. During the first 24 postoperative hours, the mean oxycodone consumption was significantly less (13 mg) than that of morphine (22 mg). The pain scores were also lower and patients were less sedated in the oxycodone group [100]. These PD data support the superiority of oxycodone in visceral pain management compared with morphine [37]. The type of surgery, laparoscopic or vaginal, may affect the severity of postoperative pain, but the type of anaesthesia, i.e. inhalation or intravenous, seems not to affect oxycodone consumption in hysterectomy patients [101, 102].
In breast surgery, the extent of surgery seems to affect the need for oxycodone. Cajanus et al. [103] evaluated the MEC of oxycodone after breast cancer surgery. In patients who did not need axillary evacuation, MEAC was 29 ng/mL, and 39 ng/mL with axillary evacuation; however, the extent of interindividual variation in the MEAC values was excessive (range between < 0.1 and 311 ng/mL) [103]. CYP2D6 and CYP3A4/5 polymorphisms and OPRM1 c.118A > G did not affect the analgesic concentration of oxycodone or the duration of analgesic action [104].
11 Cancer Pain and Cachexic Patients
Opioids are the mainstay of therapy in treating moderate to severe cancer pain. Pain is common in cancer patients—in the advanced stage of disease the pain prevalence is 75%. Moreover, 5–10% of cancer survivors have severe chronic pain [105].
Opioids are associated with a high incidence of opioid-induced bowel dysfunction (OIBD). Evidence indicates that in selected patients, a CR oxycodone/naloxone tablet can be useful in reducing OIBD; positive effects on OIBD are seen after 7 days of use. Moreover, combining naloxone with oxycodone seems to have a carry-on effect on OIBD [106, 107].
In long-term treatment, CR formulations provide predictable plasma concentrations. As the PK of oxycodone are dose-independent, exposure to oxycodone after administration of a CR tablet is dose-related—after doses of 5, 10, 20 and 40 mg, Cmax is approximately 5, 10–12, 21–23 and 39–47 ng/mL, respectively. After multiple doses of 10 and 40 mg, Cmax is 15 and 57 ng/mL, respectively, and trough concentrations are dose-related, i.e. 6–7 and 25 ng/mL after 10 and 40 mg CR tablets, respectively. Compared with IR formulations, the Cmax for CR formulations is significantly lower and Tmax is later, but AUC values and trough concentrations before the next dose are similar. Combining naloxone with oxycodone in the same CR tablet seems to not affect the PK of oxycodone [5, 80, 108, 109].
Compared with acute pain management and the treatment of non-malignant pain, in opioid-tolerant cancer patients oxycodone doses and plasma concentrations can be excessive, but still dose-related, i.e. 350 ng/mL after 200 mg/day and up to 500 ng/mL after 400 mg/day [110].
Cancer cachexia influences drug metabolism, including reduced CYP450 activity [111]. Cachectic patients experience a decrease in CYP3A enzymatic activity but no major change in CYP2D6 activity. This results in lower plasma concentrations of the main metabolite noroxycodone, and increased oxycodone and oxymorphone exposure [111,112,114]. Cachexia is also characterised by hypoalbuminemia [115]. In the study by Naito et al. [112], there was a correlation between the severity of cachexia and the PK of oxycodone; the median NOR:OXY ratio at 12 h after oxycodone dosing decreased from 1.33 in patients with C-reactive protein (CRP) < 10 mg/L and serum albumin > 35 g/L, to 0.26 in patients with CRP > 10 mg/L and albumin < 35 g/L, who were later classified as having more severe cachexia. There was also a weak inverse correlation between serum albumin and the protein-free oxycodone fraction (r = − 0.3). The median serum albumin concentration was 35 g/L and the median protein-bound oxycodone was 37%, similar to that reported in healthy subjects [7, 112, 116].
The effect of the different CYP2D6 phenotypes (PMs, IMs, NMs and UMs) on the PK of oxycodone has been evaluated in cancer patients. Similar to the non-cancer population [18], NM and UM cancer patients have comparable oxycodone concentrations, but oxymorphone concentrations are higher than in PMs. Nevertheless, no difference in adverse effects was noted between phenotypes [117].
Heiskanen et al. [110] evaluated whether oral fluid concentrations can be used to estimate oxycodone exposure in cancer patients. The mean oral fluid:plasma ratio was 14.9 and there was great interindividual variation in the concentrations. The data indicated that oral fluid is not an appropriate media for estimating oxycodone exposure [110].
12 Oxycodone in Neuropathic Pain
Neuropathic pain is caused by damage or dysfunction in peripheral nerves or in the CNS. It often becomes chronic and can substantially reduce quality of life. Unfortunately, neuropathic pain is challenging to treat efficiently. Traditional analgesics often lack sufficient efficacy, and antidepressants and anticonvulsants are often used to treat neuropathic pain [118]. Opioids can be used to treat neuropathic pain and are effective in some patients, although there is concern regarding intolerable adverse effects in long-term use.
Experimental studies indicate that oxycodone alleviates symptoms of neuropathic pain. In a recent study on vincristine-induced peripheral neuropathic pain rats, both oxycodone and morphine as a single injection attenuated allodynia and hyperalgesia, but oxycodone was more potent on static mechanical hyperalgesia and dynamic mechanical hyperesthesia. Moreover, oxycodone maintained a significant analgesic efficacy after repeated doses, but multiple morphine injections led to an attenuation of analgesic efficacy [119]. Supporting data on the superiority of oxycodone was observed in a mouse femur bone cancer model. In the study by Nakamura et al. [120], some decrease in oxycodone-induced G-protein activation in brain in tumour-implanted mice was noted, but the morphine effect was significantly and more severely attenuated. No difference was found in µ-opioid receptor activation in the spinal cord, and binding into µ-opioid receptors was similar for both compounds. Analgesic potency was also greater for oxycodone. After intracerebroventricular administration, oxycodone dose-dependently ameliorated pain-related responses, while analgesic effects of morphine were less apparent [120].
In humans, oxycodone has been evaluated in postherpetic neuralgia and diabetic neuropathy. In a crossover study comparing CR oxycodone 45 mg/day and placebo in postherpetic neuralgia, oxycodone provided significantly lower pain and disability scores, but also led to a greater number of opioid-related adverse effects [121]. Similar superior pain relief has been reported in diabetic neuropathy compared with placebo [122] and benztropine [123]. In the study by Watson et al., the mean daily dose of CR oxycodone was 40 mg and benztropine was 1.2 mg during 4–6 weeks of follow-up [123]. Combining CR oxycodone with gabapentin in patients who were still experiencing moderate or severe diabetic neuropathy pain, reduced pain by 33% from baseline, significantly more than combining placebo with gabapentin treatment [124]. Nausea, somnolence and constipation have been common adverse effects in these oxycodone groups [121,122,124]. In an observational, retrospective study on spinal cord injury pain, oxycodone combined with an anticonvulsant seemed to decrease pain and increase quality of life without severe adverse effects [125].
13 Oxycodone in Neuraxial Anaesthesia
Opioids are highly effective in spinal analgesia. Morphine and sufentanil are two opioids approved for intraspinal use, but fentanyl is also commonly used, although not formally approved, for this administration route. Hydrophilic morphine provides more long-lasting analgesia than lipophilic sufentanil. Oxycodone is another hydrophilic opioid. It also has other physiochemical properties similar to morphine, but has received less attention in neuraxial use [77].
In early experimental studies, oxycodone was found to be less potent than morphine after intrathecal administration, with a potency ratio of 1:14. The onset of analgesia was faster, but the duration of analgesia was shorter, with oxycodone compared with morphine [36, 126]. These experimental data were supported by clinical studies showing that in epidural analgesia, a two- to tenfold higher dose of oxycodone is needed to provide similar analgesia as morphine [127, 128].
Contrary to early reports, recent data indicate that epidural oxycodone is a highly effective administration route for providing analgesia in postoperative pain management. A recent study shows that laparotomy patients receiving an epidural injection of oxycodone require less rescue analgesia and have lower pain scores than those receiving oxycodone intravenously [129]. CSF samples from patients receiving epidural oxycodone show that the AUC of oxycodone is > 100-fold higher, and Cmax values 320-fold higher, than in CSF samples from patients receiving oxycodone intravenously [41]. Moreover, oxycodone concentrations in CSF after epidural oxycodone were 1000- to 10,000-fold higher than those of oxymorphone, and only negligible concentrations of noroxymorphone could be detected in CSF. This suggests that oxycodone provides the analgesic effect rather than the active metabolites [41, 129]. In post caesarean section pain, 3 mg of epidural oxycodone or morphine provided similar pain relief at rest, but dynamic pain control was inferior with oxycodone. This indicates that in epidural administration, the potency ratio between oxycodone and morphine is less than 1:1 [130].
Tissue toxicity is a concern in the intrathecal administration of compounds. In human neuroblastoma cells and mouse motoneuronal cells, it was shown that the neurotoxicity of oxycodone is similar or less than that with morphine [131]; however, further data are needed on the optimal dose and safety of epidural use of oxycodone. No human data on spinal administration of oxycodone are available.
14 Transmucosal Oxycodone
Mucous membrane drug permeability is based on the relative thickness and degree of keratinisation, as well as the blood supply of mucous membranes. The extent of drug delivery is also affected by the physicochemical properties of the administered drug. Oral mucosal membrane delivery bypasses gastrointestinal and hepatic presystemic elimination [132].
Oxycodone is a weak base (pKa 8.5) and, at higher pH, the majority of oxycodone is unionised (95% at pH 9.0), whereas, at a lower pH, more oxycodone was protonated. In an experimental study, the effect of pH on the PK of sublingual oxycodone spray was evaluated. Bioavailability was higher for a formulation with a pH of 9.0 (70%) than a formulation with a pH of 4.0 (45%) [132].
In an early study on healthy adult subjects, sublingual oxycodone was found to have a low bioavailability of < 20% [133]. However, this poor absorption has not been confirmed in later studies. In adults, nasal oxycodone 0.1 mg/kg was readily absorbed, with a median Cmax of 13 ng/mL (range 9–17), Tmax of 25 min (range 20–240) and bioavailability of 46% (range 0.3–67) [134].
Transmucosal administration of oxycodone is feasible in children who do not undergo intravenous administration as any skin-penetrating injections may cause unnecessary suffering. In children aged 6–93 months, buccal mucosal absorption of oxycodone solution was reliable, with a relatively high bioavailability of 55% (22–84) [83]. Oral mucosal membranes vary in thickness and keratinisation; the sublingual mucosa of the oral cavity is more permeable than the buccal mucosa [135]. In children, the PK of sublingual oxycodone 0.2 mg/kg seem to be superior compared with buccal administration; Cmax is 38% higher [22 ng/mL (range 5.5–42) vs. 16 ng/mL (range 5.4–39)] and AUC 31% higher (5500 ng·min/mL vs. 4200 ng·min/mL). An analgesic concentration of oxycodone 12 ng/mL was also achieved faster (45 vs. 52 min) and was sustained for a longer period of time (163 vs. 133 min) after sublingual compared with buccal administration. The NOR:OXY ratio was 0.78 in both groups [83].
In recreational use, oxycodone CR tablets have been crushed and snorted to the nasal mucosa. After tampering with CR formulations, the bioavailability of snorted oxycodone was high (75–78%) [136]. The new tamper-resistant oxycodone CR formulations make recreational use of oxycodone via the mucosal route inconvenient and irritating [137, 138]. In addition, the tamper-resistant formulation results in a reduced rate and extent of oxycodone absorption during the first hours after administration, with the hope of less drug-liking [138].
15 Abuse-Deterrent Formulations
Although somewhat less than that of heroin and fentanyl [139], the abuse and misuse liability of oxycodone is high. Potential routes for abuse are ingestion, injection, snorting and smoking. Abuse potential might be increased with physical manipulation, e.g. by cutting, breaking, chewing, crushing, or dissolving the formulation. To reduce opioid abuse, CR and deterrent oxycodone formulations have been developed [140].
Unfortunately, the CR oxycodone formulation is appealing to drug abusers since it contains a higher amount of opioid [137, 141]. The original CR tablets were manipulated to achieve IR properties and thus higher drug-liking. With regard to opioid abuse and misuse, the most interesting PK parameters are Cmax and Tmax. After oral administration of 40 mg of intact CR oxycodone, Cmax was 47 ng/mL and Tmax 4.5 h. These CR properties were lost when the drug was crushed; a higher Cmax (80 ng/mL) was reached in 1.7 h [97, 142]. This rapid effect was similar with IR oxycodone and drug-liking was also similar.
An abuse-deterrent formulation of CR oxycodone has been developed. After crushing and after intranasal administration, 30 mg of the current formulation of oxycontin (Pudrue Pharma, Stamford, CY, USA) had a lower Cmax (29 ng/mL vs. 60 ng/mL) and later Tmax (2.6 h vs. 1.1 h) compared with the crushed original CR formulation. Drug-liking was also less with the abuse-deterrent formulation [137, 138].
One approach to prevent abuse is to combine an antagonist with oxycodone in the same formulation. The Troxyca ER capsule (Pfizer Inc, New York, NY, USA) contains pellets, whereas oxycodone is surrounded by sequestered naltrexone at a ratio of 100:12. When taken as directed, the naltrexone remains sequestered and patients receive oxycodone in an ER manner. When the pellets are crushed, the sequestered naltrexone is released to counteract the effects of oxycodone [143, 144]. However, crushed Troxyca ER 60/7.2 mg has similar PK as crushed IR oxycodone 60 mg, with a Cmax of 112 vs. 87 ng/mL, Tmax of 0.6 vs. 1 h, and t½ of 4.4 vs. 4.2 h after oral administration. In comparison, intact Troxyca ER 60/7.2 mg has a Cmax of 29 ng/mL, Tmax of 12 h, and t½ of 9.3 h [142]. After snorting, the crushed Troxyca ER 30/3.6 mg formulation has a lower Cmax than crushed IR oxycodone 30 mg (56 ng/mL vs. 80 ng/mL) and later Tmax (1.6 vs. 0.5 h), but similar t½ (4.1 vs. 4.2 h). Despite the similar PK, the crushed Troxyca ER formulation has lower ‘drug-liking and high’ scores after both oral and nasal routes than similar doses of the crushed IR oxycodone formulation [143, 144].
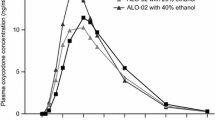
Coadministration of ethanol with abuse-deterrent ER oxycodone formulations may increase oxycodone exposure. When an oxycodone-naloxone capsule was administered with 20% ethanol 240 mL, the PK parameters were similar to administration with water, but when coadministered with 40% ethanol 240 mL, Cmax was higher (15 ng/mL vs. 11 ng/mL), Tmax was shorter (8 vs. 12 h) and AUC was modestly higher (221 vs. 191 ng·h/mL) [145].
CR oxycodone combined with naloxone (2:1) is one of the most widely used tablet formulations of oxycodone. PK parameters are similar after intravenous administration of oxycodone alone or combined with naloxone, but drug-liking is significantly less and is similar to placebo when oxycodone is coadministered with naloxone [146].
Another deterrent method is to use a physical or chemical barrier that provides resistance to mechanical alteration. Xtampza ER (Collegium Pharmaceutical, Inc., Canton, MA, USA) is a microsphere-in-capsule formulation, designed to retain its CR properties following tampering [142]. Crushing does not significantly change the PK parameters of Xtampza ER; after 40 mg orally, Cmax was 65 ng/mL for the intact Xtampza ER capsule, and 60 ng/mL for the crushed capsule, while Tmax was 4 h and 4 h, respectively. For the crushed IR oxycodone tablets, these values were 79 ng/mL and 2 h, respectively. After intranasal administration of crushed Xtampza ER 40 mg, Cmax was 30 ng/mL and Tmax was 5 h, both significantly different than after crushed IR oxycodone (Cmax 61 ng/mL and Tmax 2.6 h, respectively) [142, 147]. Drug-liking after crushed Xtampza ER is correspondingly low [148].
One abuse-deterrent IR oxycodone product has also been approved. RoxyBond (Inspirion Delivery Sciences, Valley Cottage, NY, USA) contains layered inactive ingredients to make it resistant to physical manipulation, but does not prevent absorption. If the formulation is dissolved, it forms a viscous material that resists passage through a needle. The physicochemical properties make the compound more difficult to prepare solutions suitable for intravenous injection. The absorption of an intact RoxyBond 30 mg tablet is slower compared with a traditional IR tablet; Cmax 58 vs. 68 ng/mL, Tmax 1.0 vs. 1.8 h, and AUC 287 vs. 305 ng·h/mL. Moreover, after intranasal administration of a crushed RoxyBond 30 mg tablet, Cmax was lower (43 ng/mL) and Tmax was longer (2.3 h). Drug-liking was also lower for intranasal crushed RoxyBond [149].
16 Drug Interactions
Oxycodone undergoes extensive hepatic conjugation and oxidative degradation to metabolites that are excreted mainly in urine as conjugated and unconjugated metabolites; the amount of free and conjugated oxycodone in urine is 9–11% [16]. Oxycodone is metabolised via CYP3A-mediated N-demethylation to noroxycodone, noroxymorphone, and α- and β-noroxycodol, and via CYP2D6-mediated O-demethylation to oxymorphone and α- and β-oxymorphol [9]. CYP3A-mediated N-demethylation to noroxycodone is the primary metabolic pathway, accounting for 45% of the dose, whereas CYP2D6-mediated O-demethylation to oxymorphone and noroxymorphone accounts for 19% of the dose. Less than 10% of oxycodone is reduced to α- and β-oxycodol by 6-ketoreduction. The enzymes responsible for the ketoreduction and glucuronidation pathways in oxycodone metabolism have not been established [5]. Due to CYP-based metabolism, oxycodone is prone to drug interactions that must be taken into account in patients currently taking or discontinuing CYP3A inducers or inhibitors and CYP2D6 inhibitors during oxycodone use (Fig. 2).
Since the CYP3A4 isoenzyme plays a major role in the metabolism of oxycodone, drugs that alter CYP3A4 activity may cause significant changes in the CL of oxycodone that could lead to profound and clinically important changes in oxycodone plasma concentrations.
Concomitant use of CYP3A inhibitors, such as clarithromycin [150] and telithromycin [151], itraconazole [152], ketoconazole [18, 153], miconazole [154], and voriconazole [155], and ritonavir [156], decrease oxycodone CL, increase both Cmax and AUC values, and induce significant PD effects. On the contrary, CYP3A inducers, such as rifampin [12] and St John’s wort [157], increase oxycodone metabolism and therefore increase CL. As a result, oxycodone plasma concentrations are lower and PD responses are attenuated.
Coadministration of CYP2D6 inhibitors such as quinidine [18, 158] and paroxetine [153, 158,159,161] affects oxycodone metabolism less than CYP3A inducers and inhibitors. However, concomitant use of CYP2D6 inhibitors with CYP3A4 inhibitors may cause more pronounced effects of oxycodone than a single compound (Table 3) [18, 154, 156, 159, 160].
These drug–drug interactions seem to similarly affect young and old adults [150, 152]. The interaction potential seems to be more significant when oxycodone is administered orally, indicating some first-pass metabolism in the intestine [11, 12, 160].
If coadministration is necessary, caution is advised when initiating oxycodone treatment in patients currently taking, starting or discontinuing CYP3A4 inhibitors or inducers. These patients should be evaluated more frequently, and dose adjustments should be considered until stable drug effects are achieved. Coadministering CYP2D6 inhibitors can decrease oxycodone CL, but, in general, have less significant effects on the PK of oxycodone.
Amitriptyline and tramadol do not affect the PK of oxycodone, thus oxycodone can be safely used in patients taking amitriptyline [11, 162].
17 Coadministration of Oxycodone with Ethanol
Ethanol is frequently coabused with opioids and detected at postmortem samples in opioid-related deaths [94, 163, 164]. Both ethanol and opioids cause respiratory depression and oversedation, potentially lethal adverse effects, and synergism can be supra-additive. Expanding the use of oxycodone has led to a higher count of unintentional overdose deaths [164]; simultaneous use of alcohol may further add to this risk [165].
In healthy opioid-naive volunteers, an oral oxycodone 20 mg IR tablet and ethanol administered intravenously, to a breath ethanol concentration of 0.5 or 1.0 g/L, caused more profound respiratory depression compared with administration of ethanol or oxycodone alone. After oxycodone, MV decreased from a baseline of 8.0 L/min to 5.8 L/min, and oxygen saturation (SpO2) decreased from 98–99% to 92–94%. When coadministered with ethanol, the extent of changes was more apparent: MV decreased to 5.1 and 4.7 L/min, and SpO2 decreased to 87–92% and 86–93% in the lower and higher ethanol groups, respectively. Similar changes were also observed in the respiratory rate. After oxycodone, the mean respiratory rate decreased to 11/min; after oxycodone + ethanol 0.5 g/L, the mean respiratory rate decreased to 10/min; and after oxycodone + ethanol 1.0 g/L, the mean respiratory rate decreased to 9/min. In this study, coadministration of ethanol and oxycodone did not induce more sedation than oxycodone alone. Elderly subjects (66–77 years of age) were more prone to respiratory depression than younger subjects (21–28 years of age). Apnoea episodes were also more common in the elderly [94].
Ethanol in combination with an ER opioid formulation can result in accelerated release of oxycodone. It is not only the ethanol exposure that seems to be important, but also the concentration. When an oxycodone-naloxone capsule was administered with 40% ethanol, Cmax was higher (15 ng/mL vs. 11 ng/mL) and Tmax was shorter (8 vs. 12 h) compared with co-ingestion with water or 20% ethanol [145].
Recent experimental data suggest that ethanol may reverse oxycodone tolerance. A single injection of ethanol 1.0 g/kg administered intraperitoneally to mice reversed oxycodone-induced antinociceptive tolerance [40]. In another mice study, coadministration of intraperitoneal ethanol 0.3 g/kg, a dose that did not cause respiratory depression alone, with a challenge dose of morphine 10 mg/kg, caused greater respiratory depression in opioid-tolerant mice than morphine alone [28]. Interestingly, these results indicate that tolerance to nociception and respiratory depression in regular oxycodone users is reversed with a low dose of ethanol, highlighting the potential risk of concomitant use of these substances. The mechanism underneath remains unclear. Ethanol did not affect the brain concentrations of oxycodone [40]. In the study by Hill et al. [28], inhibition of protein kinase C with calphostin C reversed tolerance to respiratory depression; however, a combination of calphostin C and ethanol did not decrease tolerance more than either of the compounds alone, suggesting that ethanol might be influential via inhibition of protein kinase C [28].
18 Discussion
Since its introduction into clinical use in 1917, the increase of oxycodone consumption was rather slow for many decades. When the CR oxycodone formulation was approved in the mid-1990s, oxycodone use has increased sharply, and 10 years ago oxycodone consumption surpassed that of morphine; in 2015, global use of oxycodone was twofold that of morphine [1]. Oxycodone has a twofold higher bioavailability after oral administration than morphine, is likely more potent for visceral pain, has superior CNS PK, and may cause less adverse effects. These are some of the reasons why oxycodone, particularly oral formulations, has become popular [15].
One of the important messages from this review is that oxycodone is prone to interactions with CYP3A4 inducers and inhibitors. During the last decade, several studies have described significant interactions that must be taken into account in clinical pharmacotherapy. The use of CYP3A inhibitors with oxycodone, such as certain macrolide antibiotics [150, 151], antifungals [18, 151,152,153,155], and the antiviral ritonavir [156], increase oxycodone exposure substantially and induce more significant opioid effects. On the contrary, CYP3A inducers, such as rifampin [12] and St John’s wort [157], increase oxycodone metabolism. As a result, oxycodone plasma concentrations are low and PD responses are attenuated [18]. CYP2D6 inhibitors [18, 153, 158,159,161] affect oxycodone metabolism less than CYP3A inducers and inhibitors, but concomitant use of CYP2D6 inhibitors with CYP3A4 inhibitors may cause more pronounced effects of oxycodone than a single compound [18, 153, 155, 159, 160].
The main effect site of opioids for their analgesic action is the CNS, in the brain and spinal cord. Thus, CNS PK are an important parameter when comparing different opioid analgesics. Experimental data on oxycodone indicate that oxycodone is actively transported through the BBB [9]. This is a likely justification for high oxycodone analgesic action, despite relatively low µ-receptor binding and activity [9, 29,30,31]. Recent data have also implied, contrary to earlier studies, that epidural oxycodone could be highly effective and feasible for epidural administration. After epidural administration, oxycodone is rapidly detected in CSF and analgesic efficacy is superior compared with intravenous oxycodone [41, 42, 129]. Cell models suggest that the neurotoxicity of oxycodone is similar or less than that of morphine, but further studies are warranted [131].
New data in vulnerable patient groups have emerged. Oxycodone crosses freely through the placenta and is excreted into the mother’s milk; however, lower doses of oxycodone may be used for labour pain and during the first days after delivery [68, 70]. Moreover, newborns should be monitored for any signs or symptoms of drowsiness, lethargy, or other signs of CNS depression, and any prolonged use of oxycodone beyond the first 3 days after delivery should only be under close supervision of the mother and child. If oxycodone causes maternal drowsiness, it is a warning sign that the newborn is also affected [72]; therefore, the dose of oxycodone and the duration of therapy during breastfeeding should be limited.
Pain in the elderly is common and non-opioid analgesics are often insufficient and, in many patients, contraindicated. In the elderly, oxycodone CL is reduced and the AUC of oxycodone is larger than in young adults. These changes are more pronounced in frail elderly patients and dose reductions to 50%, from standard doses, as well as dosing interval prolongation, are needed [14, 60, 90, 92].
Children, including preterm neonates and infants, need effective analgesia and evaluated medicine [78]. The PK of oxycodone are not fully matured in neonates. In preterm neonates, hepatic CL was low, Vd was relatively high, and thus t½ was longer in extremely preterm neonates (up to 12.5 h) and with high between-subject variation [85, 87]. Oxycodone CL matures during the first 6 months following birth, and, in older infants, the PK of oxycodone are fairly similar to adults. Population PK models enabled dosing recommendations to be determined for preterm neonates to older children [81, 86].
Misuse and addiction are concerns related to oxycodone, as with any other opioid analgesic. New abuse-deterrent oxycodone formulations have been developed, and these formulations may delay oxycodone release from the tablet, providing steady concentrations and avoiding peaks and troughs [140]. In chronic opioid use, these tamper-resistant formulations should be preferred in order to achieve steady analgesia and avoid leakage of opioids resulting in misuse and disuse.
19 Conclusions
Oxycodone has high analgesic efficacy in severe acute or long-term pain conditions. With careful dose selection and close patient follow-up, oxycodone is well-tolerated in different patient groups. Analgesia is based on the parent drug, and as concentrations of active metabolites are very low, they are less likely to contribute. As oxycodone is mainly metabolised in the liver, the effect of interactions must be known. Preliminary data indicate that oxycodone could also be feasible for intrathecal use and transmucosal sublingual administration is feasible in acute pain management.
References
Pain and Policy Studies Group. Opioid Consumption Data. Available at: https://ppsg-chart.medicine.wisc.edu/. Accessed 1 Oct 2018.
Falk E. Eukodal, ein neues Narkoticum. Munchener Medizinische Wochenschrift. 1917;20:381–4.
Pöyhiä R. Opioids in anaesthesia: a questionnaire survey in Finland. Eur J Anaesthesiol. 1994;11(3):221–30.
Pharmacokinetic studies in man. Available at: https://www.ema.europa.eu/documents/scientific-guideline/pharmacokinetic-studies-man_en.pdf. Accessed 1 Oct 2018.
Lalovic B, Kharasch E, Hoffer C, Risler L, Liu-Chen LY, Shen DD. Pharmacokinetics and pharmacodynamics of oral oxycodone in healthy human subjects: role of circulating active metabolites. Clin Pharmacol Ther. 2006;79(5):461–79.
Pöyhiä R, Vainio A, Kalso E. A review of oxycodone’s clinical pharmacokinetics and pharmacodynamics. J Pain Symptom Manage. 1993;8(2):63–7.
Pöyhiä R, Seppälä T. Liposolubility and protein binding of oxycodone in vitro. Pharmacol Toxicol. 1994;74(1):23–7.
McDonald J, Lambert DG. Opioid receptors. BJA Education. 2015;15(5):219–24.
Kokki H, Kokki M. Central nervous system penetration of the opioid oxycodone. In: Preedy VR (ed). Neuropathology of drug addictions and substance misuse, vol. 3, 1st ed. New York: Academic; 2016. ISBN:9780128006344.
Pöyhiä R, Olkkola KT, Seppälä T, Kalso E. The pharmacokinetics of oxycodone after intravenous injection in adults. Br J Clin Pharmacol. 1991;32(4):516–8.
Pöyhiä R, Seppälä T, Olkkola KT, Kalso E. The pharmacokinetics and metabolism of oxycodone after intramuscular and oral administration to healthy subjects. Br J Clin Pharmacol. 1992;33(6):617–21.
Nieminen TH, Hagelberg NM, Saari TI, Pertovaara A, Neuvonen M, Laine K, et al. Rifampin greatly reduces the plasma concentrations of intravenous and oral oxycodone. Anesthesiology. 2009;110(6):1371–8.
Mandema JW, Kaiko RF, Oshlack B, Reder RF, Stanski DR. Characterization and validation of a pharmacokinetic model for controlled-release oxycodone. Br J Clin Pharmacol. 1996;42(6):747–56.
Kokki M, Välitalo P, Rasanen I, Aaltomaa S, Ojanperä I, Eskelinen M, et al. Absorption of different oral dosage forms of oxycodone in the elderly: a cross-over clinical trial in patients undergoing cystoscopy. Eur J Clin Pharmacol. 2012;68(10):1357–63.
Kalso E. Oxycodone. J Pain Symptom Manage. 2005;29:47–56.
Söderberg Löfdal KC, Andersson ML, Gustafsson LL. Cytochrome P450-mediated changes in oxycodone pharmacokinetics/pharmacodynamics and their clinical implications. Drugs. 2013;73(6):533–43.
Zwisler ST, Enggaard TP, Noehr-Jensen L, Pedersen RS, Mikkelsen S, Nielsen F, et al. The hypoalgesic effect of oxycodone in human experimental pain models in relation to the CYP2D6 oxidation polymorphism. Basic Clin Pharmacol Toxicol. 2009;104(4):335–44.
Samer CF, Daali Y, Wagner M, Hopfgartner G, Eap CB, Rebsamen MC, et al. The effects of CYP2D6 and CYP3A activities on the pharmacokinetics of immediate release oxycodone. Br J Pharmacol. 2010;160(4):907–18.
Samer CF, Daali Y, Wagner M, Hopfgartner G, Eap CB, Rebsamen MC, et al. Genetic polymorphisms and drug interactions modulating CYP2D6 and CYP3A activities have a major effect on oxycodone analgesic efficacy and safety. Br J Pharmacol. 2010;160(4):919–30.
Zwisler ST, Enggaard TP, Mikkelsen S, Brosen K, Sindrup SH. Impact of the CYP2D6 genotype on post-operative intravenous oxycodone analgesia. Acta Anaesthesiol Scand. 2010;54(2):232–40.
Stamer UM, Zhang L, Book M, Lehmann LE, Stuber F, Musshoff F. CYP2D6 genotype dependent oxycodone metabolism in postoperative patients. PLoS One. 2013;8(3):e60239.
Ruan X, Mancuso KF, Kaye AD. Revisiting oxycodone analgesia: a review and hypothesis. Anesthesiol Clin. 2017;35(2):e163–74.
Olesen AE, Upton R, Foster DJ, Staahl C, Christrup LL, Arendt-Nielsen L, et al. A pharmacokinetic and pharmacodynamic study of oral oxycodone in a human experimental pain model of hyperalgesia. Clin Pharmacokinet. 2010;49(12):817–27.
Olesen AE, Kristensen K, Staahl C, Kell S, Wong GY, Arendt-Nielsen L, et al. A population pharmacokinetic and pharmacodynamic study of a peripheral kappa-opioid receptor agonist CR665 and oxycodone. Clin Pharmacokinet. 2013;52(2):125–37.
Boström E, Simonsson US, Hammarlund-Udenaes M. In vivo blood-brain barrier transport of oxycodone in the rat: indications for active influx and implications for pharmacokinetics/pharmacodynamics. Drug Metab Dispos. 2006;34(9):1624–31.
Boström E, Hammarlund-Udenaes M, Simonsson US. Blood-brain barrier transport helps to explain discrepancies in in vivo potency between oxycodone and morphine. Anesthesiology. 2008;108(3):495–505.
Villesen HH, Foster DJ, Upton RN, Somogyi AA, Martinez A, Grant C. Cerebral kinetics of oxycodone in conscious sheep. J Pharm Sci. 2006;95(8):1666–76.
Hill R, Dewey WL, Kelly E, Henderson G. Oxycodone-induced tolerance to respiratory depression: reversal by ethanol, pregabalin and protein kinase C inhibition. Br J Pharmacol. 2018;175(12):2492–503.
Okura T, Hattori A, Takano Y, Sato T, Hammarlund-Udenaes M, Terasaki T, et al. Involvement of the pyrilamine transporter, a putative organic cation transporter, in blood-brain barrier transport of oxycodone. Drug Metab Dispos. 2008;36(10):2005–13.
Shimomura K, Okura T, Kato S, Couraud PO, Schermann JM, Terasaki T, et al. Functional expression of a proton-coupled organic cation (H +/OC) antiporter in human brain capillary endothelial cell line hCMEC/D3, a human blood-brain barrier model. Fluids Barriers CNS. 2013;10(1):8.
Sadiq MW, Borgs A, Okura T, Shimomura K, Kato S, Deguchi Y, et al. Diphenhydramine active uptake at the blood-brain barrier and its interaction with oxycodone in vitro and in vivo. J Pharm Sci. 2011;100(9):3912–23.
Mihajlica N, Betsholtz C, Hammarlund-Udenaes M. Pharmacokinetics of pericyte involvement in small-molecular drug transport across the blood-brain barrier. Eur J Pharm Sci. 2018;122:77–84.
Gustafsson S, Lindström V, Ingelsson M, Hammarlund-Udenaes M, Syvänen S. Intact blood-brain barrier transport of small molecular drugs in animal models of amyloid beta and alpha-synuclein pathology. Neuropharmacology. 2018;128:482–91.
Boström E, Simonsson US, Hammarlund-Udenaes M. Oxycodone pharmacokinetics and pharmacodynamics in the rat in the presence of the P-glycoprotein inhibitor PSC833. J Pharm Sci. 2005;94(5):1060–6.
Hassan HE, Myers AL, Lee IJ, Coop A, Eddington ND. Oxycodone induces overexpression of P-glycoprotein (ABCB1) and affects paclitaxel’s tissue distribution in Sprague Dawley rats. J Pharm Sci. 2007;96(9):2494–506.
Pöyhiä R, Kalso EA. Antinociceptive effects and central nervous system depression caused by oxycodone and morphine in rats. Pharmacol Toxicol. 1992;70(2):125–30.
Kalso E, Pöyhiä R, Onnela P, Linko K, Tigerstedt I, Tammisto T. Intravenous morphine and oxycodone for pain after abdominal surgery. Acta Anaesthesiol Scand. 1991;35(7):642–6.
Lilius T, Kangas E, Niemi M, Rauhala P, Kalso E. Ketamine and norketamine attenuate oxycodone tolerance markedly less than that of morphine: from behaviour to drug availability. Br J Anaesth. 2018;120(4):818–26.
Jokinen V, Lilius TO, Laitila J, Niemi M, Rauhala PV, Kalso EA. Pregabalin enhances the antinociceptive effect of oxycodone and morphine in thermal models of nociception in the rat without any pharmacokinetic interactions. Eur J Pain. 2016;20(2):297–306.
Jacob JC, Poklis JL, Akbarali HI, Henderson G, Dewey WL. Ethanol reversal of tolerance to the antinociceptive effects of oxycodone and hydrocodone. J Pharmacol Exp Ther. 2017;362(1):45–52.
Kokki M, Välitalo P, Kuusisto M, Ranta VP, Raatikainen K, Hautajärvi H, et al. Central nervous system penetration of oxycodone after intravenous and epidural administration. Br J Anaesth. 2014;112(1):133–40.
Yamamoto Y, Välitalo PA, Wong YC, Huntjens DR, Proost JH, Vermeulen A, et al. Prediction of human CNS pharmacokinetics using a physiologically-based pharmacokinetic modeling approach. Eur J Pharm Sci. 2018;112:168–79.
Sadiq MW, Boström E, Keizer R, Björkman S, Hammarlund-Udenaes M. Oxymorphone active uptake at the blood-brain barrier and population modeling of its pharmacokinetic-pharmacodynamic relationship. J Pharm Sci. 2013;102(9):3320–31.
Lemberg KK, Siiskonen AO, Kontinen VK, Yli-Kauhaluoma JT, Kalso EA. Pharmacological characterization of noroxymorphone as a new opioid for spinal analgesia. Anesth Analg. 2008;106(2):463–70.
McMillan DM, Tyndale RF. CYP-mediated drug metabolism in the brain impacts drug response. Pharmacol Ther. 2018;184:189–200.
Lemberg KK, Kontinen VK, Siiskonen AO, Viljakka KM, Yli-Kauhaluoma JT, Korpi ER, et al. Antinociception by spinal and systemic oxycodone: why does the route make a difference? In vitro and in vivo studies in rats. Anesthesiology. 2006;105(4):801–12.
McMillan DM, Miksys S, Tyndale RF. Rat brain CYP2D activity alters in vivo central oxycodone metabolism, levels and resulting analgesia. Addict Biol. Epub 21 Dec 2017. https://doi.org/10.1111/adb.12590.
Marcelli D, Brand K, Ponce P, Milkowski A, Marelli C, Ok E, et al. Longitudinal changes in body composition in patients after initiation of hemodialysis therapy: results from an international cohort. J Ren Nutr. 2016;26(2):72–80.
Kirvelä M, Lindgren L, Seppälä T, Olkkola KT. The pharmacokinetics of oxycodone in uremic patients undergoing renal transplantation. J Clin Anesth. 1996;8(1):13–8.
Malhotra BK, Schoenhard GL, de Kater AW, Friedmann N. The pharmacokinetics of oxycodone and its metabolites following single oral doses of Remoxy(R), an abuse-deterrent formulation of extended-release oxycodone, in patients with hepatic or renal impairment. J Opioid Manag. 2015;11(2):157–69.
Tallgren M, Olkkola KT, Seppälä T, Hockerstedt K, Lindgren L. Pharmacokinetics and ventilatory effects of oxycodone before and after liver transplantation. Clin Pharmacol Ther. 1997;61(6):655–61.
Kaiko RF. Pharmacokinetics and pharmacodynamics of controlled-release opioids. Acta Anaesthesiol Scand. 1997;41:166–74.
Leuppi-Taegtmeyer A, Duthaler U, Hammann F, Schmid Y, Dickenmann M, Amico P, et al. Pharmacokinetics of oxycodone/naloxone and its metabolites in patients with end-stage renal disease during and between haemodialysis sessions. Nephrol Dial Transplant. Epub 5 Sep 2018. https://doi.org/10.1093/ndt/gfy285.
Mogil JS. Sex differences in pain and pain inhibition: multiple explanations of a controversial phenomenon. Nat Rev Neurosci. 2012;13(12):859–66.
Verriotis M, Jones L, Whitehead K, Laudiano-Dray M, Panayotidis I, Patel H, et al. The distribution of pain activity across the human neonatal brain is sex dependent. Neuroimage. 2018;178:69–77.
Kaiko RF, Benziger DP, Fitzmartin RD, Burke BE, Reder RF, Goldenheim PD. Pharmacokinetic-pharmacodynamic relationships of controlled-release oxycodone. Clin Pharmacol Ther. 1996;59(1):52–61.
Franke RM, Morton T, Devarakonda K. Pooled post hoc analysis of population pharmacokinetics of oxycodone and acetaminophen following a single oral dose of biphasic immediate-release/extended-release oxycodone/acetaminophen tablets. Drug Des Devel Ther. 2015;9:4587–97.
Morton T, Franke R, Devarakonda K. Pooled post hoc analysis of population pharmacokinetics of oxycodone and acetaminophen following multiple oral doses of biphasic immediate-release/extended-release oxycodone/acetaminophen tablets. Pain Pract. 2016;16(6):730–6.
Charles B, Hardy J, Anderson H, Tapuni A, George R, Norris R. Should the dosage of controlled-release oxycodone in advanced cancer be modified on the basis of patient characteristics? Support Care Cancer. 2014;22(2):325–30.
Liukas A, Kuusniemi K, Aantaa R, Virolainen P, Neuvonen M, Neuvonen PJ, et al. Elimination of intravenous oxycodone in the elderly: a pharmacokinetic study in postoperative orthopaedic patients of different age groups. Drugs Aging. 2011;28(1):41–50.
Elder NM, Atayee RS, Best BM, Ma JD. Observations of urinary oxycodone and metabolite distributions in pain patients. J Anal Toxicol. 2014;38(3):129–34.
Andreassen TN, Klepstad P, Davies A, Bjordal K, Lundstrom S, Kaasa S, et al. Influences on the pharmacokinetics of oxycodone: a multicentre cross-sectional study in 439 adult cancer patients. Eur J Clin Pharmacol. 2011;67(5):493–506.
Soldin OP, Mattison DR. Sex differences in pharmacokinetics and pharmacodynamics. Clin Pharmacokinet. 2009;48(3):143–57.
Drugs and Lactation Database (LactMed). Oxycodone. https://toxnet.nlm.nih.gov/cgi-bin/sis/search2/f?./temp/~q7l2nJ:1. Accessed 1 Oct 2018.
Pariente G, Leibson T, Carls A, Adams-Webber T, Ito S, Koren G. Pregnancy-associated changes in pharmacokinetics: a systematic review. PLoS Med. 2016;13(11):e1002160.
Dallmann A, Ince I, Meyer M, Willmann S, Eissing T, Hempel G. Gestation-specific changes in the anatomy and physiology of healthy pregnant women: an extended repository of model parameters for physiologically based pharmacokinetic modeling in pregnancy. Clin Pharmacokinet. 2017;56(11):1303–30.
Kinnunen M, Kokki H, Hautajärvi H, Huhta H, Ranta VP, Räsänen J, et al. Oxycodone pharmacokinetics and fetal exposure after intravenous or epidural administration to the ewe. Acta Obstet Gynecol Scand. 2018;97(10):1200–5.
Kokki M, Franco MG, Raatikainen K, Välitalo P, Sankilampi U, Heinonen S, et al. Intravenous oxycodone for pain relief in the first stage of labour–maternal pharmacokinetics and neonatal exposure. Basic Clin Pharmacol Toxicol. 2012;111(3):182–8.
Atkinson HC, Begg EJ, Darlow BA. Drugs in human milk. Clinical pharmacokinetic considerations. Clin Pharmacokinet. 1988;14(4):217–40.
Seaton S, Reeves M, McLean S. Oxycodone as a component of multimodal analgesia for lactating mothers after Caesarean section: relationships between maternal plasma, breast milk and neonatal plasma levels. Aust N Z J Obstet Gynaecol. 2007;47(3):181–5.
Evans KC, Evans RG, Royal R, Esterman AJ, James SL. Effect of caesarean section on breast milk transfer to the normal term newborn over the first week of life. Arch Dis Child Fetal Neonatal Ed. 2003;88:380–2.
Lam J, Kelly L, Ciszkowski C, Landsmeer ML, Nauta M, Carleton BC, et al. Central nervous system depression of neonates breastfed by mothers receiving oxycodone for postpartum analgesia. J Pediatr. 2012;160(1):33–7.
Lam J, Kelly L, Matok I, Ross CJ, Carleton BC, Hayden MR, et al. Putative association of ABCB1 2677G > T/A with oxycodone-induced central nervous system depression in breastfeeding mothers. Ther Drug Monit. 2013;35(4):466–72.
Friedrichsdorf SJ, Giordano J, Desai Dakoji K, Warmuth A, Daughtry C, Schulz CA. Chronic pain in children and adolescents: diagnosis and treatment of primary pain disorders in head, abdomen, muscles and joints. Children (Basel). 2016;3(4):10.
Kokki H. Nonsteroidal anti-inflammatory drugs for postoperative pain: a focus on children. Paediatr Drugs. 2003;5(2):103–12.
Kokki H, Lintula H, Vanamo K, Heiskanen M, Eskelinen M. Oxycodone vs placebo in children with undifferentiated abdominal pain: a randomized, double-blind clinical trial of the effect of analgesia on diagnostic accuracy. Arch Pediatr Adolesc Med. 2005;159(4):320–5.
Kokki H, Kokki M, Sjövall S. Oxycodone for the treatment of postoperative pain. Expert Opin Pharmacother. 2012;13(7):1045–58.
Lindell-Osuagwu L, Hakkarainen M, Sepponen K, Vainio K, Naaranlahti T, Kokki H. Prescribing for off-label use and unauthorized medicines in three paediatric wards in Finland, the status before and after the European Union Paediatric Regulation. J Clin Pharm Ther. 2014;39(2):144–53.
SPC. Oxynorm. http://spc.nam.fi/indox/nam/html/nam/humspc/2/92692.pdf. Accessed 1 Oct 2018.
SPC. Oxycontin. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/022272s027lbl.pdf. Accessed 1 Oct 2018.
El-Tahtawy A, Kokki H, Reidenberg BE. Population pharmacokinetics of oxycodone in children 6 months to 7 years old. J Clin Pharmacol. 2006;46(4):433–42.
Kokki H, Rasanen I, Lasalmi M, Lehtola S, Ranta VP, Vanamo K, et al. Comparison of oxycodone pharmacokinetics after buccal and sublingual administration in children. Clin Pharmacokinet. 2006;45(7):745–54.
Kokki H, Rasanen I, Reinikainen M, Suhonen P, Vanamo K, Ojanperä I. Pharmacokinetics of oxycodone after intravenous, buccal, intramuscular and gastric administration in children. Clin Pharmacokinet. 2004;43(9):613–22.
Korjamo T, Tolonen A, Ranta VP, Turpeinen M, Kokki H. Metabolism of oxycodone in human hepatocytes from different age groups and prediction of hepatic plasma clearance. Front Pharmacol. 2012;2:87.
Pokela ML, Anttila E, Seppälä T, Olkkola KT. Marked variation in oxycodone pharmacokinetics in infants. Paediatr Anaesth. 2005;15(7):560–5.
Välitalo P, Kokki M, Ranta VP, Olkkola KT, Hooker AC, Kokki H. Maturation of oxycodone pharmacokinetics in neonates and infants: a population pharmacokinetic model of three clinical trials. Pharm Res. 2017;34(5):1125–33.
Kokki M, Heikkinen M, Välitalo P, Hautajärvi H, Hokkanen J, Pitkänen H, et al. Maturation of oxycodone pharmacokinetics in neonates and infants: oxycodone and its metabolites in plasma and urine. Br J Clin Pharmacol. 2017;83(4):791–800.
Olkkola KT, Hamunen K, Seppälä T, Maunuksela EL. Pharmacokinetics and ventilatory effects of intravenous oxycodone in postoperative children. Br J Clin Pharmacol. 1994;38(1):71–6.
Balyan R, Mecoli M, Venkatasubramanian R, Chidambaran V, Kamos N, Clay S, et al. CYP2D6 pharmacogenetic and oxycodone pharmacokinetic association study in pediatric surgical patients. Pharmacogenomics. 2017;18(4):337–48.
Liukas A, Kuusniemi K, Aantaa R, Virolainen P, Neuvonen M, Neuvonen PJ, et al. Plasma concentrations of oral oxycodone are greatly increased in the elderly. Clin Pharmacol Ther. 2008;84(4):462–7.
Saari TI, Ihmsen H, Neuvonen PJ, Olkkola KT, Schwilden H. Oxycodone clearance is markedly reduced with advancing age: a population pharmacokinetic study. Br J Anaesth. 2012;108(3):491–8.
Villesen HH, Banning AM, Petersen RH, Weinelt S, Poulsen JB, Hansen SH, et al. Pharmacokinetics of morphine and oxycodone following intravenous administration in elderly patients. Ther Clin Risk Manag. 2007;3(5):961–7.
Olkkola KT, Kontinen VK, Saari TI, Kalso EA. Does the pharmacology of oxycodone justify its increasing use as an analgesic? Trends Pharmacol Sci. 2013;34(4):206–14.
van der Schrier R, Roozekrans M, Olofsen E, Aarts L, van Velzen M, de Jong M, et al. Influence of ethanol on oxycodone-induced respiratory depression: a dose-escalating study in young and elderly individuals. Anesthesiology. 2017;126(3):534–42.
Jokela R, Ahonen J, Valjus M, Seppala T, Korttila K. Premedication with controlled-release oxycodone does not improve management of postoperative pain after day-case gynaecological laparoscopic surgery. Br J Anaesth. 2007;98(2):255–60.
Kokki M, Broms S, Eskelinen M, Rasanen I, Ojanpera I, Kokki H. Analgesic concentrations of oxycodone–a prospective clinical PK/PD study in patients with laparoscopic cholecystectomy. Basic Clin Pharmacol Toxicol. 2012;110(5):469–75.
Webster LR, Bath B, Medve RA, Marmon T, Stoddard GJ. Randomized, double-blind, placebo-controlled study of the abuse potential of different formulations of oral oxycodone. Pain Med. 2012;13(6):790–801.
Queckenberg C, Fuhr U. Influence of posture on pharmacokinetics. Eur J Clin Pharmacol. 2009;65(2):109–19.
Kennedy JM, van Rij AM. Drug absorption from the small intestine in immediate postoperative patients. Br J Anaesth. 2006;97(2):171–80.
Lenz H, Sandvik L, Qvigstad E, Bjerkelund CE, Raeder J. A comparison of intravenous oxycodone and intravenous morphine in patient-controlled postoperative analgesia after laparoscopic hysterectomy. Anesth Analg. 2009;109(4):1279–83.
Pokkinen SM, Yli-Hankala A, Kalliomaki ML. The effects of propofol vs. sevoflurane on post-operative pain and need of opioid. Acta Anaesthesiol Scand. 2014;58(8):980–5.
Pokkinen SM, Kalliomäki ML, Yli-Hankala A, Nieminen K. Less postoperative pain after laparoscopic hysterectomy than after vaginal hysterectomy. Arch Gynecol Obstet. 2015;292(1):149–54.
Cajanus K, Neuvonen M, Koskela O, Kaunisto MA, Neuvonen PJ, Niemi M, et al. Analgesic plasma concentrations of oxycodone after surgery for breast cancer: which factors matter? Clin Pharmacol Ther. 2018;103(4):653–62.
Cajanus K, Kaunisto MA, Tallgren M, Jokela R, Kalso E. How much oxycodone is needed for adequate analgesia after breast cancer surgery: effect of the OPRM1 118A > G polymorphism. J Pain. 2014;15(12):1248–56.
Fallon M, Giusti R, Aielli F, Hoskin P, Rolke R, Sharma M, et al.; ESMO Guidelines Committee. Management of cancer pain in adult patients: ESMO clinical practice guidelines. Ann Oncol. 2018;29:166–91.
Dupoiron D, Stachowiak A, Loewenstein O, Ellery A, Kremers W, Bosse B, et al. A phase III randomized controlled study on the efficacy and improved bowel function of prolonged-release (PR) oxycodone-naloxone (up to 160/80 mg daily) vs oxycodone PR. Eur J Pain. 2017;21(9):1528–37.
Kokki M, Kuronen M, Naaranlahti T, Nyyssönen T, Pikkarainen I, Savolainen S, et al. Opioid-induced bowel dysfunction in patients undergoing spine surgery: comparison of oxycodone and oxycodone-naloxone treatment. Adv Ther. 2017;34(1):236–51.
Kuusniemi K, Zollner J, Sjovall S, Huhtala J, Karjalainen P, Kokki M, et al. Prolonged-release oxycodone/naloxone in postoperative pain management: from a randomized clinical trial to usual clinical practice. J Int Med Res. 2012;40(5):1775–93.
SPC. TARGINIQ™ ER. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/205777lbl.pdf. Accessed 1 Oct 2018.
Heiskanen T, Langel K, Gunnar T, Lillsunde P, Kalso EA. Opioid concentrations in oral fluid and plasma in cancer patients with pain. J Pain Sympt Manage. 2015;50(4):524–32.
Rivory LP, Slaviero KA, Clarke SJ. Hepatic cytochrome P450 3A drug metabolism is reduced in cancer patients who have an acute-phase response. Br J Cancer. 2002;87(3):277–80.
Naito T, Tashiro M, Yamamoto K, Ohnishi K, Kagawa Y, Kawakami J. Impact of cachexia on pharmacokinetic disposition of and clinical responses to oxycodone in cancer patients. Eur J Clin Pharmacol. 2012;68(10):1411–8.
Naito T, Tashiro M, Ishida T, Ohnishi K, Kawakami J. Cancer cachexia raises the plasma concentration of oxymorphone through the reduction of CYP3A but not CYP2D6 in oxycodone-treated patients. J Clin Pharmacol. 2013;53(8):812–8.
Sato H, Naito T, Ishida T, Kawakami J. Relationships between oxycodone pharmacokinetics, central symptoms, and serum interleukin-6 in cachectic cancer patients. Eur J Clin Pharmacol. 2016;72(12):1463–70.
Fearon KC, Falconer JS, Slater C, McMillan DC, Ross JA, Preston T. Albumin synthesis rates are not decreased in hypoalbuminemic cachectic cancer patients with an ongoing acute-phase protein response. Ann Surg. 1998;227(2):249–54.
Leow KP, Wright AW, Cramond T, Smith MT. Determination of the serum protein binding of oxycodone and morphine using ultrafiltration. Ther Drug Monit. 1993;15(5):440–7.
Andreassen TN, Eftedal I, Klepstad P, Davies A, Bjordal K, Lundstrom S, et al. Do CYP2D6 genotypes reflect oxycodone requirements for cancer patients treated for cancer pain? A cross-sectional multicentre study. Eur J Clin Pharmacol. 2012;68(1):55–64.
Hurley RW, Adams MC, Benzon HT. Neuropathic pain: treatment guidelines and updates. Curr Opin Anaesthesiol. 2013;26(5):580–7.
Thibault K, Calvino B, Rivals I, Marchand F, Dubacq S, McMahon SB, et al. Molecular mechanisms underlying the enhanced analgesic effect of oxycodone compared to morphine in chemotherapy-induced neuropathic pain. PLoS One. 2014;9(3):e91297.
Nakamura A, Hasegawa M, Minami K, Kanbara T, Tomii T, Nishiyori A, et al. Differential activation of the mu-opioid receptor by oxycodone and morphine in pain-related brain regions in a bone cancer pain model. Br J Pharmacol. 2013;168(2):375–88.
Watson CP, Babul N. Efficacy of oxycodone in neuropathic pain: a randomized trial in postherpetic neuralgia. Neurology. 1998;50(6):1837–41.
Gimbel JS, Richards P, Portenoy RK. Controlled-release oxycodone for pain in diabetic neuropathy: a randomized controlled trial. Neurology. 2003;60(6):927–34.
Watson CP, Moulin D, Watt-Watson J, Gordon A, Eisenhoffer J. Controlled-release oxycodone relieves neuropathic pain: a randomized controlled trial in painful diabetic neuropathy. Pain. 2003;105(1–2):71–8.
Hanna M, O’Brien C, Wilson MC. Prolonged-release oxycodone enhances the effects of existing gabapentin therapy in painful diabetic neuropathy patients. Eur J Pain. 2008;12(6):804–13.
Barrera-Chacon JM, Mendez-Suarez JL, Jauregui-Abrisqueta ML, Palazon R, Barbara-Bataller E, Garcia-Obrero I. Oxycodone improves pain control and quality of life in anticonvulsant-pretreated spinal cord-injured patients with neuropathic pain. Spinal Cord. 2011;49(1):36–42.
Plummer JL, Cmielewski PL, Reynolds GD, Gourlay GK, Cherry DA. Influence of polarity on dose-response relationships of intrathecal opioids in rats. Pain. 1990;40(3):339–47.
Bäcklund M, Lindgren L, Kajimoto Y, Rosenberg PH. Comparison of epidural morphine and oxycodone for pain after abdominal surgery. J Clin Anesth. 1997;9(1):30–5.
Yanagidate F, Dohi S. Epidural oxycodone or morphine following gynaecological surgery. Br J Anaesth. 2004;93(3):362–7.
Piirainen P, Kokki H, Hautajärvi H, Ranta VP, Kokki M. The analgesic efficacy and pharmacokinetics of epidural oxycodone after gynaecological laparotomy: a randomized, double-blind, double-dummy comparison with intravenous administration. Br J Clin Pharmacol. 2018;84(9):2088–96.
Sng BL, Kwok SC, Mathur D, Ithnin F, Newton-Dunn C, Assam PN, et al. Comparison of epidural oxycodone and epidural morphine for post-caesarean section analgesia: a randomised controlled trial. Indian J Anaesth. 2016;60(3):187–93.
Kokki M, Pesonen M, Vehviläinen P, Litmala O, Pasanen M, Kokki H. Cytotoxicity of oxycodone and morphine in human neuroblastoma and mouse motoneuronal cells: a comparative approach. Drugs R D. 2016;16(2):155–63.
Al-Ghananeem AM, Malkawi AH, Crooks PA. Effect of pH on sublingual absorption of oxycodone hydrochloride. AAPS PharmSciTech. 2006;7(1):163–7.
Weinberg DS, Inturrisi CE, Reidenberg B, Moulin DE, Nip TJ, Wallenstein S, et al. Sublingual absorption of selected opioid analgesics. Clin Pharmacol Ther. 1988;44(3):335–42.
Takala A, Kaasalainen V, Seppälä T, Kalso E, Olkkola KT. Pharmacokinetic comparison of intravenous and intranasal administration of oxycodone. Acta Anaesthesiol Scand. 1997;41(2):309–12.
Kurosaki Y, Takatori T, Nishimura H, Nakayama T, Kimura T. Regional variation in oral mucosal drug absorption: permeability and degree of keratinization in hamster oral cavity. Pharm Res. 1991;8(10):1297–301.
Lofwall MR, Moody DE, Fang WB, Nuzzo PA, Walsh SL. Pharmacokinetics of intranasal crushed OxyContin and intravenous oxycodone in nondependent prescription opioid abusers. J Clin Pharmacol. 2012;52(4):600–6.
Perrino PJ, Colucci SV, Apseloff G, Harris SC. Pharmacokinetics, tolerability, and safety of intranasal administration of reformulated OxyContin((R)) tablets compared with original OxyContin® tablets in healthy adults. Clin Drug Investig. 2013;33(6):441–9.
Harris SC, Perrino PJ, Smith I, Shram MJ, Colucci SV, Bartlett C, et al. Abuse potential, pharmacokinetics, pharmacodynamics, and safety of intranasally administered crushed oxycodone HCl abuse-deterrent controlled-release tablets in recreational opioid users. J Clin Pharmacol. 2014;54(4):468–77.
Comer SD, Sullivan MA, Whittington RA, Vosburg SK, Kowalczyk WJ. Abuse liability of prescription opioids compared to heroin in morphine-maintained heroin abusers. Neuropsychopharmacology. 2008;33(5):1179–91.
Walter C, Knothe C, Lotsch J. Abuse-deterrent opioid formulations: pharmacokinetic and pharmacodynamic considerations. Clin Pharmacokinet. 2016;55(7):751–67.
Nalamachu SR, Shah B. Abuse of immediate-release opioids and current approaches to reduce misuse, abuse, and diversion. Postgrad Med. Epub 26 Jul 2018. https://doi.org/10.1080/00325481.2018.1502569.
Gudin J, Levy-Cooperman N, Kopecky EA, Fleming AB. Comparing the effect of tampering on the oral pharmacokinetic profiles of two extended-release oxycodone formulations with abuse-deterrent properties. Pain Med. 2015;16(11):2142–51.
Setnik B, Bramson C, Bass A, Levy-Cooperman N, Malhotra B, Matschke K, et al. Intranasal administration of crushed ALO-02 (extended-release oxycodone with sequestered naltrexone): a randomized, controlled abuse-potential study in nondependent recreational opioid users. J Clin Pharmacol. 2015;55(12):1351–61.
Setnik B, Bass A, Bramson C, Levy-Cooperman N, Malhotra B, Matschke K, et al. Abuse potential study of ALO-02 (extended-release oxycodone surrounding sequestered naltrexone) compared with immediate-release oxycodone administered orally to nondependent recreational opioid users. Pain Med. 2017;18(6):1077–88.
Malhotra BK, Matschke K, Wang Q, Bramson C, Salageanu J. Effects of ethanol on the pharmacokinetics of extended-release oxycodone with sequestered naltrexone (ALO-02). Clin Drug Investig. 2015;35(4):267–74.
Colucci SV, Perrino PJ, Shram M, Bartlett C, Wang Y, Harris SC. Abuse potential of intravenous oxycodone/naloxone solution in nondependent recreational drug users. Clin Drug Investig. 2014;34(6):421–9.
Kopecky EA, Fleming AB, Noonan PK, Varanasi RK, Grima M, Saim S, et al. Impact of physical manipulation on in vitro and in vivo release profiles of oxycodone DETERx(R): an extended-release, abuse-deterrent formulation. J Opioid Manag. 2014;10(4):233–46.
Kopecky EA, Fleming AB, Levy-Cooperman N, O’Connor M, M Sellers E. Oral human abuse potential of oxycodone DETERx® (Xtampza® ER). J Clin Pharmacol. 2017;57(4):500–12.
SPC, ROXYBON. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209777lbl.pdf. Accessed 1 Oct 2018.
Liukas A, Hagelberg NM, Kuusniemi K, Neuvonen PJ, Olkkola KT. Inhibition of cytochrome P450 3A by clarithromycin uniformly affects the pharmacokinetics and pharmacodynamics of oxycodone in young and elderly volunteers. J Clin Psychopharmacol. 2011;31(3):302–8.
Grönlund J, Saari T, Hagelberg N, Martikainen IK, Neuvonen PJ, Olkkola KT, et al. Effect of telithromycin on the pharmacokinetics and pharmacodynamics of oral oxycodone. J Clin Pharmacol. 2010;50(1):101–8.
Saari TI, Grönlund J, Hagelberg NM, Neuvonen M, Laine K, Neuvonen PJ, et al. Effects of itraconazole on the pharmacokinetics and pharmacodynamics of intravenously and orally administered oxycodone. Eur J Clin Pharmacol. 2010;66(4):387–97.
Kummer O, Hammann F, Moser C, Schaller O, Drewe J, Krahenbuhl S. Effect of the inhibition of CYP3A4 or CYP2D6 on the pharmacokinetics and pharmacodynamics of oxycodone. Eur J Clin Pharmacol. 2011;67(1):63–71.
Grönlund J, Saari TI, Hagelberg N, Neuvonen PJ, Olkkola KT, Laine K. Miconazole oral gel increases exposure to oral oxycodone by inhibition of CYP2D6 and CYP3A4. Antimicrob Agents Chemother. 2011;55(3):1063–7.
Hagelberg NM, Nieminen TH, Saari TI, Neuvonen M, Neuvonen PJ, Laine K, et al. Voriconazole drastically increases exposure to oral oxycodone. Eur J Clin Pharmacol. 2009;65(3):263–71.
Nieminen TH, Hagelberg NM, Saari TI, Neuvonen M, Neuvonen PJ, Laine K, et al. Oxycodone concentrations are greatly increased by the concomitant use of ritonavir or lopinavir/ritonavir. Eur J Clin Pharmacol. 2010;66(10):977–85.
Nieminen TH, Hagelberg NM, Saari TI, Neuvonen M, Laine K, Neuvonen PJ, et al. St John’s wort greatly reduces the concentrations of oral oxycodone. Eur J Pain. 2010;14(8):854–9.
Heiskanen T, Olkkola KT, Kalso E. Effects of blocking CYP2D6 on the pharmacokinetics and pharmacodynamics of oxycodone. Clin Pharmacol Ther. 1998;64(6):603–11.
Grönlund J, Saari TI, Hagelberg NM, Neuvonen PJ, Olkkola KT, Laine K. Exposure to oral oxycodone is increased by concomitant inhibition of CYP2D6 and 3A4 pathways, but not by inhibition of CYP2D6 alone. Br J Clin Pharmacol. 2010;70(1):78–87.
Grönlund J, Saari TI, Hagelberg NM, Neuvonen PJ, Laine K, Olkkola KT. Effect of inhibition of cytochrome P450 enzymes 2D6 and 3A4 on the pharmacokinetics of intravenous oxycodone: a randomized, three-phase, crossover, placebo-controlled study. Clin Drug Investig. 2011;31(3):143–53.
Lemberg KK, Heiskanen TE, Neuvonen M, Kontinen VK, Neuvonen PJ, Dahl ML, et al. Does co-administration of paroxetine change oxycodone analgesia: an interaction study in chronic pain patients. Scand J Pain. 2010;1(1):24–33.
Curry SC, Watts DJ, Katz KD, Bikin D, Bukaveckas BL. The effect of single-dose tramadol on oxycodone clearance. J Emerg Med. 2007;33(4):407–11.
Häkkinen M, Launiainen T, Vuori E, Ojanperä I. Comparison of fatal poisonings by prescription opioids. Forensic Sci Int. 2012;222(1–3):327–31.
Pilgrim JL, Yafistham SP, Gaya S, Saar E, Drummer OH. An update on oxycodone: lessons for death investigators in Australia. Forensic Sci Med Pathol. 2015;11(1):3–12.
Thompson JG, Vanderwerf S, Seningen J, Carr M, Kloss J, Apple FS. Free oxycodone concentrations in 67 postmortem cases from the Hennepin County medical examiner’s office. J Anal Toxicol. 2008;32(8):673–9.
Reder RF, Oshlack B, Miotto JB, Benziger DD, Kaiko RF. Steady-state bioavailability of controlled-release oxycodone in normal subjects. Clin Ther. 1996;18(1):95–105.
Pesonen A, Suojaranta-Ylinen R, Hammarén E, Tarkkila P, Seppälä T, Rosenberg PH. Comparison of effects and plasma concentrations of opioids between elderly and middle-aged patients after cardiac surgery. Acta Anaesthesiol Scand. 2009;53(1):101–8.
Kokki M, Broms S, Eskelinen M, Neuvonen PJ, Halonen T, Kokki H. The analgesic concentration of oxycodone with co-administration of paracetamol: a dose-finding study in adult patients undergoing laparoscopic cholecystectomy. Basic Clin Pharmacol Toxicol. 2012;111(6):391–5.
Piirainen A, Kokki H, Immonen S, Eskelinen M, Häkkinen MR, Hautajärvi H, et al. A dose-finding study of dexketoprofen in patients undergoing laparoscopic cholecystectomy: a randomized clinical trial on effects on the analgesic concentration of oxycodone. Drugs R D. 2015;15(4):319–28.
Choi BM, Lee YH, An SM, Lee SH, Lee EK, Noh GJ. Population pharmacokinetics and analgesic potency of oxycodone. Br J Clin Pharmacol. 2017;83(2):314–25.
Purdy M, Kinnunen M, Kokki M, Anttila M, Eskelinen M, Hautajärvi H, et al. A prospective, randomized, open label, controlled study investigating the efficiency and safety of 3 different methods of rectus sheath block analgesia following midline laparotomy. Medicine (Baltimore). 2018;97(7):e9968.
Nieminen TH, Hagelberg NM, Saari TI, Neuvonen M, Neuvonen PJ, Laine K, et al. Grapefruit juice enhances the exposure to oral oxycodone. Basic Clin Pharmacol Toxicol. 2010;107(4):782–8.
Acknowledgements
Open access funding provided by University of Eastern Finland (UEF) including Kuopio University Hospital.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The research of Mari Kinnunen, Panu Piirainen, Hannu Kokki and Merja Kokki has received funding from the governmental VTR fund, the Hospital District of Northern Savo, Kuopio, Finland (HK, MKo); Olvi Säätiö, Iisalmi, Finland (MKi, PP, HK, MKo); and the Finnish Cultural Foundation, Helsinki, Finland (MKi, MKo). The authors have no financial relationships to disclose that are relevant to this article.
Conflict of interest
Mari Kinnunen, Panu Piirainen, Hannu Kokki, Pauliina Lammi and Merja Kokki have no conflicts of interest to declare.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kinnunen, M., Piirainen, P., Kokki, H. et al. Updated Clinical Pharmacokinetics and Pharmacodynamics of Oxycodone. Clin Pharmacokinet 58, 705–725 (2019). https://doi.org/10.1007/s40262-018-00731-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-018-00731-3