
Abstract
Age-related macular degeneration (AMD) and diabetic retinopathy (DR) are vascular diseases with high prevalence, ranking among the leading causes of blindness and vision loss worldwide. Despite being effective, current treatments for AMD and DR are burdensome for patients and clinicians, resulting in suboptimal compliance and real risk of vision loss. Thus, there is an unmet need for long-lasting alternatives with improved safety and efficacy. Adeno-associated virus (AAV) is the leading vector for ocular gene delivery, given its ability to enable long-term expression while eliciting relatively mild immune responses. Progress has been made in AAV-based gene therapies for not only inherited retinal diseases but also acquired conditions with preclinical and clinical studies of AMD and DR showing promising results. These studies have explored several pathways involved in the disease pathogenesis, as well as different strategies to optimise gene delivery. These include engineered capsids with enhanced tropism to particular cell types, and expression cassettes incorporating elements for a targeted and controlled expression. Multiple-acting constructs have also been investigated, in addition to gene silencing and editing. Here, we provide an overview of strategies employing AAV-mediated gene delivery to treat AMD and DR. We discuss preclinical efficacy studies and present the latest data from clinical trials for both diseases.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
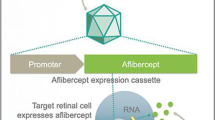
Several strategies have been employed to improve gene delivery with adeno-associated virus in preclinical and clinical studies for age-related macular degeneration and diabetic retinopathy. |
Current clinical trials have reported promising outcomes. |
Preclinical studies have shown encouraging results with a variety of approaches, including multigenic constructs, engineered vectors, inducible systems, tissue-specific promoters and regulated expression platforms. |
1 Introduction
Gene therapy has emerged as a powerful strategy to treat ocular diseases by offering the possibility of long-term treatment for challenging and untreatable diseases [1]. The well-defined compartmentalised anatomy of the eye and its immune-privileged environment make it an attractive target [2]. In addition, the eye’s easy access and the optical transparency of visual media allow the evaluation of new interventions by non-invasive techniques [3]. These factors, combined with the number of identified genes associated with retinopathies and the availability of well-characterised animal models of ocular diseases, contribute to a dominance of ophthalmic conditions within the gene therapy field [2, 4]. The first clinical trials with ocular gene therapy were reported in 2008, with three independent studies investigating the safety of adeno-associated virus (AAV) serotype 2 vectors expressing retinal pigment epithelium-specific 65-kDa protein (RPE65), administered through subretinal injection in three patients with Leber congenital amaurosis (LCA) [5,6,7]. These primary data, along with other studies conducted over the following decade, led to the US FDA approval in 2017 of voretigene neparvovec (Luxturna), a single-dose gene therapy consisting of AAV2 vectors expressing human RPE65 to treat LCA [8]. Several other clinical trials using gene therapy to treat eye disorders have been conducted, with AAV being the most used vector, accounting for about 70% of all trials (https://clinicaltrials.gov; search term ‘eye diseases’ OR ‘ocular diseases’ OR ‘ophthalmic diseases’ AND ‘gene therapy’ OR ‘AAV’; accessed 17 March 2023).
The utilisation of AAV vectors is a breakthrough in gene delivery research. AAV belongs to the genus Dependoparvovirus and was first described as adenovirus-associated defective virus particles by Atchison et al., who identified them in simian adenovirus type 15 cultures and observed their inability to replicate alone [9]. AAV is a non-enveloped small (25 nm) virus carrying a 4.7 Kb long single-stranded DNA [10, 11]. Its genome is packaged within an icosahedral capsid containing two open reading frames encoding rep and cap genes, which are replaced with therapeutic genes in recombinant vectors (rAAV) [12,13,14]. AAV is considered non-pathogenic, and its replication depends on co-infection with a helper virus, such as adenovirus or herpesvirus. The rAAV, devoid of the rep and cap genes, are non-replicative and their cellular tropism is dependent on proteins within their capsid, the sequence of which is defined by their serotype. The immune response induced by AAV is relatively mild, and various cell types can be transduced, including dividing and nondividing cells [15, 16]. Thus, different transduction performances, immunogenicity, and cellular tropism are observed with the multiplicity of AAV serotypes available [17]. Despite the benefits outlined, the maximum cargo capacity of about 5 kb and the pre-existence/development of anti-AAV neutralising antibodies (nABs) limit the application and success of AAV-based gene therapies [11, 18]. The serotype most commonly employed in ocular gene delivery is the AAV2, which is known to transduce primarily retinal pigmented epithelium (RPE) cells and also Müller cells, ganglion cells, and photoreceptors [19,20,21,22,23]. Optimised tropism towards specific eye tissues and enhanced transduction can be achieved with hybrid (pseudotyped) and engineered capsids, resulting in highly specific vectors [1, 24]. Pseudotyping involves generating recombinant AAV using the rep gene and inverted terminal repeats (ITRs) from one serotype, commonly AAV2, and the cap gene of a different serotype (e.g., AAV8). The resulting hybrid AAV (AAV2/8) has its packaged expression cassette being flanked by the ITRs of AAV2 and its viral capsid defined by the cap gene of AAV8 [1]. Capsid engineering can also be used as a strategy to circumvent the immune response, which is affected by vector titre, species, delivery route, type of transgene, and the ratio of empty capsids [11, 25]. Therefore, the understanding of AAV virology and immunogenicity, together with capsid and construct optimisation, has allowed the development of numerous AAV-based platforms targeting eye disorders, both hereditary and nonhereditary [2]. The latter is of great relevance to the field since, unlike inherited diseases, which are mostly rare conditions, nonhereditary-acquired disorders are highly prevalent, representing an excellent opportunity to provide a population-wide impact of AAV-mediated gene therapies.
Retinal and choroidal vascular diseases are among the leading causes of blindness and visual impairment worldwide. They are highly prevalent and affect people of all ages, representing a significant socioeconomic problem [26]. Current treatment of most of these chronic conditions is effective, albeit burdensome for patients and clinicians, particularly in the long-term. Thus, cost, low compliance and consequent therapeutic failure limit the efficacy of available therapies, emphasising the unmet need for safe and effective longer-acting alternatives with better regimens [27]. AAV-mediated expression of molecules targeting pathways involved in the pathogenesis of these diseases has emerged as a powerful strategy to overcome the outlined challenges. Moreover, as technology advances and disease mechanisms are better understood, new targets are identified and more therapies are investigated, increasing the chances of finding translatable options [3, 28]. In addition, different strategies have been explored to optimise ocular gene delivery with AAV and reduce potential adverse effects associated with the therapy [1]. Therefore, clinical and preclinical progress has been made with AAV-based gene therapies for retinal and choroidal vascular diseases, including age-related macular degeneration (AMD), diabetic retinopathy (DR), and diabetic macular edema (DME).
This review will provide an overview of AAV-based strategies that have shown promise for the treatment of AMD, DR and DME in vivo. Given the potential benefits a gene therapy with optimised tropism or regulatable expression could offer for these conditions, this review will cover both constitutive and controllable expression platforms, as well as modified capsids, multigenic constructs and cell-specific promoters.
2 Age-Related Macular Degeneration (AMD)
AMD is the leading cause of legal blindness in the developed world, accounting for about 9% of all cases globally. It is estimated that in 2040 a total of 288 million people in the world will have AMD [29]. AMD is a multifactorial disease characterised by the development and accumulation of extracellular deposits (drusen) in the subretinal space, progressive degeneration of the RPE, photoreceptors and adjacent tissues, with resultant central vision loss [30, 31]. Several risk factors have been associated with AMD, including ageing, genetic susceptibility, and environmental risks, such as diet, hypertension, hyperlipidaemia, obesity, and smoking [30, 32]. AMD can be classified into two forms—dry AMD and wet AMD (wAMD). In the early and intermediate stages of the disease, the accumulation of drusen is observed, along with atrophy of photoreceptors and choriocapillaris, in addition to loss of the RPE at the macular region, with consequent progressive central vision loss [30]. The Beckman Initiative for Macular Research clinically classifies as intermediate AMD if large drusen (> 125 um) and/or pigmentary irregularities associated with medium (> 63 µm and ≤ 125 µm) or large drusen are identified. The risk of advancing to the late stage increases if large drusen areas are associated with pigmentary changes [33]. Late-stage AMD manifests as geographic atrophy (GA), also known as dry AMD, and/or neovascular AMD. Choroidal neovascularisation (CNV) is the aberrant growth of choroidal vessels invading the retina, a process that can occur at any point of AMD and characterises the neovascular form. Leakage or rupture of these vessels characterises the exudative stage of AMD, also called wet AMD, a process accompanied by severe deterioration of vision [30]. Although dry AMD represents 90% of all cases, no worldwide treatments are available for this form of the disease, except for two intravitreal complement inhibitors recently approved by the FDA in February and August 2023 to treat GA secondary to AMD [34, 35]. On the other hand, since the role of vascular endothelial growth factor (VEGF) in wAMD pathogenesis is well established, the development of intravitreal anti-VEGF therapies was a milestone in AMD management, representing an effective alternative for these patients [1, 31]. However, it is a costly treatment and requires repeated intravitreal injections, which is associated with complications and a substantial burden for patients [36]. Consequently, treatment outcomes vary, and not all patients achieve or maintain stable good vision long-term [30]. Therefore, long-acting therapies with improved safety and efficacy have been investigated, including AAV-based gene therapies [1].
2.1 Adeno-Associated Virus (AAV)-Mediated Gene Therapies Delivering Endogenous Inhibitors
The most common AAV-mediated approach is to deliver genes encoding factors associated with angiogenesis to treat wAMD. These include antiangiogenic proteins, such as endostatin, angiostatin and protein pigment endothelial-derived factor (PEDF), as well as VEGF receptors, inhibitors, and antibodies. AAV-mediated delivery of the gene encoding the soluble VEGF receptor 1 (sFLT-1), a VEGF antagonist, has been investigated for over a decade, with primary studies showing safety in vivo and efficacy in rodent and non-human primate (NHP) models of CNV, using both subretinal and intravitreal routes [37,38,39,40,41,42]. Positive results observed in these animal studies led to clinical trials (NCT01024998 and NCT01494805) (Table 1) in which patients received intravitreal (NCT01024998) or subretinal (NCT01494805) injections containing AAV2 vectors encoding different forms of sFLT-1. The application of the same serotype in both subretinal and intravitreal delivery of sFLT-1 is a result of the broad tropism of AAV2 to retinal cells and its ability to bind to heparan sulphate proteoglycan at the inner limiting membrane, which facilitates retinal penetration from the vitreous [43]. The first trial started in 2010 and evaluated four doses (2E8, 2E9, 6E9, 2E10 viral genome; vg) of AAV2 vectors packaging FLT-1 domain 2 for a 52-week follow-up, eventually extended to 4 years. Despite promising results regarding safety and tolerability, important variations in sFLT-1 expression were observed, possibly attributed to serum AAV2 antibodies, which are highly prevalent in the general population [25, 44]. Further trials with this therapy were not conducted. However, a similar phase I/II clinical study was started in 2011, investigating the safety and efficacy of subretinal injection of AAV2 vectors delivering a natural form of sFLT-1, at 1E10 and 1E11 vg, for 3 years. The gene therapy was safe and well tolerated. However, visual acuity outcomes, assessed as a secondary endpoint in the phase IIa trial, were insufficient to suggest efficacy for wAMD, with no further studies reported [45, 46].
Multiple strategies have been employed in preclinical studies evaluating AAV-mediated delivery of the sFLT-1 gene. Lee et al. achieved efficient retinal transduction and attenuation of murine CNV with 5E7 vg of AAV2 vectors encoding a truncated form of sFLT-1. The therapy was administered by intravitreal injections administered 5 days after laser coagulation, and the antiangiogenic effect was comparable with bevacizumab. These results are promising, considering that fewer toxic effects and immune reactions are expected with lower doses and that intravitreal delivery is advantageous for patients and clinicians [47]. In addition, study designs in which the treatment is administered after disease induction provide a better representation of clinical scenarios. A recent study conducted by Yuan et al. included additional controls to optimise sFLT-1 gene delivery. The gene therapy employed a modified AAV2/8 containing a single-point mutation of tyrosine (Y) to phenylalanine (F) at residue 733 on the AAV8 capsid (Y733F). Substitution of surface-exposed tyrosine residues to phenylalanine decreases capsid tyrosine phosphorylation. This approach enables the vector to escape proteasome degradation, facilitating its nuclear transport and enhancing transduction efficiency [48]. Yuan et al. used the AAV2/8 (Y733F) vector to deliver a construct containing an RPE-specific promoter, the RPE65, located downstream of hypoxia response elements containing hypoxia-inducing factor 1 (HIF-1) binding sites, in order to achieve targeted and physiologically regulated expression of sFLT-1. Treatment with 8E10 vg was administered intravitreally in mice further submitted to chemically-induced hypoxia (26 days later) or laser-induced CNV (21 days later). The hypoxia-regulated expression of sFLT-1 in the RPE was demonstrated, along with its ability to reduce CNV areas and VEGF receptor 2 (VEGFR2) mRNA levels [49].
Similar clinical findings were previously reported by Biswal et al., who utilised the hypoxia-induced elements to regulate the expression of human endostatin XVIII in RPE cells. For this, AAV2 vectors (1E9 vg) harbouring HIF-1 response elements upstream of the RPE65 promoter and the endostatin genes were delivered by subretinal injection in mice later submitted to the laser-induced CNV model [50]. Endostatin has well-known antiangiogenic properties, which have been explored in a clinical trial for wAMD using lentivirus vectors [51]. Therefore, sFLT1, endostatin, and other endogenous inhibitors of angiogenesis are promising targets for AAV-mediated gene therapies for wAMD. Yet, strategies including modifications in vectors capsid, alteration of expressed protein structure, and insertion of promoters and regulatable elements, may lead to more effective expression and perhaps improve clinical outcomes.
2.2 AAV-Mediated Gene Therapies Targeting Vascular Endothelial Growth Factor
Grishanin et al. employed AAV2.7m8, an engineered vector with optimised pan-retinal transduction, to deliver aflibercept by intravitreal injection in NHP with CNV. Aflibercept is an approved drug for wAMD consisting of a recombinant protein comprising domains of (VEGFR1) and 2 fused to the Fc portion of a human antibody [52]. Three different doses of vector were initially evaluated in a pharmacokinetic study, in which signs of inflammation were observed in all tested doses within 56 days of the intravitreal injections [53]. However, the higher dose, 2E12 vg/eye, was later assessed in a long-term safety study, which showed that the therapy was well tolerated, long-term expression (13 months) was achieved, and suppression of CNV at both 56 days and 13 months after administration was detected [54]. This formulation, named ADVM-022 or Ixo-vec, was assessed in a phase I clinical trial for wAMD (OPTIC [NCT03748784] and OPTIC-EXT [NCT04645212]) (Table 1) and a phase II trial (INFINITY [NCT04418427]) for DME, both with patients receiving 2E11 or 6E11 vg per eye through a single intravitreal injection [55]. The wAMD study was completed in 2022, and a final 2-year analysis was presented in the 2023 Annual Macula Society Meeting reported mainly mild to moderate adverse events, mostly associated with a dose-depended inflammation that was responsive to corticosteroids. Uveitis and dry AMD were also reported as severe adverse events associated with the therapy. An 81–98% decrease in the annual rate of anti-VEGF injections was reported, with 53% of patients treated with the lowest dose remaining supplemental injection-free after 2 years [56]. There is an ongoing phase II trial (LUNA [NCT05536973]) assessing 2E11 vg and a lower dose (6E10 vg) along with an improved corticosteroid regimen [56]. In contrast, ADVM-022 will no longer be developed for DME due to severe adverse reactions reported in patients receiving the higher dose [57], which led to the reassessment of the study. Although these adverse events were observed only in DME patients, further studies with this therapy for wAMD will focus on lower doses.
The 7m8 capsid used in the above trials was discovered by directed evolution, a strategy commonly employed to select capsids with improved tropism to specific cell types [58]. Kotterman et al. performed the directed evolution method in primates to isolate AAV R100, a variant of AAV2 with higher transduction efficiency after intravitreal delivery [59]. The functionality and safety of R100 vectors were confirmed in NHP induced with CNV and pretreated with an intravitreal injection of R100 vectors carrying anti-VEGF scFv [59]. The AAV R100 capsid was further utilised to develop a therapy named 4D-150, encoding microRNA (miRNA) targeting VEGF-C and a codon-optimised sequence of aflibercept. Preclinical results presented at the 2021 Annual American Society of Gene and Cell Therapy Meeting showed improvement in CNV in NHP treated with 1E11, 3E11 and 1E12 vg/eye of 4D-150, at 6 weeks prior to disease induction. In addition, both aflibercept and miRNA VEGF-C were expressed for at least 12 months in NHP [60]. These results led to a phase I/II clinical trial (PRISM [NCT05197270]) (Table 1) in which participants with wAMD were treated with 6E9, 1E10, 3E10 vg of 4D-150 intravitreally and monitored for 24 months. Interim data from July 2023 reported a 100% reduction in the mean annual rate of anti-VEGF injections at 36 weeks in patients treated with 3E10 vg of 4D-150 [61]. The gene therapy was well tolerated in all three cohorts, with no reported inflammation (grade ≥ 1) or toxicity associated with the treatment [61]. The study includes a phase II dose-expansion part, which has recently completed enrolment. Furthermore, recruitment for an additional trial investigating 4D-150 in patients with DME is expected to start in 2023 [61]. While the 7m8 capsid was generated in mice, R100 vectors evolved in NHP retinas, which might yield a better performance in human tissues, possibly requiring lower doses to reach efficient transduction, a desirable feature when developing AAV-based therapies.
The AAV serotype plays an important role in tissue tropism, also affected by molecular and anatomical differences across species [43, 62]. AAV8 has previously shown better transduction performance in the outer retina, compared with AAV2, when injected by the subretinal route in NHP [63]. However, while AAV8 seems to be more efficient when administered by subretinal injection in NHP, only AAV2 was able to transduce the inner retina from the vitreous in rats [43]. Nevertheless, CNV development led to enhanced transduction for both serotypes when injected into mice intravitreally [64]. AAV8 vectors have also shown good transduction efficiency in rats’ retinas when delivered by suprachoroidal injections, which could be an attractive route for diseases affecting the choroid, such as uveitis and wAMD.
This was demonstrated by Ding et al., who investigated in rats the suprachoroidal delivery of 1.2E8 vg of RGX-314 (also named ABBV-RGX-314), a gene therapy comprised of AAV2/8 vectors harbouring an anti-VEGF antibody fragment similar to ranibizumab [65]. Suppression of VEGF-induced vasodilation and leakage was reported in treated animals at 2 weeks and these results were comparable with that seen with subretinal delivery of the same formulation [65]. This therapy was previously tested for subretinal delivery and showed promising results in transgenic mice expressing VEGF in the retina [66]. The successful preclinical studies led to a phase I/IIa clinical trial (NCT03066258) (Table 1) in which subjects with wAMD received 3E9, 1E10, 6E10, 1.6E11 or 2.5E11 vg/eye, by subretinal route, and were monitored for up to 104 weeks. Results presented in the 2022 American Academy of Ophthalmology showed that RGX-314 was generally well tolerated at all doses. Yet, one severe drug-related adverse event was reported in a patient treated with the highest dose and nine other adverse events considered non-drug-related were reported in four patients. Overall, treatment with subretinal RGX-314 decreased the frequency of anti-VEGF injections during the 4-year follow-up, especially for the three higher doses [67]. Moreover, patients enrolled in the long-term follow-up study receiving 6E10 and 1.6E11 vg/eye showed long-term treatment effects and improved visual acuity [67]. Subsequently, a phase IIb/III (ATMOSPHERE™) study and phase III (ASCENT™) study (NCT04704921; NCT05407636) (Table 1) were designed and are currently recruiting, as well as a 5-year follow-up with all patients (NCT03999801) (Table 1). A further trial (NCT04832724) (Table 1) comparing the pharmacodynamics of the formulation used in the previous studies with one produced by a commercial-ready manufacturing process is also underway, with preliminary results showing similar safety and clinical profile for both methods [68].
RGX-314 is additionally being assessed for suprachoroidal delivery in patients with wAMD and DR. For wAMD, the phase II AAVIATE™ study and the subsequent 5-year follow-up (NCT04514653; NCT05210803) (Table 1) are evaluating patients treated with suprachoroidal RGX-314 at 2.5E11, 5E11 or 1E12 vg/eye. Results presented at the 2023 meeting of the American Society of Retinal Surgeons demonstrated good tolerability with all doses after 6 months. Additionally, an 85% reduction in treatment burden was observed with the highest dose and no significant differences were found in clinical outcomes of patients positive for NAb [69]. It is important to mention that the study was expanded to add a cohort including two regimens of prophylactic corticosteroids with the highest dose, with early results indicating a meaningful reduction in intraocular inflammation with the preventive therapy [69]. Interestingly, the phase I/IIb trial assessing subretinal RGX-314 in wAMD enrolled participants regardless of their nAB titers and did not report corticosteroid therapy throughout the study (NCT03066258). These data align with previous studies reporting a greater immune privilege in the subretinal space in comparison with other ocular routes [70]. Furthermore, both subretinal and suprachoroidal RGX-314 might benefit from lower seroprevalence of nABs against AAV8 in different populations and human vitreous [25].
In this context, She et al. chose AAV2/8 to deliver by subretinal injection a novel VEGF trap, similar to aflibercept but containing domains 1 and 2 of VEGFR1 and domain 3 of VEGFR2 [71]. The study evaluated 4E6, 4E7, 4E8, and 4E9 vg/eye of the therapy injected 4 weeks before CNV induction in mice and found a relatively low minimum effective dose, ranging from 4E7 to 4E8 vg/eye. Similarly, Hughes et al. sought to improve safety and reduce unwanted immune responses by utilising AAV2/8 vectors delivering single-chain fragment variable (scFv) anti-VEGF antibodies, which consist of the minimum part of an antibody necessary for antigen binding [72]. The scFv format should induce less immune response compared with antibodies containing the Fc domain and improve AAV packaging. Subretinal administration of AAV2/8 encoding anti-VEGF scFv (8E8 vg) at 3 and 8 weeks before CNV induction led to preservation of function and reduced CNV in mice, with results comparable with intravitreal injected anti-VEGF IgG1. However, anti-VEGF scFc was found in both serum and sham eyes of AAV-treated animals, indicating systemic absorption. It could lead to production of nABs in addition to potentially serious systemic antiangiogenic activity. The authors attributed this outcome to the blood–retinal barrier (BRB) rupture due to the disease and the subretinal injection, which was administered through the choroid. It highlights the relevance of other factors besides vector serotype, such as administration route and expressed protein, when assessing AAV-based therapies for such complex diseases as AMD. Moreover, due to the multifactorial and chronic nature of AMD, it is questionable whether one single therapeutic target can control the disease [73].
In this regard, Askou et al. developed a multigenic expression vector consisting of AAV5 encoding an RPE-specific promoter, VDM2, driving the expression of PEDF and an miRNA cluster targeting VEGFA [74]. PEDF is an endogenous angiogenesis inhibitor expressed in the retina and other cell populations within the eye, known to be involved in inflammation, neuroprotection, and angiogenesis [74]. The therapy was assessed in the CNV model after pretreatment with 4.2E9 vg by subretinal injection, administered 50 days before laser administration. The dual-acting therapy led to reduced VEGFA levels and CNV area, although the latter was not significantly different from treatment with each gene alone. Still, better results were seen with the bicistronic vector, and the expression of molecules inhibiting the VEGF signalling by different mechanisms seems to be an effective approach for wAMD.
Indeed, favourable safety and efficacy outcomes reported in the 4D-150 clinical trial support the multigenic anti-VEGF approach. Intravitreal delivery of VEGF A, B, and C inhibitors through an engineered capsid, as in 4D-150, led to a reduction of treatment burden without causing intraocular inflammation at doses lower than those used in similar studies with single gene platforms, such as ADVM-022 and RGX-314 [56, 67, 75]. Moreover, the strategy used in 4D-150 seems to counteract the need for high doses with intravitreal delivery, often associated with intraocular inflammation, as seen in other studies using this route [56, 76].
2.3 AAV-Mediated Gene Therapies Targeting the Complement Cascade
Li et al., on the other hand, proposed a multigenic therapy targeting multiple pathways involved in wAMD, complement activation and angiogenesis [77]. The study employed a VEGF inhibitor domain containing the same sequence of aflibercept, a complement inhibitor domain derived from soluble complement receptor 1, and a dual decoy receptor (ACVP1). The latter consisted of a VEGF binding motif, a complement binding motif, and the C-terminal of Fc. These molecules were packaged into AAV2 vectors and intravitreally injected (7.5E8 vg) in mice submitted to laser burns 21 days later to induce CNV. Improvement in CNV was observed in all AAV-treated groups, with better results found in animals treated with the dual inhibitor system, suggesting that targeting these two pathways may be beneficial for wAMD treatment. It is known that polymorphisms in multiple genes encoding complement pathway proteins are associated with increased risks for AMD, with most cases linked to the complement factor H (CFH) and CFH-related genes [78]. CFH is a soluble inhibitor of the complement alternative pathway, which has been suggested to be a common target in both wet and dry AMD [79]. In fact, the only therapies recently approved by the FDA for GA secondary to AMD target the complement pathway by inhibiting complement C3 and C5, central proteins in classical, lectin and alternative pathways [34, 80, 81].
In 2011, Cashman et al. explored the role of complement activation in both types of AMD and developed a gene therapy consisting of adenovirus vectors mediating the expression of soluble CD59, a complement activation regulatory protein [82]. CD59 inhibits the formation of the membrane attachment complex (MAC), which plays a pathological role in AMD, correlating with disease severity and RPE damage [83]. The therapeutic effect of soluble CD59 in experimental CNV, reported by Cashman et al., led to clinical studies with this protein, targeting dry and wet AMD, using AAV2 as vectors. Hence, AAV2 encoding soluble CD59, named HMR59, were intravitreally injected at 3.56E10, 1.07E11, and 3.56E11 vg/eye in patients with dry AMD during a phase I trial (HMR1001 [NCT03144999]) (Table 1) completed in December 2019 [84]. In 2020, Janssen Pharmaceuticals acquired rights to HMR59 and renamed the gene therapy as JNJ-1887 and JNJ-81201887 [85]. Results presented at the American Academy of Ophthalmology 2022 Annual Meeting showed that the therapy was well tolerated during the 2-year follow-up and decreased lesion growth rate, with no progression for the neovascular form detected. Five patients developed mild intraocular inflammation and were either monitored until resolution or treated with corticosteroids [85, 86]. Janssen Pharmaceuticals is currently enrolling patients with GA secondary to dry AMD for a phase IIb trial (PARASOL [NCT05811351]) (Table 1). The study will compare two doses of HMR59 with placebo injections and patients will receive oral and periocular corticosteroid therapy. A phase I trial with intravitreal HMR59 for wAMD (HMR1002 [NCT03585556]) (Table 1) commenced in September 2018 and finished in 2022. This study included an initial anti-VEGF injection 7 days prior to HMR59 (3.56E11 and 1.071E12 vg) and a short course of oral prednisolone. Patients were monitored up to 24 months. Primary outcomes reported in 2019 showed that 18.2% of patients treated with the lower dose did not require retreatment in the first 6 months, but additional anti-inflammatory intervention was still needed in three patients [76]. Safety data updates were shared during the 2023 Association for Research in Vision and Ophthalmology (ARVO) meeting, indicating that the mild to moderate ocular inflammation reported in 16% of patients was resolved with the prophylactic therapy [84]. No further data regarding this therapy have been released, but if successful, this therapy may benefit patients exhibiting CNV and GA simultaneously in the same eye, a condition reported in several clinical studies [87]. In addition, anti-VEGF therapy has been associated with GA development, making it even more challenging to treat patients with this combined phenotype [87].
Long-lasting expression of anti-complement factors affecting MAC formation was also explored by Dreismann et al., who proposed the use of AAV encoding complement factor I (CFI), an important regulator of the alternative pathway, to treat AMD [88]. This study initially demonstrated the expression of CFI in murine RPE and photoreceptors after subretinal injection of 1E07 vg of AAV2 vectors encoding CFI (GT005). The efficient CFI expression in vitro and in vivo led to a phase I/II clinical trial (FOCUS [ NCT03846193]) (Table 1) assessing the safety and efficacy of 2E10, 5E10, and 2E11 vg of GT005 by subretinal transvitreal injection or via a proprietary subretinal delivery device (Orbit™; 5E10 and 2E11 only) in patients with GA secondary to dry AMD. Preliminary data announced in the 2022 ARVO meeting showed that the therapy had been well tolerated. No serious adverse events were reported, and most injection-related reactions were mild. AAV2 nABs did not change in 95% of patients at week 5, while 55% had pre-existing anti-AAV2 antibodies. Additionally, increased levels of CFI were found in the vitreous of most patients, as well as factors indicating reduced complement activity [89]. This study is ongoing and is estimated to be completed in 2027. Phase II trials and a subsequent follow-up study (HORIZON [NCT04566445], EXPLORE [NCT04437368], and ORACLE [NCT05481827]) (Table 1) are also underway and are estimated to finish in 2025 and 2028.
Despite the greater prevalence of dry AMD compared with the neovascular type, research efforts, along with clinical and preclinical studies, predominantly target the latter. This discrepancy highlights the urgent unmet need for developing effective treatment options for the dry form, which would highly benefit from AAV-mediated gene delivery [90].
2.4 AAV-Mediated Gene Therapies Targeting Inflammation and Oxidative Stress
Evidence of the influence of innate immunity and oxidative stress in retinal degeneration has led to the investigation of several targets implicated in these processes for both wet and dry AMD. Wang et al. used scAAV2 vectors to deliver Ras-related protein 1A (Rap1A), a protein important to maintain the integrity of RPE and attain its barrier properties [91]. The expression of Rap1A was driven by an RPE-specific promoter, VMD2. The efficacy of the therapy was investigated in mice treated with 5E8 vg subretinally, injected 5 weeks before establishment of the laser-induced CNV model. Reduced levels of inflammation, angiogenesis and apoptosis markers were detected in RPE/choroid tissues, in addition to attenuated CNV lesions. The positive findings reported with Rap1A gene delivery bring attention to other pathways, apart from VEGF involved in the pathologic angiogenesis occurring in AMD.
In contrast, Young et al. targeted dry AMD and proposed inhibiting inflammasome signalling pathways in retinal cells by delivering cytokines ensuing from inflammasome activation [92]. The study employed AAV2 with four tyrosine-to-phenylalanine and one tyrosine-to-valine mutation [AAV2quad(Y-F) + T491V] to enhance RGC and bipolar cells transduction while sparing photoreceptors and RPE. The vector was used to deliver genes encoding a cell-penetrating sequence linked to a caspase activator and recruitment domain (CARD), a soluble caspase-1 inhibitor. This therapy was tested in two models of RPE-choroid oxidative damage—the acute model induced by NaIO3 injection and the chronic oxidative stress by RPE-specific Sod2 deletion, both in mice. Treatment with an intravitreal injection of 3E10 vg/eye administered 1 month before disease induction improved retinal function in the acute model of oxidative stress. The same treatment in mice carrying the RPE-specific deletion of Sod2 decreased interleukin (IL)-1β expression in the retina and improved retinal degeneration at later stages of the disease [92]. Furthermore, another study tested this therapy in liver-X-receptor alpha (LXRα-/-) knockout mice, which exhibit phenotypic features of early dry AMD. Animals treated with 0.8–1.2E10 vg/eye, administered by intravitreal injection, showed preserved RPE-dependent ERG curves and decreased production of inflammatory cytokines in the retina, RPE and choroid [93]. These studies indeed showed the therapeutic potential of CARD protein, expressed through AAV vectors, in regulating pathological processes involved in dry AMD, such as inflammation and oxidative stress. However, this therapy did not promote photoreceptor protection in the early dry AMD model and when administered at an early stage in the chronic oxidative stress model [92, 93]. This result highlights the importance of assessing the best moment for therapeutic intervention in preclinical studies and emphasises the relevance of having well-characterised models replicating different manifestations and stages of the disease at a feasible time course [94]. It also encourages testing the therapy in different models, considering that no single model recapitulates all features of human AMD [94].
In this context, Biswal et al. investigated the best timing for antioxidant gene therapy in the RPE-specific Sod2 knockout model of dry AMD by comparing AAV1-mediated administration of the Sod2 gene administered at the early or late stages of the disease by subretinal injections [95]. The antioxidant therapy could prevent retinal degeneration only when administered before electrophysiological and morphological tissue damage [95]. Interestingly, this group later injected AAV1 vectors (1E9 vg) encoding a modified form of erythropoietin (EPO-R76R), a hormone with antioxidant and neuroprotective effects, in mice with the same experimental model, but at a disease stage between the two previously investigated [96, 97]. In this case, the therapy was able to protect RPE and photoreceptors from oxidative stress, again showing that earlier intervention with antioxidants might result in better protection, in addition to demonstrating the potential of EPO-R76R.
Hitherto, we have discussed therapies involving gene augmentation or addition, utilising AAV to deliver a therapeutic gene targeting specific aspects of the disease pathogenesis [27]. Nonetheless, gene-editing technology has evolved and has broadened the range of candidate therapies for AMD. Huang et al. developed a dual-rAAV2/1 system delivering clustered regularly interspaced short palindromic repeats (CRISPR)/Cas 9 from Streptococcus pyogenes (SpCas9) to edit and deplete VEGFR2 and thereby mitigate pathological angiogenesis [98]. The intercellular adhesion molecule 2 (pICAM2) promoter was used to drive the expression of the SpCas9 and direct the VEGFR2 mutation to endothelial cells. Mice treated with 3.75E9 vg intravitreally, administered immediately after or 7 days post CNV induction, exhibited a 30% decrease in VEGFR2 expression, along with reduced CNV areas, with no toxicity associated [98]. However, owing to the large size of the SpCas9 gene, a dual-AAV system is usually required to deliver the SpCas9 gene into one vector and the sgRNA with the donor template DNA in the other [99]. Hence, high vector doses are usually needed to achieve efficient co-infection and thereby enable gene editing [100]. Koo et al. overcame the need for dual-AAV platforms by using LbCpf1 CRISPR, which has the advantage of having a smaller gene size compared with SpCas9. This system was packaged into AAV2/9 vectors and intravitreally injected in mice (2E10 vg) prior to laser burns for CNV. The therapy aims to induce mutations in VEGFA and HIF1a [101]. Although improvements in CNV comparable with aflibercept were observed, the employment of a single-AAV approach did not result in lower doses compared with other studies using the SpCas9 system [74, 98]. Nevertheless, gene editing systems stand as a promising technology that may overcome some common challenges faced when using traditional gene therapy. Still, off-target effects, bioethical and safety concerns, in addition to technical difficulties, limit the application of this tool and hinder its clinical translation [100]. However, off-target events can be minimised with other delivery approaches, including non-viral vectors [102]. Although clinical trials using gene editing to treat AMD are yet to be conducted, current studies targeting other diseases, including inherited retinal disorders, may pave the way to a broader application of this innovative technique [103].
3 Diabetic Retinopathy and Diabetic Macular Edema
DR is a microvascular complication of diabetes mellitus (DM) type 1 and 2, present in about 30% of all patients [104]. DM is one of the most significant public health concerns, estimated to be affecting over 570 million people by 2025 [105]. It consists of a group of conditions characterised by impaired glucose metabolism caused by insulin secretion deficiency and/or its biological function disorder, resulting in hyperglycaemia and several vascular complications [106]. DR is the most common complication of DM, estimated to be present in more than 103 million people worldwide, and ranking as one of the leading causes of preventable blindness in the working-age population [107]. In its severe stage, DR highly impacts the quality of life of patients due to its association with social isolation, reduced physical activity, and dependence on daily activities, resulting in a significant economic and healthcare burden [108, 109]. The disease can be classified as non-proliferative (mild, moderate, severe, or very severe) or proliferative (early, high-risk, or severe), with the latter corresponding to the late stage, characterised by neovascularisation [109]. At any stage of both proliferative and non-proliferative DR, patients can develop DME. DME is a complication of DR resulting from outer BRB disruption due to damaged endothelial tight junctions, allowing extravasation and accumulation of fluid in the macula and consequently vision loss [105, 110]. The first clinical stage, mild non-proliferative DR, is identified by the formation of microaneurysms or small haemorrhage spots in the retina, which may lead to exudation and DME, and/or become more diffuse and severe, reaching a preproliferative stage or moderate/severe nonproliferative DR. This stage is characterised by capillary occlusion and non-perfused areas, typically evidenced as ‘cotton wool’ spots (soft exudates), venous dilatation and intraretinal microvascular abnormalities.
As the disease progresses to proliferative DR, neovascularisation and vitreous or preretinal haemorrhage are observed, which can lead to retinal detachment and subsequent blindness [104, 106]. The disease has a slow onset and gradual progression, affected by several risk factors, including glycaemic control, hypertension, hyperlipidaemia, obesity, duration of DM, puberty, and pregnancy [111]. Ocular and genetic factors such as cataract surgery, myopic refractive error, and heritability have also been associated with DR [104, 112, 113]. Hyperglycaemia is the main contributor to DR development and progression by driving different metabolic pathways that result in oxidative stress, inflammation, microvascular dysfunction, and mitochondrial damage. In turn, these processes upregulate factors associated with BRB disruption and production of proangiogenic factors and hormones, leading to proliferative DR and DME [114, 115]. Other events, including neurodegeneration and activation of the renin-angiotensin system (RAS), have also been implicated in DR development [104]. Although pathways triggered by hyperglycaemia underlie the pathophysiology of DR, retinal insults persist even after blood glucose is controlled, a phenomenon known as metabolic memory [115]. The molecular mechanism driving this phenomenon remains unclear, but the accumulation of reactive oxygen species and its resulting epigenetic modifications are thought to play crucial roles in this process [115, 116].
The management of this multifactorial and complex disease includes surgical interventions such as panretinal laser photocoagulation and vitrectomy, as well as administration of anti-VEGF and anti-inflammatory agents [117, 118]. However, as invasive procedures are associated with attendant adverse effects, the use and success of these interventions can be limited [104, 115]. Furthermore, these therapies only ameliorate vision temporarily and are not able to fully attenuate disease progression or reverse the ocular damage, prompting the investigation of new strategies, including gene therapies [118, 119]. The most common approaches in gene therapies for DR and DME target factors involved in neovascularisation, vascular hyperpermeability, and degeneration [119].
3.1 AAV-Mediated Gene Therapies Targeting Angiogenesis
AAV-mediated expression of molecules interfering with VEGF pathways is a common approach to treat DR, as it was with wAMD, given the important role played by VEGF in ocular angiogenesis [28]. In this regard, sFLT-1 has been investigated in different models of neovascularisation and retinopathies [120,121,122,123]. Early studies showed that AAV2 vectors delivering sFLT-1 by the intravitreal route inhibited retinal neovascularisation in the oxygen-induced retinopathy (OIR) model in mice [120, 122]. Likewise, sFLT-1 expressed through AAV5 vectors by subretinal injections prevented DR in spontaneously diabetic non-obese Torii (SDT) rats [121]. Diaz-Lezama et al. reported a slightly different approach, evaluating AAV2 vectors delivering either sFLT-1 or vasoinhibins, a family of peptides produced in the retina with antiangiogenic and anti-permeability effects [123]. Unlike previous studies, this therapy was injected in rats 2 weeks after diabetes induction, when BRB breakdown had been detected. The animals with streptozotocin (STZ)-induced diabetes were treated with an intravitreal injection (2.8E9 vg) of sFLT-1 or vasoinhibins vectors, and 4 weeks later exhibited restored BRB. More interestingly, this work showed that the diabetic condition enhanced the retinal transduction of AAV2 administered intravitreally compared with healthy subjects [123]. Although this may be beneficial for the clinical application of AAV-mediated therapies in DR patients, more caution should be taken when translating therapeutic candidates tested before disease onset in the preclinical phase.
Indeed, as previously discussed, important differences were observed in DME and wAMD patients treated with AAV2.7m8 encoding aflibercept (ADVM-022) at the same dose. Whereas dose-limiting toxicity was reported in DME patients, positive results with wAMD warranted further investigation in a phase II trial, highlighting the influence of the diabetic condition on the ocular safety profile of AAV therapies [57]. The aforementioned study started in 2020 and planned to monitor patients with DME treated with intravitreal ADVM-022 at 6E11 and 2E11 vg/eye (INFINITY [NCT04418427]) (Table 2) for 96 weeks. In May 2021, the study was unmasked due to a suspected unexpected serious adverse reaction (SUSAR) of hypotony in the eye treated with the highest dose, with additional patients in this cohort exhibiting this reaction later. However, the lowest dose was not associated with these events and decreased the need for supplemental anti-VEGF injection, with 61% of patients remaining injection-free at week 24. Nevertheless, Adverum is no longer developing this therapy for DME and has attributed these adverse events to the comorbid nature of diabetes, including renal impairment and vascular diseases [57, 124].
Meanwhile, the ALTITUDE™ clinical trial, treating patients with DR without centre-involved DME with a suprachoroidal injection of RGX-314 (2.5E11 and 5E11 vg/eye), reported good tolerability at 6 months, with no associated drug-related severe adverse events (NCT04567550, NCT05296447) (Table 2) [125]. RGX-314 consists of AAV2/8 vectors encoding an anti-VEGF antibody fragment [65]. Patients in the ALTITUDE trial did not receive prophylactic anti-inflammatory treatment, but topical corticosteroids were required in three subjects exhibiting mild intraocular inflammation within the first 6 months. These data were reported at the 2023 meeting of the American Society of Retinal Surgeons and also stated improvement of two steps or more in the DR severity scale (DRSS) relative to baseline in 20% of all patients, and any improvement in DRSS in 54%, with no subject showing worsening of two or more steps [125]. The trial has been expanded to add two cohorts with a higher dose of 1E12 vg/eye along with a short course of corticosteroid prophylactic therapy [125]. Interim safety data from the latest cohorts report no RGX-314-related severe adverse events and the absence of intraocular inflammation after corticosteroid therapy [125]. The estimated completion date of the ALTITUDE trial is 2024, with the follow-up phase finishing in 2028. Both ADVM-022 and RGX-314 have been previously assessed for wAMD and are the only AAV-based gene therapies reaching clinical trials for DR to date. Likewise, 4D-150, also investigated for wAMD, has recently received FDA Investigational New Drug clearance to initiate a phase II trial in DR patients, including a dose-confirmation and a dose-expansion study (SPECTRA [NCT05930561]) (Table 2) [126]. The difficulty in translating gene therapies for DR is likely related to the complex pathogenesis of the disease, which directly affects patient selection and endpoint standardisation in clinical studies [119].
However, promising outcomes have been reported in preclinical research investigating different targets, including those involved in ocular angiogenesis. For instance, Cahoon and colleagues used AAV2 to deliver a soluble, stable, and more potent form of angiopoietin 1 (Ang 1), the cartilage oligomeric matrix protein Ang1 (COMP-Ang1) [127]. Ang 1 is a vascular growth factor that regulates vascular permeability and neovascularisation [127, 128]. A single intravitreal injection of this therapy at 2E9 vg was administered to diabetic Ins2Akita mice at an early stage of DR and improved vascular structure, visual function, and BRB integrity [127]. The same group later injected this therapy in animals with vascular damage resulting from the same disease model but at a more advanced stage [129]. Despite preventing proliferative vascular retinopathy, this therapy could not ameliorate neuroretina degeneration and visual function, suggesting the involvement of other processes, besides vascularity, in neuronal cell loss. This study emphasises how difficult it is to recapitulate later stages of DR given the high mortality rate of diabetic animals. Furthermore, it corroborates the findings reported by Diaz-Lezama et al. because the same dose considered safe when injected before disease onset led to inflammation when administered after the vascular breakdown [123].
In this context, Biswal et al. proposed a prophylactic gene therapy for DR patients at risk of developing neovascularisation [130]. The authors utilised scAAV2 vectors delivering a cassette containing hypoxia responsive elements and the glial fibrillary acidic protein (GFAP) promoter for a controlled expression of endostatin in Müller cells. The therapy was assessed in the OIR model before exposure to high oxygen levels. The intravitreal treatment of 2E9 vg led to reduction in VEGF levels, with a 93% decrease in the murine neovascular area [130]. The study also demonstrated the efficiency of the hypoxia regulatory element, although the specificity of the Müller cell-specific promoter was not assessed. The restricted and regulated expression of molecules such as endostatin stands as an attractive strategy for DR, in which overexpression may result in toxicity. Therefore, cell-specific promoters and elements allowing a controlled expression of the therapeutic gene offer additional safety to gene therapies and are relevant features to be considered in potential candidates for DR treatment. Huang et al. also included a promoter specific to endothelial cells (pICAM2) in their gene editing therapy targeting VEGFR2, as discussed in the AMD section, which additionally showed positive results in the OIR model. In this case, the therapy administered after hyperoxia led to reduced CNV areas and VEGFR2 expression [98].
3.2 AAV-Mediated Gene Therapies Targeting Other Pathways
Despite the success of anti-VEGF therapy in ophthalmology, approximately 15–25% of DR patients do not respond to it. Therefore, therapies targeting additional pathways involved in DR pathogenesis, such as neurodegeneration, inflammation, and the RAS, are needed [104, 118]. In this context, a research group has explored the protective axis of RAS and demonstrated the therapeutic potential of AAV-mediated expression of angiotensin-converting enzyme-2 (ACE2) and angiotensin 1–7 (Ang 1–7) in models of DR [131, 132]. Their latest work sought to restore RAS balance in mice with STZ-induced diabetes by overexpressing ACE2 through AAV2 vectors [131]. ACE2 is the main enzyme of the vasoprotective axis of the RAS, responsible for converting angiotensin 2 (Ang 2) into Ang 1–7, which has vasodilatory, antiproliferative, and apoptotic effects [131]. Animals treated with an intravitreal injection of about 1E9 vg of AAV2-ACE2, before and after disease induction, showed attenuated DR with improved retinal vasculature and inflammation, supporting the use of RAS regulators in DR [131].
Other approaches have aimed to restore the BRB, whose breakdown occurs in the macular edema seen in DR and other ocular pathologies [133, 134]. Vacca et al. proposed a therapy consisting of AAV ShH10 expressing Dystrophin protein 71 (Dp71), a cytoskeletal protein mainly localised in Müller cells with a putative role in BRB integrity [134]. The shH10 AAV capsid was a variant selected to efficiently transduce Müller cells through intravitreal injection [135]. Intravitreal treatment with 1.8E10 vg of this construct in mice with increased BRB permeability due to Dp71 knockdown resulted in reabsorption of retinal edema and restoration of BRB homeostasis [134]. Protection of BRB integrity was also observed by Yang et al. after silencing the Nogo-B protein gene by siRNA, delivered through AAV PHP.eB vectors in rats with STZ-induced DR [136]. The AAV PHP.eB is a variant of AAV9 developed for enhanced transduction in the central and peripheral nervous system [137]. The Nogo-B protein seems to play an essential role in BRB regulation, being proposed as an inducer of vascular permeability in experimental DR whilst acting as a protector under normal conditions [136]. The preliminary findings reported by Yang et al. and Vacca et al. are encouraging and should spur further research into BRB regulators for DR gene therapies [134, 136].
Likewise, recent studies have reported promising early results with gene therapies targeting factors involved in inflammation and neurodegeneration for DR treatment. Jiang et al. used AAV8 vectors to express the D-amino acid oxidase (DAAO) enzyme, whose substrate is increased by inflammation and correlates to retinal ganglion cell death [138]. DAAO overexpression in rats treated with 4E10 vg, administered intravitreally 1 week before STZ administration, prevented RGC loss and protected from neurovascular abnormalities observed in early DR [138]. Employing the same animal model, Lee et al. investigated the effect of mammalian target of rapamycin (mTOR)-inhibiting short hairpin RNA (shRNA), delivered by AAV2 vectors, in mice with early DR [139]. The mTOR pathway regulates angiogenesis and inflammation, with its inhibition resulting in anti-inflammatory and antiangiogenic effects, as previously reported in animal models of laser-induced CNV and OIR [140, 141]. In STZ-induced diabetes, mTOR signalling inhibition reduced cell loss, vascular permeability, and retinal thinning. It was achieved with 5E7 vg of the gene therapy, administered intravitreally 1 month before STZ injection [139]. Indeed, targets interfering with different pathways involved in DR pathogenesis stand as promising strategies and may pave the way to more effective gene therapies. Nonetheless, a more in-depth understanding of the disease pathogenesis is still required to identify new targets and pathways, allowing the development of successful and translatable gene therapies for DR.
4 Conclusion
The progress made with AAV-mediated gene therapies in inherited retinal diseases has paved the way for developing promising treatments for acquired conditions such as AMD and DR. This group of heterogenous and multifactorial diseases are among the leading causes of blindness and visual loss globally, even with effective treatment options available, highlighting the demand for better alternatives. Their complex pathogenesis and lack of complete understanding of the disease mechanism make it challenging to find effective solutions. However, AAV-mediated delivery of proteins affecting pathways implicated in these disorders has emerged as a promising approach, with positive outcomes already seen in current clinical trials. Preclinical studies have investigated several strategies, including modified capsids and constructs containing elements for targeted and controlled expression of a broad range of molecules. Despite the therapeutic effect shown with the proposed therapies, most studies assessed their protective effect, with treatments applied before disease induction and tissue damage. Hence, the efficacy of such therapies in a diseased setting remains to be addressed. Still, a controllable and more specific expression seems suitable to overcome drawbacks reported in most studies. Furthermore, due to the multifactorial nature of these disorders, a multigenic strategy may be needed to tackle the variety of pathways involved. Thus, the versatility of AAV-based gene therapies supports their application in retinal and choroidal vascular diseases, such as AMD and DR. Nevertheless, further development is required before these strategies are translated into clinical practice, especially considering that they have the potential to create lifelong and unforeseen complications and they must outperform current treatments while being affordable for payors and patients. In the meantime, accumulating further in-depth knowledge of disease mechanisms, AAV biology, and gene expression might ease the development and approval of promising therapies that could potentially change many lives.
References
Rodrigues GA, Shalaev E, Karami TK, et al. Pharmaceutical development of AAV-based gene therapy products for the eye. Pharm Res. 2018;36(2):29. https://doi.org/10.1007/s11095-018-2554-7.
Lee JH, Wang JH, Chen J, et al. Gene therapy for visual loss: opportunities and concerns. Prog Retin Eye Res. 2019;68:31–53. https://doi.org/10.1016/j.preteyeres.2018.08.003.
Petit L, Khanna H, Punzo C. Advances in gene therapy for diseases of the eye. Hum Gene Ther. 2016;27(8):563–79. https://doi.org/10.1089/hum.2016.040.
Botto C, Rucli M, Tekinsoy MD, et al. Early and late stage gene therapy interventions for inherited retinal degenerations. Prog Retin Eye Res. 2022;86: 100975. https://doi.org/10.1016/j.preteyeres.2021.100975.
Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2231–9. https://doi.org/10.1056/NEJMoa0802268.
Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2240–8. https://doi.org/10.1056/NEJMoa0802315.
Hauswirth WW, Aleman TS, Kaushal S, et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19(10):979–90. https://doi.org/10.1089/hum.2008.107.
Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, phase 3 trial. Lancet. 2017;390(10097):849–60. https://doi.org/10.1016/S0140-6736(17)31868-8.
Atchison RW, Casto BC, Hammon WM. Adenovirus-associated defective virus particles. Science. 1965;149(3685):754–6. https://doi.org/10.1126/science.149.3685.754.
Rose JA, Berns KI, Hoggan MD, Koczot FJ. Evidence for a single-stranded adenovirus-associated virus genome: formation of a DNA density hybrid on release of viral DNA. Proc Natl Acad Sci U S A. 1969;64(3):863–9. https://doi.org/10.1073/pnas.64.3.863.
Bucher K, Rodriguez-Bocanegra E, Dauletbekov D, Fischer MD. Immune responses to retinal gene therapy using adeno-associated viral vectors—implications for treatment success and safety. Prog Retin Eye Res. 2021;83: 100915. https://doi.org/10.1016/j.preteyeres.2020.100915.
Srivastava A, Lusby EW, Berns KI. Nucleotide sequence and organization of the adeno-associated virus 2 genome. J Virol. 1983;45(2):555–64. https://doi.org/10.1128/JVI.45.2.555-564.1983.
Im D-S, Muzyczka N. The AAV origin binding protein Rep68 is an ATP-dependent site-specific endonuclease with DNA helicase activity. Cell. 1990;61(3):447–57.
McLaughlin SK, Collis P, Hermonat PL, Muzyczka N. Adeno-associated virus general transduction vectors: analysis of proviral structures. J Virol. 1988;62(6):1963–73. https://doi.org/10.1128/JVI.62.6.1963-1973.1988.
Khanani AM, Thomas MJ, Aziz AA, et al. Review of gene therapies for age-related macular degeneration. Eye (Lond). 2022;36(2):303–11. https://doi.org/10.1038/s41433-021-01842-1.
Naso MF, Tomkowicz B, Perry WL 3rd, Strohl WR. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs. 2017;31(4):317–34. https://doi.org/10.1007/s40259-017-0234-5.
Srivastava A. In vivo tissue-tropism of adeno-associated viral vectors. Curr Opin Virol. 2016;21:75–80. https://doi.org/10.1016/j.coviro.2016.08.003.
Whitehead M, Osborne A, Yu-Wai-Man P, Martin K. Humoral immune responses to AAV gene therapy in the ocular compartment. Biol Rev Camb Philos Soc. 2021;96(4):1616–44. https://doi.org/10.1111/brv.12718.
Auricchio A, Kobinger G, Anand V, et al. Exchange of surface proteins impacts on viral vector cellular specificity and transduction characteristics: the retina as a model. Hum Mol Genet. 2001;10(26):3075–81.
Yang GS, Schmidt M, Yan Z, et al. Virus-mediated transduction of murine retina with adeno-associated virus: effects of viral capsid and genome size. J Virol. 2002;76(15):7651–60. https://doi.org/10.1128/jvi.76.15.7651-7660.2002.
Surace EM, Auricchio A, Reich SJ, et al. Delivery of adeno-associated virus vectors to the fetal retina: impact of viral capsid proteins on retinal neuronal progenitor transduction. J Virol. 2003;77(14):7957–63. https://doi.org/10.1128/jvi.77.14.7957-7963.2003.
Weber M, Rabinowitz J, Provost N, et al. Recombinant adeno-associated virus serotype 4 mediates unique and exclusive long-term transduction of retinal pigmented epithelium in rat, dog, and nonhuman primate after subretinal delivery. Mol Ther. 2003;7(6):774–81. https://doi.org/10.1016/s1525-0016(03)00098-4.
Han IC, Cheng JL, Burnight ER, et al. Retinal tropism and transduction of adeno-associated virus varies by serotype and route of delivery (Intravitreal, Subretinal, or Suprachoroidal) in rats. Hum Gene Ther. 2020;31(23–24):1288–99. https://doi.org/10.1089/hum.2020.043.
Lugin ML, Lee RT, Kwon YJ. Synthetically engineered adeno-associated virus for efficient, safe, and versatile gene therapy applications. ACS Nano. 2020;14(11):14262–83. https://doi.org/10.1021/acsnano.0c03850.
Andrzejewski S, Moyle PM, Stringer BW, Steel JC, Layton CJ. Neutralisation of adeno-associated virus transduction by human vitreous humour. Gene Ther. 2021;28(5):242–55. https://doi.org/10.1038/s41434-020-0162-8.
Blindness GBD, Vision Impairment C, Vision Loss Expert Group of the Global Burden of Disease S. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to VISION 2020: the Right to Sight: an analysis for the Global Burden of Disease Study. Lancet Glob Health. 2021;9(2):e144–60. https://doi.org/10.1016/S2214-109X(20)30489-7.
Tan TE, Fenner BJ, Barathi VA, et al. Gene-based therapeutics for acquired retinal disease: opportunities and progress. Front Genet. 2021;12: 795010. https://doi.org/10.3389/fgene.2021.795010.
Lin F-L, Wang P-Y, Chuang Y-F, et al. Gene therapy intervention in neovascular eye disease: a recent update. Mol Ther. 2020;28(10):2120–38. https://doi.org/10.1016/j.ymthe.2020.06.029.
Wong WL, Su X, Li X, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014;2(2):e106–16. https://doi.org/10.1016/S2214-109X(13)70145-1.
Fleckenstein M, Keenan TDL, Guymer RH, et al. Age-related macular degeneration. Nat Rev Dis Primers. 2021;7(1):31. https://doi.org/10.1038/s41572-021-00265-2.
Thomas CN, Sim DA, Lee WH, et al. Emerging therapies and their delivery for treating age-related macular degeneration. Br J Pharmacol. 2022;179(9):1908–37. https://doi.org/10.1111/bph.15459.
Rim PHH, de Vasconcellos JPC, de Melo MB, et al. Correlation between genetic and environmental risk factors for age-related macular degeneration in Brazilian patients. PLoS ONE. 2022;17(6): e0268795. https://doi.org/10.1371/journal.pone.0268795.
Ferris FL 3rd, Wilkinson CP, Bird A, et al. Clinical classification of age-related macular degeneration. Ophthalmology. 2013;120(4):844–51. https://doi.org/10.1016/j.ophtha.2012.10.036.
Pavluk L. FDA Approves SYFOVRE™ (pegcetacoplan injection) as the First and Only Treatment for Geographic Atrophy (GA), a Leading Cause of Blindness. Apellis Press Release. 2023. https://investors.apellis.com/news-releases/news-release-details/fda-approves-syfovretm-pegcetacoplan-injection-first-and-only. Accessed 12 April 2023.
Neufeld J. Iveric Bio Receives U.S. FDA Approval for IZERVAY™ (avacincaptad pegol intravitreal solution), a New Treatment for Geographic Atrophy. 2023.
Lau PE, Jenkins KS, Layton CJ. Current evidence for the prevention of endophthalmitis in anti-VEGF intravitreal injections. J Ophthalmol. 2018;2018:8567912. https://doi.org/10.1155/2018/8567912.
Lai YK, Shen WY, Brankov M, et al. Potential long-term inhibition of ocular neovascularisation by recombinant adeno-associated virus-mediated secretion gene therapy. Gene Ther. 2002;9(12):804–13. https://doi.org/10.1038/sj.gt.3301695.
Lai CM, Estcourt MJ, Wikstrom M, et al. rAAV.sFlt-1 gene therapy achieves lasting reversal of retinal neovascularization in the absence of a strong immune response to the viral vector. Invest Ophthalmol Vis Sci. 2009;50(9):4279–87. https://doi.org/10.1167/iovs.08-3253.
Lai CM, Shen WY, Brankov M, et al. Long-term evaluation of AAV-mediated sFlt-1 gene therapy for ocular neovascularization in mice and monkeys. Mol Ther. 2005;12(4):659–68. https://doi.org/10.1016/j.ymthe.2005.04.022.
Lukason M, DuFresne E, Rubin H, et al. Inhibition of choroidal neovascularization in a nonhuman primate model by intravitreal administration of an AAV2 vector expressing a novel anti-VEGF molecule. Mol Ther. 2011;19(2):260–5. https://doi.org/10.1038/mt.2010.230.
Lai CM, Estcourt MJ, Himbeck RP, et al. Preclinical safety evaluation of subretinal AAV2.sFlt-1 in non-human primates. Gene Ther. 2012;19(10):999–1009. https://doi.org/10.1038/gt.2011.169.
Maclachlan TK, Lukason M, Collins M, et al. Preclinical safety evaluation of AAV2-sFLT01- a gene therapy for age-related macular degeneration. Mol Ther. 2011;19(2):326–34. https://doi.org/10.1038/mt.2010.258.
Dalkara D, Kolstad KD, Caporale N, et al. Inner limiting membrane barriers to AAV-mediated retinal transduction from the vitreous. Mol Ther. 2009;17(12):2096–102. https://doi.org/10.1038/mt.2009.181.
Heier JS, Kherani S, Desai S, et al. Intravitreous injection of AAV2-sFLT01 in patients with advanced neovascular age-related macular degeneration: a phase 1, open-label trial. Lancet. 2017;390(10089):50–61. https://doi.org/10.1016/S0140-6736(17)30979-0.
Rakoczy EP, Lai CM, Magno AL, et al. Gene therapy with recombinant adeno-associated vectors for neovascular age-related macular degeneration: 1 year follow-up of a phase 1 randomised clinical trial. Lancet. 2015;386(10011):2395–403. https://doi.org/10.1016/S0140-6736(15)00345-1.
Constable IJ, Pierce CM, Lai CM, et al. Phase 2a randomized clinical trial: safety and post hoc analysis of subretinal rAAV.sFLT-1 for wet age-related macular degeneration. EBioMedicine. 2016;14:168–75. https://doi.org/10.1016/j.ebiom.2016.11.016.
Lee SHS, Kim HJ, Shin OK, et al. Intravitreal injection of AAV expressing soluble VEGF receptor-1 variant induces anti-VEGF activity and suppresses choroidal neovascularization. Invest Ophthalmol Vis Sci. 2018;59(13):5398–407. https://doi.org/10.1167/iovs.18-24926.
Zhong L, Li B, Mah CS, Govindasamy L, Agbandje-McKenna M, Cooper M, et al. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. PNAS. 2008;105(22):6.
Yuan Y, Kong W, Liu XM, Shi GH. A hypoxia-regulated retinal pigment epithelium-specific gene therapy vector reduces choroidal neovascularization in a mouse model. Curr Gene Ther. 2022;22(5):417–26. https://doi.org/10.2174/1566523222666220405135135.
Biswal MR, Prentice HM, Smith GW, et al. Cell-specific gene therapy driven by an optimized hypoxia-regulated vector reduces choroidal neovascularization. J Mol Med (Berl). 2018;96(10):1107–18. https://doi.org/10.1007/s00109-018-1683-0.
Campochiaro PA, Lauer AK, Sohn EH, et al. Lentiviral Vector gene transfer of endostatin/angiostatin for macular degeneration (GEM) study. Hum Gene Ther. 2017;28(1):99–111. https://doi.org/10.1089/hum.2016.117.
Grishanin R, Vuillemenot B, Sharma P, et al. Preclinical evaluation of ADVM-022, a novel gene therapy approach to treating wet age-related macular degeneration. Mol Ther. 2019;27(1):118–29. https://doi.org/10.1016/j.ymthe.2018.11.003.
Kiss S, Grishanin R, Nguyen A, et al. Analysis of aflibercept expression in NHPs following intravitreal administration of ADVM-022, a potential gene therapy for nAMD. Mol Ther Methods Clin Dev. 2020;18:345–53. https://doi.org/10.1016/j.omtm.2020.06.007.
Kiss S, Oresic Bender K, Grishanin RN, et al. Long-term safety evaluation of continuous intraocular delivery of aflibercept by the intravitreal gene therapy candidate ADVM-022 in nonhuman primates. Transl Vis Sci Technol. 2021;10(1):34. https://doi.org/10.1167/tvst.10.1.34.
Gelfman CM, Grishanin R, Bender KO, et al. Comprehensive preclinical assessment of ADVM-022, an intravitreal anti-VEGF gene therapy for the treatment of neovascular AMD and diabetic macular edema. J Ocul Pharmacol Ther. 2021;37(3):181–90. https://doi.org/10.1089/jop.2021.0001.
Kiss S, editor. Ixoberogene soroparvovec (Ixo-vec) Intravitreal gene therapy for neovascular age-related macular degeneration. In: 46th Annual Macula Society Meeting; 2023 February 15–18, 2023.
Figueroa A. Adverum provides update on ADVM-022 and the INFINITY trial in patients with diabetic macular edema. 2021
Becker J, Fakhiri J, Grimm D. Fantastic AAV gene therapy vectors and how to find them-random diversification, rational design and machine learning. Pathogens. 2022. https://doi.org/10.3390/pathogens11070756.
Kotterman M, Beliakoff G, Croze R et al. 2021. doi:https://doi.org/10.1101/2021.06.24.449775.
Francis P, editor. A multi-mechanistic anti-angiogenic AAV gene therapy product candidate, 4D-150, for the treatment of wet age-related macular degeneration (Wet AMD) and Diabetic Macular Edema (DME): intravitreal biodistribution, transgene expression, safety and efficacy in non-human primates. In: 24th Annual American Society of Gene & Cell Therapy (ASGCT) Meeting; 2021 May 11–14, 2021.
Smith K. 4DMT Presents Additional Positive Interim Data from Intravitreal 4D-150 Phase 1/2 PRISM Clinical Trial in Patients with Wet AMD at ASRS 2023. 4dmoleculartherapeutics.com2023.
Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019;18(5):358–78. https://doi.org/10.1038/s41573-019-0012-9.
Vandenberghe LH, Bell P, Maguire AM, et al. Dosage thresholds for AAV2 and AAV8 photoreceptor gene therapy in monkey. Sci Transl Med. 2011;3(88):88ra54. https://doi.org/10.1126/scitranslmed.3002103.
Lee SH, Kim YS, Nah SK, et al. Transduction patterns of adeno-associated viral vectors in a laser-induced choroidal neovascularization mouse model. Mol Ther Methods Clin Dev. 2018;9:90–8. https://doi.org/10.1016/j.omtm.2018.01.008.
Ding K, Shen J, Hafiz Z, et al. AAV8-vectored suprachoroidal gene transfer produces widespread ocular transgene expression. J Clin Invest. 2019;129(11):4901–11. https://doi.org/10.1172/JCI129085.
Liu Y, Fortmann SD, Shen J, et al. AAV8-antiVEGFfab ocular gene transfer for neovascular age-related macular degeneration. Mol Ther. 2018;26(2):542–9. https://doi.org/10.1016/j.ymthe.2017.12.002.
Campochiaro PA, editor. Gene Therapy for Neovascular AMD: subretinal RGX-314: phase I/IIa long-term follow-up results up to 4 years. In: 2022 American Academy of Ophthalmology 2022 2022.
Abbey A, editor. Subretinal delivery of investigational ABBV-RGX-314 for neovascular AMD: a phase II pharmacodynamic study. In: 2023 Anual Meeting of The American Society of Retina Specialists; 2023 30/07/2023; Seattle.
Boyer D, editor. Suprachoroidal delivery of investigational ABBV-RGX-314 for neovascular AMD: results from the phase II AAVIATE study. In: 2023 Annual Meeting of the American Society of Retinal Surgeons; 2023 30/07/2023; Seattle.
Desrosiers M, Dalkara D. Neutralizing antibodies against adeno-associated virus (AAV): measurement and influence on retinal gene delivery. Methods Mol Biol. 2018;1715:225–38. https://doi.org/10.1007/978-1-4939-7522-8_16.
She K, Su J, Wang Q, et al. Delivery of nVEGFi using AAV8 for the treatment of neovascular age-related macular degeneration. Mol Ther Methods Clin Dev. 2022;24:210–21. https://doi.org/10.1016/j.omtm.2022.01.002.
Hughes CP, O’Flynn NMJ, Gatherer M, et al. AAV2/8 Anti-angiogenic gene therapy using single-chain antibodies inhibits murine choroidal neovascularization. Mol Ther Methods Clin Dev. 2019;13:86–98. https://doi.org/10.1016/j.omtm.2018.11.005.
Bordet T, Behar-Cohen F. Ocular gene therapies in clinical practice: viral vectors and nonviral alternatives. Drug Discov Today. 2019;24(8):1685–93. https://doi.org/10.1016/j.drudis.2019.05.038.
Askou AL, Alsing S, Benckendorff JNE, et al. Suppression of choroidal neovascularization by AAV-based dual-acting antiangiogenic gene therapy. Mol Ther Nucleic Acids. 2019;16:38–50. https://doi.org/10.1016/j.omtn.2019.01.012.
Phase 1/2 Clinical trial of intravitreal 4D-150 in patients with wet age-related macular degeneration. PRISM Interim Safety & Efficacy Data. 4DMT website: 4DMT.
Dugel PU. CLINICAL TRIAL DOWNLOAD: Data on a gene therapy for dry and wet AMD. A phase 1 clinical trial program is targeting both disease states. Retinal Phys. 2020;16:7.
Li Y, Zhu P, Verma A, et al. A novel bispecific molecule delivered by recombinant AAV2 suppresses ocular inflammation and choroidal neovascularization. J Cell Mol Med. 2017;21(8):1555–71. https://doi.org/10.1111/jcmm.13086.
Cipriani V, Lores-Motta L, He F, et al. Increased circulating levels of Factor H-Related Protein 4 are strongly associated with age-related macular degeneration. Nat Commun. 2020;11(1):778. https://doi.org/10.1038/s41467-020-14499-3.
Schnabolk G, Parsons N, Obert E, et al. Delivery of CR2-fH using AAV vector therapy as treatment strategy in the mouse model of choroidal neovascularization. Mol Ther Methods Clin Dev. 2018;9:1–11. https://doi.org/10.1016/j.omtm.2017.11.003.
Pfau M, Schmitz-Valckenberg S, Ribeiro R, et al. Association of complement C3 inhibitor pegcetacoplan with reduced photoreceptor degeneration beyond areas of geographic atrophy. Sci Rep. 2022;12(1):17870. https://doi.org/10.1038/s41598-022-22404-9.
Patel SS, Lally DR, Hsu J, et al. Avacincaptad pegol for geographic atrophy secondary to age-related macular degeneration: 18-month findings from the GATHER1 trial. Eye (Lond). 2023. https://doi.org/10.1038/s41433-023-02497-w.
Cashman SM, Ramo K, Kumar-Singh R. A non membrane-targeted human soluble CD59 attenuates choroidal neovascularization in a model of age related macular degeneration. PLoS ONE. 2011. https://doi.org/10.1371/journal.pone.0019078.
Ramo K, Cashman SM, Kumar-Singh R. Evaluation of adenovirus-delivered human CD59 as a potential therapy for AMD in a model of human membrane attack complex formation on murine RPE. Invest Ophthalmol Vis Sci. 2008;49(9):4126–36. https://doi.org/10.1167/iovs.08-2025.
Lad EM, Chao DL, Pepio A, et al. Pooled safety analysis of a single intravitreal injection of JNJ-1887 (gene therapy, AAVCAGsCD59) in patients with age-related macular degeneration (AMD). Investig Ophthalmol Vis Sci. 2023;64(8):732.
Silvent J. Janssen announces late-breaking data from two gene therapy programs at the American Academy of Ophthalmology 2022 Annual Meeting. Johnson & Johnson 2022.
Cohen MN, editor. Study results: gene Tx for geographic atrophy. In: American Academy of Ophthalmology 2022 Annual Meeting; 2022.
Kaszubski P, Ben Ami T, Saade C, Smith RT. Geographic atrophy and choroidal neovascularization in the same eye: a review. Ophthalmic Res. 2016;55(4):185–93. https://doi.org/10.1159/000443209.
Dreismann AK, McClements ME, Barnard AR, et al. Functional expression of complement factor I following AAV-mediated gene delivery in the retina of mice and human cells. Gene Ther. 2021;28(5):265–76. https://doi.org/10.1038/s41434-021-00239-9.
Nielsen JS, editor. Preliminary results from a first-in-human phase I/II gene therapy trial (FOCUS) of subretinally delivered GT005, an investigational AAV2 vector, in patients with geographic atrophy secondary to age-related macular degeneration. Association for Research in Vision and Ophthalmology; 2022.
Qin S, Dong N, Yang M, et al. Complement inhibitors in age-related macular degeneration: a potential therapeutic option. J Immunol Res. 2021;2021:1–15. https://doi.org/10.1155/2021/9945725.
Wang H, Kunz E, Stoddard GJ, Hauswirth WW, Hartnett ME. Optimal inhibition of choroidal neovascularization by scAAV2 with VMD2 promoter-driven active Rap1a in the RPE. Sci Rep. 2019;9(1):15732. https://doi.org/10.1038/s41598-019-52163-z.
Young BM, Jones K, Massengill MT, et al. Expression of a CARD slows the retinal degeneration of a geographic atrophy mouse model. Mol Ther Methods Clin Dev. 2019;14:113–25. https://doi.org/10.1016/j.omtm.2019.06.001.
Choudhary M, Ildefonso CJ, Lewin AS, Malek G. Gene delivery of a caspase activation and recruitment domain improves retinal pigment epithelial function and modulates inflammation in a mouse model with features of dry age-related macular degeneration. J Ocul Pharmacol Ther. 2022;38(5):359–71. https://doi.org/10.1089/jop.2022.0002.
Pennesi ME, Neuringer M, Courtney RJ. Animal models of age related macular degeneration. Mol Aspects Med. 2012;33(4):487–509. https://doi.org/10.1016/j.mam.2012.06.003.
Biswal MR, Han P, Zhu P, et al. Timing of antioxidant gene therapy: implications for treating dry AMD. Invest Ophthalmol Vis Sci. 2017;58(2):1237–45. https://doi.org/10.1167/iovs.16-21272.
Biswal MR, Wang Z, Paulson RJ, et al. Erythropoietin gene therapy delays retinal degeneration resulting from oxidative stress in the retinal pigment epithelium. Antioxidants (Basel). 2021. https://doi.org/10.3390/antiox10060842.
Layton CJ, Wood JP, Chidlow G, Osborne NN. Neuronal death in primary retinal cultures is related to nitric oxide production, and is inhibited by erythropoietin in a glucose-sensitive manner. J Neurochem. 2005;92(3):487–93. https://doi.org/10.1111/j.1471-4159.2004.02876.x.
Huang X, Zhou G, Wu W, et al. Genome editing abrogates angiogenesis in vivo. Nat Commun. 2017;8(1):112. https://doi.org/10.1038/s41467-017-00140-3.
Xu CL, Ruan MZC, Mahajan VB, Tsang SH. Viral delivery systems for CRISPR. Viruses. 2019. https://doi.org/10.3390/v11010028.
Asmamaw MM. Viral vectors for the in vivo delivery of CRISPR components: advances and challenges. Front Bioeng Biotechnol. 2022;10: 895713. https://doi.org/10.3389/fbioe.2022.895713.
Koo T, Park SW, Jo DH, et al. CRISPR-LbCpf1 prevents choroidal neovascularization in a mouse model of age-related macular degeneration. Nat Commun. 2018;9(1):1855. https://doi.org/10.1038/s41467-018-04175-y.
Salman A, Kantor A, McClements ME, et al. Non-viral delivery of CRISPR/Cas cargo to the retina using nanoparticles: current possibilities, challenges, and limitations. Pharmaceutics. 2022. https://doi.org/10.3390/pharmaceutics14091842.
Suh S, Choi EH, Raguram A, Liu DR, Palczewski K. Precision genome editing in the eye. Proc Natl Acad Sci U S A. 2022;119(39): e2210104119. https://doi.org/10.1073/pnas.2210104119.
Wong TY, Cheung CM, Larsen M, Sharma S, Simo R. Diabetic retinopathy. Nat Rev Dis Primers. 2016;2:16012. https://doi.org/10.1038/nrdp.2016.12.
Lin X, Xu Y, Pan X, et al. Global, regional, and national burden and trend of diabetes in 195 countries and territories: an analysis from 1990 to 2025. Sci Rep. 2020;10(1):14790. https://doi.org/10.1038/s41598-020-71908-9.
Kupis M, Samelska K, Szaflik J, Skopinski P. Novel therapies for diabetic retinopathy. Cent Eur J Immunol. 2022;47(1):102–8. https://doi.org/10.5114/ceji.2022.112993.
Teo ZL, Tham YC, Yu M, et al. Global prevalence of diabetic retinopathy and projection of burden through 2045: systematic review and meta-analysis. Ophthalmology. 2021;128(11):1580–91. https://doi.org/10.1016/j.ophtha.2021.04.027.
Roberts-Martinez Aguirre I, Rodriguez-Fernandez P, Gonzalez-Santos J, et al. Exploring the quality of life related to health and vision in a group of patients with diabetic retinopathy. Healthcare (Basel). 2022. https://doi.org/10.3390/healthcare10010142.
Bunch KL, Abdelrahman AA, Caldwell RB, Caldwell RW. Novel therapeutics for diabetic retinopathy and diabetic macular edema: a pathophysiologic perspective. Front Physiol. 2022;13: 831616. https://doi.org/10.3389/fphys.2022.831616.
Kattar A, Concheiro A, Alvarez-Lorenzo C. Diabetic eye: associated diseases, drugs in clinic, and role of self-assembled carriers in topical treatment. Expert Opin Drug Deliv. 2021;18(11):1589–607. https://doi.org/10.1080/17425247.2021.1953466.
Ting DS, Cheung GC, Wong TY. Diabetic retinopathy: global prevalence, major risk factors, screening practices and public health challenges: a review. Clin Exp Ophthalmol. 2016;44(4):260–77. https://doi.org/10.1111/ceo.12696.
Graham PS, Kaidonis G, Abhary S, et al. Genome-wide association studies for diabetic macular edema and proliferative diabetic retinopathy. BMC Med Genet. 2018;19(1):71. https://doi.org/10.1186/s12881-018-0587-8.
Gurung RL, FitzGerald LM, McComish BJ, Verma N, Burdon KP. Identifying genetic risk factors for diabetic macular edema and the response to treatment. J Diabetes Res. 2020;2020:5016916. https://doi.org/10.1155/2020/5016916.
Wang JH, Ling D, Tu L, et al. Gene therapy for diabetic retinopathy: are we ready to make the leap from bench to bedside? Pharmacol Ther. 2017;173:1–18. https://doi.org/10.1016/j.pharmthera.2017.01.003.
Liu S, Ju Y, Gu P. Experiment-based interventions to diabetic retinopathy: present and advances. Int J Mol Sci. 2022. https://doi.org/10.3390/ijms23137005.
Kowluru RA, Mohammad G. Epigenetics and mitochondrial stability in the metabolic memory phenomenon associated with continued progression of diabetic retinopathy. Sci Rep. 2020;10(1):6655. https://doi.org/10.1038/s41598-020-63527-1.
Le NT, Kroeger ZA, Lin WV, Khanani AM, Weng CY. Novel treatments for diabetic macular edema and proliferative diabetic retinopathy. Curr Diab Rep. 2021;21(10):43. https://doi.org/10.1007/s11892-021-01412-5.
Smit-McBride Z, Morse LS. MicroRNA and diabetic retinopathy-biomarkers and novel therapeutics. Ann Transl Med. 2021;9(15):1280. https://doi.org/10.21037/atm-20-5189.
Wang JH, Roberts GE, Liu GS. Updates on gene therapy for diabetic retinopathy. Curr Diab Rep. 2020;20(7):22. https://doi.org/10.1007/s11892-020-01308-w.
Bainbridge JW, Mistry A, De Alwis M, et al. Inhibition of retinal neovascularisation by gene transfer of soluble VEGF receptor sFlt-1. Gene Ther. 2002;9(5):320–6. https://doi.org/10.1038/sj.gt.3301680.
Ideno J, Mizukami H, Kakehashi A, et al. Prevention of diabetic retinopathy by intraocular soluble flt-1 gene transfer in a spontaneously diabetic rat model. Int J Mol Med. 2007;19(1):75–9.
Pechan P, Rubin H, Lukason M, et al. Novel anti-VEGF chimeric molecules delivered by AAV vectors for inhibition of retinal neovascularization. Gene Ther. 2009;16(1):10–6. https://doi.org/10.1038/gt.2008.115.
Diaz-Lezama N, Wu Z, Adan-Castro E, et al. Diabetes enhances the efficacy of AAV2 vectors in the retina: therapeutic effect of AAV2 encoding vasoinhibin and soluble VEGF receptor 1. Lab Invest. 2016;96(3):283–95. https://doi.org/10.1038/labinvest.2015.135.
Boyer D, editor. Intravitreal Gene Therapy for Diabetic Macular Edema with ADVM-022: Prospective, Randomized Phase 2 INFINITY Trial. In: American Academy of Ophthalmology 2021 Annual Meeting; 2021 November 13-16, 2021.
Dilsher S. Dhoot, editor. Suprachoroidal delivery of investigational ABBV-RGX-314 for diabetic retinopathy: the Phase II ALTITUDE® Study. In: 2023 American Society of Retinal Surgeons; 2023; Seattle.
Smith K. 4D Molecular therapeutics announces FDA clearance of IND application for 4D-150 genetic medicine for the treatment of diabetic macular edema. 2023. https://ir.4dmoleculartherapeutics.com/news-releases/news-release-details/4d-molecular-therapeutics-announces-fda-clearance-ind-2/. Accessed 15 April 2023.
Cahoon JM, Rai RR, Carroll LS, et al. Intravitreal AAV2.COMP-Ang1 prevents neurovascular degeneration in a murine model of diabetic retinopathy. Diabetes. 2015;64(12):4247–59. https://doi.org/10.2337/db14-1030.
Whitehead M, Osborne A, Widdowson PS, Yu-Wai-Man P, Martin KR. Angiopoietins in diabetic retinopathy: current understanding and therapeutic potential. J Diabetes Res. 2019;2019:5140521. https://doi.org/10.1155/2019/5140521.
Carroll LS, Uehara H, Fang D, et al. Intravitreal AAV2.COMP-Ang1 attenuates deep capillary plexus expansion in the aged diabetic mouse retina. Invest Ophthalmol Vis Sci. 2019;60(7):2494–502. https://doi.org/10.1167/iovs.18-26182.
Biswal MR, Prentice HM, Dorey CK, Blanks JC. A hypoxia-responsive glial cell-specific gene therapy vector for targeting retinal neovascularization. Invest Ophthalmol Vis Sci. 2014;55(12):8044–53. https://doi.org/10.1167/iovs.14-13932.
Dominguez JM 2nd, Hu P, Caballero S, et al. Adeno-associated virus overexpression of angiotensin-converting enzyme-2 reverses diabetic retinopathy in type 1 diabetes in mice. Am J Pathol. 2016;186(6):1688–700. https://doi.org/10.1016/j.ajpath.2016.01.023.
Verma A, Shan Z, Lei B, et al. ACE2 and Ang-(1–7) confer protection against development of diabetic retinopathy. Mol Ther. 2012;20(1):28–36. https://doi.org/10.1038/mt.2011.155.
Daruich A, Matet A, Moulin A, et al. Mechanisms of macular edema: beyond the surface. Prog Retin Eye Res. 2018;63:20–68. https://doi.org/10.1016/j.preteyeres.2017.10.006.
Vacca O, Charles-Messance H, El Mathari B, et al. AAV-mediated gene therapy in Dystrophin-Dp71 deficient mouse leads to blood-retinal barrier restoration and oedema reabsorption. Hum Mol Genet. 2016;25(14):3070–9. https://doi.org/10.1093/hmg/ddw159.
Klimczak RR, Koerber JT, Dalkara D, Flannery JG, Schaffer DV. A novel adeno-associated viral variant for efficient and selective intravitreal transduction of rat Muller cells. PLoS ONE. 2009;4(10): e7467. https://doi.org/10.1371/journal.pone.0007467.
Yang Q, Zhang C, Xie H, et al. Silencing Nogo-B improves the integrity of blood-retinal barrier in diabetic retinopathy via regulating Src, PI3K/Akt and ERK pathways. Biochem Biophys Res Commun. 2021;581:96–102. https://doi.org/10.1016/j.bbrc.2021.10.024.
Chan KY, Jang MJ, Yoo BB, et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci. 2017;20(8):1172–9. https://doi.org/10.1038/nn.4593.
Jiang H, Zhang H, Jiang X, Wu S. Overexpression of D-amino acid oxidase prevents retinal neurovascular pathologies in diabetic rats. Diabetologia. 2021;64(3):693–706. https://doi.org/10.1007/s00125-020-05333-y.
Lee SHS, Lee JY, Choi JS, et al. mTOR inhibition as a novel gene therapeutic strategy for diabetic retinopathy. PLoS ONE. 2022;17(6): e0269951. https://doi.org/10.1371/journal.pone.0269951.
Park TK, Lee SH, Choi JS, et al. Adeno-associated viral vector-mediated mTOR inhibition by short hairpin RNA suppresses laser-induced choroidal neovascularization. Mol Ther Nucleic Acids. 2017;8:26–35. https://doi.org/10.1016/j.omtn.2017.05.012.
Lee SHS, Chang H, Kim HJ, et al. Effects of stuffer DNA on the suppression of choroidal neovascularization by a rAAV expressing a mTOR-Inhibiting shRNA. Mol Ther Methods Clin Dev. 2019;14:171–9. https://doi.org/10.1016/j.omtm.2019.06.004.
Acknowledgements
This study was supported by Layton Vision Foundation, Australia. All figures were created with BioRender.com. The authors would like to thank Dr. Sergei Kozlov for the insightful discussions.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions.
Conflicts of interest
Christopher J. Layton owns shares in Ocugene, a gene therapy company. Jason C. Steel has been a consultant for Ocugene and owns intellectual property in breast cancer gene therapy. Brenda F. M. Castro has no relevant financial or non-financial interests to disclose.
Ethics Approval
Not applicable.
Patient Consent to Participate/Publish
Not applicable.
Availability of Data and Material
Not applicable.
Code Availability
Not applicable.
Author Contributions
Conception and design of the work: BFMC, JCS, CJL. Writing – original draft: BFMC, CJL. Writing – review and editing: BFMC, JCS, CJL. Supervision, funding acquisition and resources: CJL. All authors read and approved the final version of the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Castro, B.F.M., Steel, J.C. & Layton, C.J. AAV-Based Strategies for Treatment of Retinal and Choroidal Vascular Diseases: Advances in Age-Related Macular Degeneration and Diabetic Retinopathy Therapies. BioDrugs 38, 73–93 (2024). https://doi.org/10.1007/s40259-023-00629-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-023-00629-y