
Abstract
Objective
SB2, a biosimilar to infliximab reference product (INF), has an identical amino acid sequence and similar physicochemical functional properties to its reference product. The primary objective of this study is to demonstrate pharmacokinetic (PK) bioequivalence between SB2 and EU-sourced INF (EU-INF), between SB2 and US-sourced INF (US-INF), and between EU-INF and US-INF.
Methods
This study was a randomized, single-blind, three-arm, parallel group study in 159 healthy subjects. All subjects received a single 5 mg/kg intravenous infusion of study drug and then were observed for 10 weeks to study PK, safety and immunogenicity. The primary PK parameters were area under the concentration-time curve (AUC) from time zero to infinity (AUCinf), AUC from time zero to the last quantifiable concentration (AUClast) and maximum concentration (C max). Bioequivalence for the primary PK parameters was to be concluded using an analysis of variance (ANOVA) if the 90 % confidence intervals (CIs) for the ratio of geometric least squares means (LSMeans) of the treatments compared were completely contained within the pre-defined equivalence margin, 0.8–1.25.
Results
All of the 90 % CIs for the geometric LSMean ratios of primary PK parameters for each comparison were within the pre-defined equivalence margin. The proportion of subjects who experienced treatment-emergent adverse events was comparable between treatments. The incidences of anti-drug antibodies between the three treatments were comparable.
Conclusion
This study demonstrated biosimilarity of SB2 to its marketed reference products of infliximab in terms of PK equivalence in healthy subjects. SB2 was generally well tolerated and showed comparable safety and immunogenicity profiles to the reference products (ClinicalTrials.gov Identifier: NCT01922336).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Single-dose pharmacokinetics of SB2 were shown to be bioequivalent to those of reference products (EU-sourced Remicade® and US-sourced Remicade®) in healthy subjects, considered a sensitive population for PK comparison. |
Safety and immunogenicity of single-dose SB2 in healthy subjects are comparable to those of reference products. |
1 Introduction
Infliximab, which is a genetically engineered chimeric human/mouse glycosylated monoclonal antibody (mAb) directed against tumour necrosis factor alpha (TNFα), acts by neutralizing the proinflammatory action and regulatory role of TNFα [1–3]. Infliximab was approved as Remicade® (Janssen Biotech Inc., Horsham, PA, USA) with the indications including rheumatoid arthritis, adult Crohn’s disease, paediatric Crohn’s disease, ankylosing spondylitis, psoriatic arthritis, ulcerative colitis, paediatric ulcerative colitis and psoriasis [4].
Recently, Remsima® (Celltrion Inc., Incheon, Korea), using Remicade® as the reference product, was approved for use as an infliximab biosimilar in the EU by the European Medicines Agency (EMA) [5]. As per EU biosimilar requirements, the approval application included a detailed and thorough characterization of the mAb and non-clinical studies of the biosimilar with the reference product [6]. This was complemented by a clinical phase I study in ankylosing spondylitis patients and a clinical phase III study in rheumatoid arthritis patients to establish and confirm clinical biosimilarity [7, 8].
Samsung Bioepis Co., Ltd is developing SB2, a biosimilar to Remicade®, that is produced by recombinant DNA technology and purified by various types of chromatography. In accordance with the regulatory agency biosimilar guidelines, the development of SB2 had involved biosimilarity studies starting with comparison of the structural characteristics, physicochemical properties and biological activities between SB2 and Remicade®, followed by demonstration of similar in vivo behaviour between SB2 and its reference products [9, 10]. Based on the in vitro and in vivo non-clinical study results, clinical studies could be conducted to compare the clinical efficacy, safety, and pharmacokinetics (PK) of SB2 with those of infliximab reference products (INF). The aim of this study was to compare the PK of SB2 and its reference products after single administration of infliximab in healthy subjects.
2 Methods
2.1 Subjects
Healthy female subjects of non-childbearing potential and healthy male subjects aged 18–55 years were eligible for participation in this study if bodyweight was between 60.0 and 94.9 kg and body mass index (BMI) was between 20.0 and 29.9 kg/m2. For inclusion, subjects had to be in good health without any infectious disease including active or latent tuberculosis as indicated by medical history, physical examination, vital signs, 12-lead electrocardiography (ECG), serology, clinical laboratory tests, QuantiFERON®-TB Gold test (QIAGEN, Venlo, The Netherlands) and urine drug screening. These screening tests were performed during a 3-week period prior to randomization. The nature and purpose of the study was fully explained to each subject and written informed consent was obtained from each subject before the subject was enroled in the study.
2.2 Study Design
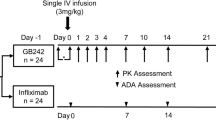
This study was a single-blind, parallel group, single-dose study with three treatment groups, which were SB2 and two infliximab reference products: EU-sourced Remicade® (EU-INF) and US-sourced Remicade® (US-INF). All subjects received a single dose of 5 mg/kg SB2, EU-INF or US-INF by intravenous (IV) infusion for 120 min on the first day of study and then were followed for 10 weeks during which the PK, safety and immunogenicity measurements were made. To avoid infusion-related reaction, premedication with IV hydrocortisone (100 mg), oral acetaminophen (1000 mg), and oral loratadine (10 mg) were administered 30 min to 1 h prior to the infusion of study drugs, which was adopted from a previous report [11]. In case of infusion-related reactions, the infusion could be temporarily discontinued or the infusion rate decreased based on assessment of the investigators.
During the treatment period, subjects were hospitalized in the PAREXEL Early Phase Clinical Unit (Berlin, Germany) from 1 day before the study drug administration until 3 days after administration for serial PK sampling and close safety monitoring. After discharge, the subjects visited the site at 6, 8, 15, 22, 29, 43, 57, and 71 days after administration. Safety was assessed by vital signs, clinical laboratory tests, 12-lead ECG, and physical examinations. Adverse events (AEs) recorded during the course of the study were categorized by system organ class and preferred terms using the MedDRA (Medical Dictionary for Regulatory Activities, version 16.0). Blood samples for immunogenicity were collected to detect anti-drug antibodies (ADAs) and neutralizing antibodies (NAbs) to infliximab at pre-dose, 29 and 71 days after dosing. Blood samples for immunogenicity evaluation were analysed using an electrochemiluminescent immunoassay for ADA detection and a functional cell-based assay for NAb detection. The percent coefficient of variation (% CV) for the negative control and high positive control for ADA detection were 33.7 and 26.4 %, respectively.
2.3 Pharmacokinetic Evaluation
Blood samples for PK analysis were collected at pre-dose, 1 h after the start of infusion, the end of infusion, 3, 6, 12, 24, 48, 72, 120 h after the start of infusion and then at weeks 1, 2, 3, 4, 6, 8 and 10 from the start of infusion. Samples were kept frozen at −70 °C or colder prior to analysis. The serum concentration of infliximab was measured using an enzyme-linked immunosorbent assay (ELISA) specific for the detection and quantification of infliximab by TNFα (R&D Systems, Product No. 210-TA-001MG/CF) coated in wells of an ELISA plate (PPD Bioanalytical Laboratory, Richmond, VA, USA). The concentration limit of quantification was from 100 to 3200 ng/mL. Inter-assay precision measured as the % CV were from 6.1 to 9.2 %, and the inter-assay accuracy expressed as the percent difference of the mean value were from 1.9 to 4.8 % within the quantification limit.
The PK parameter calculations were based on actual sampling times and non-compartmental analysis methods. The maximum concentration (C max) and time to reach C max (T max) were obtained directly from the observed values. The terminal elimination rate constant (λz) was estimated at terminal phase by linear regression after loge-transformation of the concentrations. The terminal half-life (t ½) was calculated as ln(2)/λz. The linear up/log down trapezoidal rule was used to obtain the area under the concentration-time curve (AUC) from time zero to the last quantifiable concentration (AUClast). AUC extrapolated to infinity (AUCinf) was calculated as AUClast + C last/λz (where C last is the last quantifiable concentration). Clearance (CL) was calculated as dose/AUCinf and volume of distribution (V d) was estimated as CL/λz. The PK parameter calculations were performed with Phoenix® WinNonlin® version 6.2 (Certara®, Princeton, NJ, USA).
2.4 Statistical Analysis
PK parameters were summarized in a descriptive manner by treatment and immunogenicity results as overall, ADA positive and ADA negative. An analysis of variance model (ANOVA) was performed for comparison of primary PK parameters (AUCinf, AUClast and C max). The difference in geometric least squares means (LSMeans) of log e -transformed primary PK parameters between SB2 and EU-IFN, between SB2 and US-IFN and between EU-IFN and US-IFN and the associated 90 % confidence intervals (CIs) were determined. Back-transformation provided the ratio of geometric means and the related 90 % CIs for the ratio of geometric LSMeans of pairwise comparison were estimated. The bioequivalence of primary PK parameters were to be concluded when the 90 % CI was within 0.8–1.25. All statistical analyses were performed using SAS® version 9.2 TS Level 2M3 (SAS-Institute Inc., Cary, NC, USA).
3 Results
3.1 Subject Disposition
A total of 319 subjects were screened, of which 159 subjects were randomized to receive one of three infliximab study drugs (SB2, EU-INF, US-INF). None of the randomized subjects discontinued from the study. The average age, height, weight and BMI were generally comparable across the three treatment groups (Table 1). Among the randomized subjects, 150 subjects were male (49 subjects in the SB2 treatment group, 51 subjects in the EU-INF group and 50 subjects in the US-INF group) and nine subjects were female (four subjects in the SB2 group, two subjects in the EU-INF group and three subjects in the US-INF group). The majority of the subjects were white. Two subjects in the SB2 treatment group were not included in the PK analysis due to the use of concomitant medication for their AE treatment that could have influenced the PK of infliximab, although their data were included in safety and immunogenicity assessments.
3.2 Pharmacokinetic Results
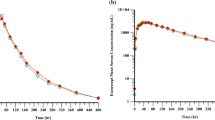
The mean serum concentration curves on semi-logarithmic scale for each treatment are presented in Fig. 1. Comparisons between SB2 and EU-INF, SB2 and US-INF, and EU-INF and US-INF showed high similarity in the mean serum concentration time profiles. In all cases, mean serum concentration time profiles reached maximum exposure between 2 and 6 h after start of infusion with a median T max of approximately 3 h for SB2 and US-INF and of approximately 2 h for EU-INF. Infliximab was slowly cleared with an average t ½ ranging from approximately 324–340 h for all treatment groups. The mean values of PK parameters (AUCinf, AUClast, C max, T max, V d, t ½ and CL) were similar between treatments (Table 2).

Mean serum concentration-time profiles after single administration of infliximabs. a All subjects’ data included in the pharmacokinetic analysis of SB2, EU-sourced infliximab reference product, and US-sourced infliximab reference product; means of all subjects and ADA subgroups for b SB2, c EU-sourced infliximab reference product, and d US-sourced infliximab reference product. Bars represent standard deviations of all subjects’ data including ADA positive and ADA negative. ADA anti-drug antibody
For the PK similarity comparisons of SB2 with each of the reference products (EU-INF or US-INF), the 90 % CI for the test-to-reference ratios of AUCinf, AUClast and C max were within the pre-defined equivalence margin of 0.8–1.25 (Table 3). The 90 % CIs of the ratios of AUCinf, AUClast and C max were also within 0.8–1.25 for the comparison of EU-INF to US-INF.
To compare the immunogenicity influences on PK of each study drug, sub-analyses based on the post-dose ADA results were performed. The mean concentration of infliximab in ADA-positive subjects compared with that of ADA-negative subjects showed that the study drugs were eliminated from blood circulation with relatively higher clearance rates in ADA-positive subjects in all three treatments (Fig. 1). The mean CL in ADA-positive subjects after SB2, EU-INF and US-INF administration were 12.7, 13.6 and 12.9 mL/h, respectively, and those in ADA-negative subjects after SB2, EU-INF and US-INF administration were 9.4, 9.5 and 9.4 mL/h, respectively (Table 2). The means for PK parameters including CL of each treatment were comparable among each ADA-positive group and -negative group.
3.3 Safety Results
A total of 124 treatment-emergent AEs (TEAEs) were reported in 71 (44.7 %) subjects. Fifty TEAEs were reported from 27 (50.9 %) subjects following SB2 administration, 36 TEAEs were reported from 21 (39.6 %) subjects after EU-INF administration and 38 TEAEs were reported from 23 (43.4 %) subjects after US-INF administration (Table 4). All reported TEAEs were of mild or moderate severity, with the majority of reported TEAEs being of mild severity, and the proportion of subjects with TEAEs was comparable across the three treatment groups. The most frequent TEAEs were nasopharyngitis and headache, and no infusion-related reaction was reported. Three serious adverse events (SAEs) were reported in two subjects from the SB2 treatment group. One subject had a concussion and a renal cyst ruptured, which were assessed not to be related to study drug, and the other subject had a Borrelia infection, which was assessed to be related to the study drug. Those two subjects recovered without any sequelae. Vitals signs, ECG parameters and laboratory data did not show any changes over time that might be considered to be related to the treatments. There were no deaths or discontinuations due to AEs during the study.
3.4 Immunogenicity Results
The overall incidence of subjects with post-dose ADA to infliximab was 47.2, 37.7 and 37.7 % in subjects treated with SB2, EU-INF and US-INF, respectively. All three subjects who were ADA positive at day 28 also had positive results from ADA test at day 71. There was no statistically significant difference in post-dose ADA incidence across the three treatment groups (p = 0.432 between SB2 and EU-INF, p = 0.432 between SB2 and US-INF and p = 1.000 between EU-INF and US-INF). The incidence of post-dose NAb was comparable across the three treatment groups (Table 5).
4 Discussion
The objective of this study was to compare the PK, safety and immunogenicity of SB2 as an infliximab biosimilar with those of infliximab reference products in healthy subjects. The clinical PK study is considered to be essential to demonstrate clinical biosimilar comparability [9], and the most sensitive population that can reduce inter-individual variation should be chosen. Healthy subjects are considered to be a more homogeneous and hence more sensitive population to study PK characteristics than patients since patients can have various disease-related factors which can influence the PK of study drugs. For example, concomitant administration of methotrexate, which is commonly used for autoimmune disease including rheumatoid arthritis and psoriasis, can reduce the clearance of infliximab compared with infliximab administered alone [12].
Equivalence of the PK between the test and reference products was based on the CIs for the primary PK variables AUCinf, AUClast and C max in relation to the accepted range of 0.8–1.25. To meet the requirements of both the EMA and FDA, comparison data between SB2 and EU-INF and between SB2 and US-INF demonstrate the evidence of equivalence in PK between SB2 and each of the two test products, respectively [9, 10]. The PK equivalence between EU-INF and US-INF would provide scientific justification to use EU-INF, which is not licensed in the USA, as the sole active comparator in a phase III clinical trial.
Although a cross-over design for comparative clinical studies greatly reduces the variability in PK, a cross-over design with infliximab was not feasible considering the long t ½. The t ½ of this study in healthy subjects is approximately 2 weeks for all studied infliximabs, which is similar to previous infliximab PK studies in patients and can be considered long enough not to apply the cross-over design [1, 12, 13]. Moreover, ADAs to the study drug from the first administration could influence the PK after a second administration [14, 15]. Therefore, a parallel design for infliximab PK comparison would be more suitable considering the study purpose.
PK sub-analyses according to the post-dose ADA status were performed to explore the relationship between immunogenicity of study drugs and PK. Within each treatment, systemic exposure of infliximab in ADA-positive subjects tended to be lower than in ADA-negative subjects. Means of AUCs in ADA-positive subjects were 23–31 % lower than those of ADA-negative subjects in each treatment. It is known that anti-infliximab antibody can have an influence on the PK of infliximab by changing the drug clearance rates [4]. A recent PK modelling study showed that mean CL was 47.1 % higher in patients positive for antibodies to infliximab compared with those who were negative [15]. There is a limitation to comparing the historical data and current study results since the previous study results were obtained from patients, while this study was performed in healthy subjects. However, the differences in mean CL between post-dose ADA-positive and ADA-negative subjects in this study were comparable with previous modelling study results (35, 43 and 37 % for SB2, EU-INF and US-INF, respectively). Though there is an immunogenicity influence on PK of infliximab, the 90 % CIs for the geometric LS Mean ratio of all primary PK parameters for each ADA-negative subject group and ADA-positive subject group between SB2 and EU-INF, SB2 and US-INF, and EU-INF and US-INF were within the 0.8–1.25 criteria (data are not shown). This PK comparison per ADA results could confirm the PK equivalence between treatments regardless of immunogenicity. Therefore, it is concluded that there is no difference in immunogenicity and the PK influence of post-dose ADA in SB2, EU-INF and US-INF.
Overall, PK equivalence between the proposed infliximab biosimilar, SB2, and reference products EU-INF or US-INF was demonstrated, and the immunogenicity impacts on PK were also similar between treatment groups in this phase I study in healthy subjects. A clinical confirmatory phase III study is in progress to compare the efficacy and safety, including immunogenicity, between SB2 and EU-INF in patients with rheumatoid arthritis.
5 Conclusion
In conclusion, this clinical study in healthy subjects showed pharmacokinetic equivalence between SB2 and its marketed reference products of infliximab. No significant difference in terms of safety and immunogenicity profiles was found across the treatment groups.
References
Maini R, St Clair EW, Breedveld F, et al. Infliximab (chimeric anti-tumour necrosis factor alpha monoclonal antibody) versus placebo in rheumatoid arthritis patients receiving concomitant methotrexate: a randomised phase III trial. Lancet. 1999;354:1932–9.
Hanauer SB, Feagan BG, Lichtenstein GR, et al. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet. 2002;359:1541–9.
Cornillie F, Shealy D, D’haens G, et al. Infliximab induces potent anti-inflammatory and local immunomodulatory activity but no systemic immune suppression in patients with Crohn’s disease. Aliment Pharmacol Ther. 2001;15:463–73.
European Medicines Agency. Remicade: EPAR—product information. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000240/WC500050888.pdf. Accessed 19 March 2015.
Thorpe R, Wadhwa M. Biosimilar monoclonal antibodies approved for use in the EU. Generics Biosimilars Initiat J. 2014;3:9–10.
European Medicines Agency. Remsima: EPAR—product information. 2015. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002576/WC500150871.pdf. Accessed 19 March 2015.
Park W, Hrycaj P, Jeka S, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72:1605–12.
Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013. doi:10.1136/annrheumdis-2012-203090.
European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf. Accessed 19 March 2015.
U.S. Food and Drug Administration. Guidance for industry: scientific considerations in demonstrating biosimilarity to a reference product. 2012. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm291128.pdf. Accessed 19 March 2015.
Cheifetz A, Smedley M, Martin S, et al. The incidence and management of infusion reactions to infliximab: a large center experience. Am J Gastroenterol. 2003;98:1315–24.
Klotz U, Teml A, Schwab M. Clinical pharmacokinetics and use of infliximab. Clin Pharmacokinet. 2007;46:645–60.
Ternant D, Aubourg A, Magdelaine-Beuzelin C, et al. Infliximab pharmacokinetics in inflammatory bowel disease patients. Ther Drug Monit. 2008;30:523–9.
Yi SJ, Kim SE, Park MK, et al. Comparative pharmacokinetics of HD203, a biosimilar of etanercept, with marketed etanercept (Enbrel®). BioDrugs. 2012;26:177–84.
Fasanmade AA, Adedokun OJ, Ford J, et al. Population pharmacokinetic analysis of infliximab in patients with ulcerative colitis. Eur J Clin Pharmacol. 2009;65:1211–28.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical Approvals
The study protocol was reviewed and approved by the Independent Ethics Committee (IEC), Berlin. The consent documents for the study were also reviewed and approved by the IEC prior to use. This pharmacokinetic phase I study was registered at ClinicalTrials.gov (identifier NCT01922336).
Conflict of Interest
D Shin, Y Kim, and YS Kim are employees of Samsung Bioepis Co., Ltd. which is the sponsor of this clinical study. T Körnicke and R Fuhr are employees of PAREXEL International GmbH, which is the contract research organization sponsored by Samsung Bioepis Co., Ltd. to conduct the clinical study. No other relationship or activities exist that could appear to have influenced the submitted work.
Funding
This study was funded by Samsung Bioepis Co., Ltd.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Shin, D., Kim, Y., Kim, Y.S. et al. A Randomized, Phase I Pharmacokinetic Study Comparing SB2 and Infliximab Reference Product (Remicade®) in Healthy Subjects. BioDrugs 29, 381–388 (2015). https://doi.org/10.1007/s40259-015-0150-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-015-0150-5