
Abstract
Objectives
The objective of this study was to construct an early economic evaluation for acalabrutinib for relapsed chronic lymphocytic leukaemia (CLL) to assist early reimbursement decision making. Scenarios were assessed to find the relative impact of critical parameters on incremental costs and quality-adjusted life-years (QALYs).
Methods
A partitioned survival model was constructed comparing acalabrutinib and ibrutinib from a UK national health service perspective. This model included states for progression-free survival (PFS), post-progression survival (PPS) and death. PFS and overall survival (OS) were parametrically extrapolated from ibrutinib publications and a preliminary hazard ratio based on phase I/II data was applied for acalabrutinib. Deterministic and probabilistic sensitivity analyses were performed, and 1296 scenarios were assessed.
Results
The base-case incremental cost-effectiveness ratio (ICER) was £61,941/QALY, with 3.44 incremental QALYs and incremental costs of £213,339. Deterministic sensitivity analysis indicated that survival estimates, utilities and treatment costs of ibrutinib and acalabrutinib and resource use during PFS have the greatest influence on the ICER. Probabilistic results under different development scenarios indicated that greater efficacy of acalabrutinib would decrease the likelihood of cost effectiveness (from 63% at no effect to 2% at maximum efficacy). Scenario analyses showed that a reduction in PFS did not lead to great QALY differences (− 8 to − 14% incremental QALYs) although it did greatly affect costs (− 47 to − 122% incremental pounds). For OS, the opposite was true (− 89 to − 93% QALYs and − 7 to − 39% pounds).
Conclusions
Acalabrutinib is not likely to be cost effective compared with ibrutinib under current development scenarios. The conflicting effects of OS, PFS, drug costs and utility during PFS show that determining the cost effectiveness of acalabrutinib without insight into all parameters complicates health technology assessment decision making. Early assessment of the cost effectiveness of new products can support development choices and reimbursement processes through effective early dialogues between stakeholders.
Similar content being viewed by others
Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
1 Introduction
Bruton’s tyrosine kinase (BTK) inhibitors represent a new line of treatment for chronic lymphocytic leukaemia (CLL), with drugs in development and one already on the market: ibrutinib. Ibrutinib has now been approved for previously treated and untreated CLL and has been shown to be clinically effective with a durable response [1,2,3]. However, ibrutinib is not entirely specific for BTK. It may also inhibit epidermal growth factor receptor (EGFR), interleukin-2–inducible T cell kinase (ITK), T-cell X chromosome kinase, and tyrosine kinase expressed in hepatocellular carcinoma (TEC) family proteins, leading to side effects such as bleeding, atrial fibrillation, rash and diarrhoea [4,5,6].
A more specific BTK inhibitor showing promise in preclinical and early clinical trials is acalabrutinib (ACP-196). In preclinical research, acalabrutinib did not inhibit EGFR, TEC, ITK or other agents [7,8,9,10]. Like ibrutinib, acalabrutinib binds covalently to Cys481 in the adenosine triphosphate (ATP) binding pocket of BTK. It shows a rapid oral absorption with a short plasma half-life, theoretically leading to less toxicity [11, 12]. Early clinical studies with acalabrutinib showed overall response rates of 95% at median follow-up of 14.3 months and mostly grade 1 or 2 adverse events without dose-limiting toxicity [13].
Based on these findings, acalabrutinib would be a valuable addition to the therapeutic options for CLL. However, patient access also relies on the decisions of health technology assessment (HTA) bodies. In the UK, the National Institute for Health and Care Excellence (NICE) reported that ibrutinib’s initial price led to a base-case incremental cost-effectiveness ratio (ICER) of £45,486 per quality-adjusted life-year (QALY) when compared with treatment with physicians’ choice [14]. They advised to reimburse ibrutinib only if the negotiated (confidential) discount would be upheld. Such evaluations by HTA bodies and concomitant price negotiations can take up to a year, delaying patient access.
To reduce delays in patient access, it is possible to start early with the assessment of added value of a new therapy. This can start during preclinical development but is more regularly executed during phase I or II clinical trials. One method is the use of early cost-utility analyses, which can clarify the relative impact of the parameters that drive cost effectiveness. Early cost-utility analyses allow decision makers and manufacturers to streamline clinical development and reimbursement processes [15,16,17,18].
The objective of this study was to construct, based on published phase I/II data, an early cost-utility analysis comparing acalabrutinib and ibrutinib for chronic lymphocytic leukaemia to assist early reimbursement decision making. Sensitivity analyses were performed, and possible development scenarios were assessed, identifying critical parameters and quantifying their relative impact on incremental costs and QALYs.
2 Methods
2.1 General
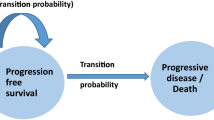
An effectively lifetime partitioned survival model comparing acalabrutinib and ibrutinib was constructed in Microsoft Excel (Microsoft, Redmond, WA, USA) from a UK national health service perspective. As portrayed in Fig. 1, included health states were progression-free survival (PFS), post-progression survival (PPS) and death. PPS was split into two sub-states: subsequent treatment (PPS-ST) and best supportive care (PPS-BSC). The modelled population was based on the only available phase I/II trial for acalabrutinib and assumed representative for the UK relapsed CLL patient population [13]. The model was based on the NICE assessment of the manufacturer’s submission for ibrutinib, appraisal number TA429 [14]. Patients moved between health states in cycles of 28 days with a time horizon of 30 years (effectively lifetime). Half-cycle corrections were applied. Costs and outcomes were both discounted by 3.5%. The model was constructed according to International Society for Pharmacoeconomics and Outcomes Research good modelling practice, and method reporting follows the Consolidated Health Economic Evaluation Reporting Standards statement for reporting standards [19].

Model structure
2.2 Treatment, Comparator and Subsequent Treatment
Ibrutinib 420 mg/day (three capsules) is administered until disease progression or until no longer tolerated by the patient. Acalabrutinib is given as 200 mg/day (two capsules). After progression, 41.9% of patients receive subsequent treatment. Subsequent treatment consists of rituximab and idelalisib. Rituximab is given in six cycles of 4 weeks, with an initial dose of 375 mg/m2 and subsequent doses of 500 mg/m2, according to the NICE guideline for CLL [20]. Idelalisib is administered until disease progression or death in a dose of 150 mg twice daily. A dose intensity of 94.8% was applied for acalabrutinib, ibrutinib and idelalisib, in accordance with findings from the RESONATE trial [2, 14]. In this study, acalabrutinib was assessed relative only to its primary comparator from the same class within the same indication (ibrutinib), as this is expected to be the main competitor in practice. Ofatumumab, physician’s choice or other treatment regimens were not assessed in this study.
2.3 Survival Data
The efficacy of ibrutinib was established in a phase III multicentre, open-label, randomised clinical trial comparing it with ofatumumab [2]. The preliminary efficacy of acalabrutinib was established in a multicentre, open-label, single-arm phase I/II trial [13].
PFS and OS individual patient data for ibrutinib was reconstructed from the reported Kaplan–Meier curves [21]. Multiple parametric survival curves were tested: an exponential, Weibull, log-logistic and lognormal distribution. The exponential curve showed physiological plausibility and overall best fit for OS as well as PFS, corresponding with the expert review group comments on the ibrutinib submission for NICE [14]. For acalabrutinib, efficacy compared with ibrutinib was established through an indirect treatment comparison based on the extracted individual patient data, providing a hazard ratio (HR) of 0.479 for PFS (95% confidence interval [CI] 0.230–0.998) and 0.391 for OS (95% CI 0.141–1.081). Because these are based on very limited data, we set the range of variation in sensitivity and scenario analyses for these HRs from 0.479 and 0.391 to 1.00, representing the full range up until no benefit for acalabrutinib. Furthermore, no assumptions were made about the distribution of this effect. The base case (which equals the maximum HR) was tested (0.479 and 0.391 for PFS and OS, respectively) as were five uniform steps up until no benefit, resulting in six scenarios for the HRs (base case/maximum, 80%, 60%, 40%, 20% and no benefit). In the probabilistic sensitivity analysis (PSA), the base assumption was that OS and PFS were independent. To test the effect of this assumption, the six scenarios for the PSA were also implemented, with the OS and PFS sharing the same random number when sampled, creating dependence. The full survival calculations, fitting criteria and ranges for all parametric models are provided in the Electronic Supplementary Material (ESM) 1.
PPS is defined as OS minus PFS and comprises patients receiving subsequent treatment as well as patients receiving best supportive care. PPS-ST was implemented by plotting a Weibull curve from the PFS given in the Kaplan–Meier graph provided by Furman et al. [22]. The Weibull curve was chosen because NICE evaluated it as being the most suited curve for this treatment. In this multicentre, randomised, double-blind, placebo-controlled, phase III study, the efficacy of rituximab and idelalisib combination therapy was assessed in patients with relapsed CLL [22]. After 80 cycles (75 months), Weibull plotted survival was < 0.01% and therefore assumed to be 0. Because transition probabilities to the PPS state are not explicitly modelled in a partitioned survival model, entry into the PPS state each cycle was calculated by subtracting a specific background mortality from the proportion of patients leaving PFS. This background mortality was retrieved from the Life Expectancy Tables from the Office for National Statistics [23]. This method was also used in the ibrutinib submission; however, the background mortality was considered fixed, whereas ours increased with increasing age.
2.4 Costs and Resource Use
Unit costs for the drug treatments were provided by the British National Formulary [24]. All costs are reported in UK pound sterling (£), year 2018 values. When cost inputs were based on different years, they were inflated with the Hospital and Community Health Services Pay and Price Index and, after discontinuation of this index in 2017, with the health-specific subset of the consumer price inflation index [25]. Daily costs for acalabrutinib treatment were assumed equal to ibrutinib in the base-case and sensitivity analyses and varied through scenario analyses, testing for 30% pricing premiums and reductions (Table 1). Costs for rituximab are only inflicted in the first six cycles of subsequent treatment, in accordance with its approved indication, and are based on an average body surface area of 1.9 m2 [14]. Full calculations for costs per cycle of treatment with acalabrutinib, ibrutinib and rituximab + idelalisib are provided in ESM 2.
Costs of grade 3 and 4 adverse events (AEs) were included according to the UK national schedule of reference costs 2015–2016 [26]. Incidences were implemented from clinical trials for acalabrutinib and from the NICE ibrutinib assessment [13, 14]. AE costs were inflicted once, in the first cycle. This matched the approach used in the NICE ibrutinib submission and is supported by the fact that onset of side effects was generally within the first half year and the duration of side effects was short [2, 14].
Annual healthcare resource use such as hospital visits or blood tests associated with routine follow-up care was included. Resource use is based on expert elicitation reported by the manufacturer in the ibrutinib submission and differs per model state (PFS, PPS-ST, PPS-BSC) and whether the patient in the PFS state was a complete responder (CR), partial responder (PR) or non-responder (NR, including stable disease and progressive disease). Treatment responses for ibrutinib were reported to be 84% PR, 6% CR and 10% NR. For acalabrutinib, 95% were PR and 5% were NR [13, 14]. Full calculations of AE costs and resource use per treatment are provided in ESM 3.
Costs for the death state were inflicted once in the cycle when death happened and equal the per patient costs of healthcare utilisation during the last 30 days of life for patients aged ≥ 65 years with any cancer reported by Bekelman et al. [27]. Costs and ranges for sensitivity analyses are stated in Table 1.
2.5 Utilities
Utility for acalabrutinib was not available and was therefore assumed equal to the ibrutinib utility of 0.799 reported in the RESONATE trial, as measured by the five-level EuroQoL-5 Dimensions (EQ-5D-5L). UK weights were used to generate patient utilities [14]. As an optimum utility for sensitivity and scenario analyses, utility was calculated according to utilities awarded to the response states [28]. AE disutility was calculated according to the incidence and utility decrement of AEs reported in clinical trials for ibrutinib and acalabrutinib [1, 2, 13]. As with AE costs, disutility according to AEs was inflicted once, in the first cycle. The full calculations for disutility due to AEs are provided in ESM 4. To get the post-progression utility, the baseline utility was corrected for the reported utility decrement of 0.098 associated with progression [14]. Base-case utilities and ranges are provided in Table 1.
2.6 Sensitivity and Scenario Analyses
Uncertainties were assessed through sensitivity and scenario analyses. In a one-way sensitivity analysis, the impact of each model input parameter was assessed individually according to their minimum and maximum value provided in Table 1. This deterministic sensitivity analysis shows the impact of the minimum and maximum values for each separate parameter on the ICER. Additionally, PSAs were performed for each of the six HR steps, thus testing cost effectiveness for different acalabrutinib efficacy scenarios. Body surface area and age were not varied in the PSA, in line with the ibrutinib submission.
From the deterministic analysis, important parameters were selected that had a profound influence on the ICER, defined by variations > 5% from the base-case ICER for the minimum and/or maximum scenario. For these critical parameters, all possible combinations of parameter values were tested in scenarios. This means that, for each value for each important parameter (the base-case, minimum and maximum values), all combinations of values for the other parameters were tested. This resulted in an overview of the impact of each parameter on incremental costs and QALYs. The calculation for this relative impact is given in ESM 5.
3 Results
The base-case ICER was £61,941/QALY, with 3.44 incremental QALYs and incremental costs of £213,339. Absolute costs and QALYs were £317,853 and 5.88 for ibrutinib and £531,192 and 9.33 for acalabrutinib, respectively. The one-way sensitivity analysis shown in Fig. 2 indicates that survival estimates, utilities and treatment costs of ibrutinib and acalabrutinib and resource use during PFS have a distinct influence on the ICER. As Fig. 2 also shows, OS and PFS have opposite effects, i.e. when OS for acalabrutinib is reduced, it increases the ICER, whereas reducing PFS leads to a smaller ICER. Higher utility and lower treatment and resource costs during PFS reduce the ICER. For ibrutinib, the opposite was true for all variables.

Relative effects of individual parameters in comparison with the base-case incremental cost-effectiveness ratio of £61,941/quality-adjusted life-year. Note that some parameters are only varied one way because the base case represents the maximum or minimum. acal acalabrutinib, AE adverse events, BSC best supportive care, C costs, ibru ibrutinib, OS overall survival, PFS progression-free survival, PPS post-progression survival, ST subsequent treatment, U utility
Results of the PSAs are shown in Fig. 3. When the efficacy of acalabrutinib grew (HR further from 1.00), the incremental costs and incremental QALYs both increased, but not simultaneously. With no effect (HRs PFS and OS = 1.00), the probability of cost effectiveness was 63% with a willingness-to-pay (WTP) threshold of £50,000/QALY. This declined gradually from 42%, to 25%, 10%, 3% and 2% (HRs 20, 40, 60, 80% and HR maximum, respectively). Thus, higher efficacy and the resulting higher QALYs led to disproportionately higher costs for acalabrutinib, when the price was equal to ibrutinib. Assuming dependence between PFS and OS did not lead to very different results. Mean ICERs were within ± 3% of mean ICERs without dependence. Probabilities of cost effectiveness with a WTP threshold of £50,000/QALY were 60%, 42%, 26%, 12%, 5% and 1% for HRs 1.00, 20%, 40%, 60%, 80% and maximum, respectively.

Cost-effectiveness planes for different hazard ratios. The dark grey dot indicates the base case
3.1 Scenarios
The deterministic analysis provided ten parameters that explained most of the variation. Of those ten, resource use costs and treatment costs during PFS were perfectly correlated with each other. Therefore, these were combined into one parameter (called costs acalabrutinib/ibrutinib) to reduce the number of scenarios. Eight parameters remained: four had base-case, minimum and maximum values (costs during PFS for both treatments and PFS and OS survival parameters for ibrutinib) and four had only two values (base-case and minimum [HRs for acalabrutinib] or base-case and maximum [utility during PFS for both treatments]). This led to a total of 34 × 24 = 1296 scenarios, which were all tested. For each parameter, the effect on incremental QALYs and incremental costs in the minimum and/or maximum scenario versus the base-case scenario was calculated for all scenarios. Figure 4 shows these effects for each of the included eight parameters. The size of the impact of a parameter on incremental costs and QALYs depended on the scenario, i.e. the values of the other input parameters. Figure 4 shows all distinctive values for each parameter. The results indicate that OS was a main driver of incremental QALYs throughout all scenarios, but it does not impact on costs proportionally. The inverse was the case for PFS, which greatly affected costs but not QALYs proportionally.

Range of variation in incremental costs and quality-adjusted life-years due to each critical parameter throughout all scenarios. Acal acalabrutinib, B base case, B/N base case/minimum, B/X base case/maximum, C costs, ibru ibrutinib, N minimum, OS overall survival, PFS progression-free survival, U utility, X maximum
A minimal OS of acalabrutinib led to a reduction in incremental QALYs (89–93% of base-case QALYs) while not greatly influencing incremental cost (reduction of 7–39% of base-case costs), leading to a higher ICER. A minimal PFS led to smaller incremental costs (reduction of 47–122% of base-case costs) but did not significantly affect QALYs (reduction of 8–14% of base-case QALYs), leading to a smaller ICER. Effects of these parameters for ibrutinib were similar but had an opposite direction of effect (i.e. small PFS led to a greater ICER). Utility had a relatively minor effect. Incremental QALY benefits due to greater utility during treatment (based on response rates as described in the Methods section) are relatively small throughout all scenarios (4.5–9.7% of base case). QALY and cost benefits due to fewer side effects were even smaller (and thus not included in scenario analyses).
4 Discussion
In this model, the base-case ICER for acalabrutinib versus ibrutinib was £61,941/QALY, with 3.44 incremental QALYs and incremental costs of £213,339. This indicated that, even with a price equal to that of ibrutinib, acalabrutinib was not cost effective with a WTP threshold of £50,000. The probability of acalabrutinib being cost effective declined with greater efficacy. This finding is explained by the fact that longer PFS led to disproportionally higher costs, even though OS was also prolonged.
In the deterministic analysis, all parameters associated with PFS and OS had significant impact on the ICER. Parameters that had little impact were all one-off parameters (AEs, death costs) and parameters associated with the PPS state. Apparently, subsequent treatment choices do not greatly affect the cost effectiveness of acalabrutinib.
A price for acalabrutinib higher than ibrutinib, with a threshold of £50,000/QALY, would not lead to a cost-effective scenario. This indicates that, even though acalabrutinib would show good survival benefits, reimbursement for a higher price remains unlikely, impeding patient access. With treatment costs set at the base case, neither ibrutinib nor acalabrutinib was cost effective in the PFS state. However, the deterministic sensitivity analysis clarified that treatment costs have a large impact on the ICER. Thus, a cost reduction may potentially lead to time spent the PFS state being cost effective, which would greatly alter the cost effectiveness of both treatments. Indeed, in the tested scenario where both drug costs were minimal (with the rest of the parameters at base case), the ICER was £44,000/QALY.
For decision purposes, the cost effectiveness of an expensive treatment in a certain health state can be roughly estimated from its treatment costs and the utility in that state. If this estimate greatly exceeds the threshold, a modelling exercise may be redundant. However, as our analysis shows, modelling may still be very useful to provide insight into the relative effects of all parameters and their relevance to the ICER. When varied between their plausible bounds, improvements in PFS and OS led to opposite effects on the ICER. The relationship between PFS, OS and the ICER is often not straightforward within the context of an incremental analysis. For example, when costs occur during PFS that are higher than the WTP threshold, the moderate QALY improvement associated with prolonged PFS may not offset these costs if prolonged PFS does not translate to prolonged OS. A positive correlation may exist, but previous publications have highlighted that these correlations are very inconsistent between and within different cancer types [29]. In this NICE decision support unit publication, it was furthermore deemed unclear how evidence supporting a correlation should be quantitatively implemented in a cost-effectiveness model. Thus, our primary assumption was independent of PFS and OS, but we ran scenario analyses assuming dependence through sampling from a shared random number. It should be noted that this was possible because an exponential curve was implemented for both survival curves. If one of the curves had been parameterised differently (e.g. Weibull), this approach would not have been viable. Correlating PFS and OS changed the shape of the cost-effectiveness plane, but it did not greatly impact the probability of the treatment being cost effective. However, the impact of correlation between PFS and OS may be greater for other drugs or in different disease areas. For early cost-effectiveness models, when information on the relation between PFS and OS is relatively sparse, we strongly advise testing the effects of correlation between survival curves in scenario analyses.
Recent research has shown that OS was included as a primary outcome in studies in only 18/68 (26%) of drug indications, whereas PFS accounted for another 31 (46%) and response rates for 11 (16%) [30]. For drug indications that lacked data on OS at time of approval, after a median follow-up of 5.4 years after market entry, only 7% were subsequently shown to extend life. Our findings emphasise that this lack of demonstrated OS benefit induces problems in reimbursement processes.
Acalabrutinib was approved by the US FDA for mantle cell lymphoma (MCL) via the accelerated approval pathway based on benefit in overall response rate. Though treatments for MCL and CLL differ, of interest is that our analysis shows that the manufacturer would get the best price in CLL when they solely show benefit through better response rates and do not prove PFS benefit. Acalabrutinib is currently being investigated in several phase II and III clinical trials for first-line and subsequent treatment in CLL, MCL and at least eight other indications varying from rheumatoid arthritis to urothelial carcinoma [10]. Our analysis shows that perverse incentives might be present in reimbursement processes. Therefore, it is essential that stakeholders engage early and discuss adequate evidence-generation plans prospectively based on scenario analyses such as the one presented here.
4.1 Strengths and Limitations
A strength of this research effort is that it establishes an indication of cost effectiveness well in advance of any reimbursement considerations for acalabrutinib. Additionally, our model is based on a previous submission to NICE and assesses the influence of each parameter in sensitivity and scenario analyses, leading to well-founded conclusions on each parameters’ relevance.
However, relying on a previous NICE submission has its caveats. The use of input parameter values provided for ibrutinib may lead to biased estimates. In the ibrutinib submissions, resource use was estimated through expert opinion. Furthermore, the public report of the NICE appraisal is redacted in many places, which made it hard to implement some of the features and numbers. For example, we had to estimate the survival curves from the published data because the parameters for the curves were redacted in the report. Additionally, the utility reported was established in patients in clinical trials that differed from those for acalabrutinib. The lack of mature data specifically for acalabrutinib led to larger uncertainties in cost-effectiveness estimates but is also an inherent limitation to early modelling. We provided extensive sensitivity and scenario analyses to limit these risks.
Additionally, as mentioned, survival benefit was extracted from published phase I/II (acalabrutinib) and III (ibrutinib) studies. Several valid methods exist to estimate individual patient data from published Kaplan–Meier curves, which all vary slightly [21, 31, 32]. We chose the method developed by Hoyle and Henley [21] but others may also have been appropriate [33]. All of them represent an approximation of individual patient data (IPD) and thus have limitations. Unfortunately, IPD is not shared by the company.
While naive comparisons between trials have limitations, they are also common in the economic evaluation of pharmaceutical products. Additionally, a previous study investigated effect sizes between phase II and phase III and found that, for solid malignancies, phase III studies yielded on average a 12.9% lower objective response rate [34]. Though our analysis is not in solid malignancies and the endpoints used from the trials are survival endpoints, it should be noted from this previous research that comparing a phase II with a phase III trial may not be appropriate. However, we have performed extensive sensitivity and scenario analyses on the HRs provided by this comparison. To represent all possible outcomes for the survival benefit of acalabrutinib in comparison with ibrutinib, we chose the lower value for sensitivity analyses as no benefit (HR = 1.00).
Partitioned survival modelling itself has limitations, because modelling PFS and OS without modelling the underlying events may lead to over or underestimation of long-term survival. However, partitioned survival modelling is a common approach in oncology and is usually accepted by HTA bodies, as it was in the case of ibrutinib. Because survival in the PPS state was time dependent, we required the proportion of patients entering PPS from PFS. Our method to retrieve these events was similar to the ibrutinib submission in that it included correcting for background mortality. Still, the lack of actual information on progression of patients is a limitation to partitioned survival models.
We also did not include subsequent treatments other than rituximab + idelalisib, but results show that the nature and costs of subsequent treatment are practically irrelevant for cost-effectiveness estimates. Last, we also did not include ofatumumab as a comparator. Though the benefits of acalabrutinib over ofatumumab may be different, it is likely that ibrutinib will be the primary comparator because it belongs to the same class.
4.2 Further Research
It was impossible to assess all scenarios when including all parameters (> 650 million scenarios). Automated analyses might provide additional insight into parameters that we excluded from scenario analysis. Finally, combining multiple disease and treatment models leads to more insight into product dynamics and lifetime cost effectiveness. Such interactive models can accommodate the complexity of value-based pricing within different indications for multiple drugs, leading to more appropriate reimbursement mechanisms.
5 Conclusion
In this early cost-utility analysis, survival benefits of acalabrutinib do not result in a cost-effective scenario compared with ibrutinib. The relative and conflicting effects of OS, PFS, drug costs and utility during PFS show that determining the cost effectiveness of acalabrutinib without insight into all parameters complicates HTA decision making. Early assessment of the cost effectiveness of new products can support development choices and reimbursement processes through effective early dialogues between stakeholders, ultimately improving patient access.
Data Availability
All data used for this study are public and referenced throughout the manuscript and its supplementary information files. The Excel model is available from the corresponding author on reasonable request.
References
Byrd JC, Furman RR, Coutre SE, Burger JA, Blum KA, Coleman M, et al. Three-year follow-up of treatment-naïve and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood. 2015;125:2497–506.
Byrd JC, Brown JR, O’Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371:213–23.
de Claro RA, McGinn KM, Verdun N, Lee S-L, Chiu H-J, Saber H, et al. FDA approval: ibrutinib for patients with previously treated mantle cell lymphoma and previously treated chronic lymphocytic leukemia. Clin Cancer Res. 2015;21:3586–90.
Levade M, David E, Garcia C, Laurent P-A, Cadot S, Michallet A-S, et al. Ibrutinib treatment affects collagen and von Willebrand factor-dependent platelet functions. Blood. 2014;124:3991–5.
Fabbro SK, Smith SM, Dubovsky JA, Gru AA, Jones JA. Panniculitis in patients undergoing treatment with the bruton tyrosine kinase inhibitor ibrutinib for lymphoid leukemias. JAMA Oncol. 2015;1:684–6.
McMullen JR, Boey EJH, Ooi JYY, Seymour JF, Keating MJ, Tam CS. Ibrutinib increases the risk of atrial fibrillation, potentially through inhibition of cardiac PI3 K-Akt signaling. Blood. 2014;124:3829–30.
Burki TK. Acalabrutinib for relapsed chronic lymphocytic leukaemia. Lancet Oncol. 2016;17:e48.
Patel V, Balakrishnan K, Bibikova E, Ayres M, Keating MJ, Wierda WG, et al. Comparison of acalabrutinib, a selective bruton tyrosine kinase inhibitor, with ibrutinib in chronic lymphocytic leukemia cells. Clin Cancer Res. 2017;23:3734–43.
Wu J, Liu C, Tsui ST, Liu D. Second-generation inhibitors of Bruton tyrosine kinase. J Hematol OncolJ Hematol Oncol. 2016;9:80.
Wu J, Zhang M, Liu D. Acalabrutinib (ACP-196): a selective second-generation BTK inhibitor. J Hematol Oncol J Hematol Oncol. 2016;9:21.
Herman SEM, Montraveta A, Niemann CU, Mora-Jensen H, Gulrajani M, Krantz F, et al. The Bruton Tyrosine Kinase (BTK) inhibitor acalabrutinib demonstrates potent on-target effects and efficacy in two mouse models of chronic lymphocytic leukemia. Clin Cancer Res. 2017;23:2831–41.
Smith CIE. From identification of the BTK kinase to effective management of leukemia. Oncogene. 2017;36:2045–53.
Byrd JC, Harrington B, O’Brien S, Jones JA, Schuh A, Devereux S, et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374:323–32.
National Institute for Health and Care Excellence. Ibrutinib final appraisal determination. Committee papers. United Kingdom. 2017.
Ijzerman MJ, Steuten LMG. Early assessment of medical technologies to inform product development and market access: a review of methods and applications. Appl Health Econ Health Policy. 2011;9:331–47.
IJzerman MJ, Koffijberg H, Fenwick E, Krahn M. Emerging use of early health technology assessment in medical product development: a scoping review of the literature. Pharmacoeconomics. 2017;35(7):727–40.
Maignen F, Osipenko L, Pinilla-Dominguez P, Crowe E. Regulatory watch: outcomes of early health technology assessment dialogues in medicinal product development. Nat Rev Drug Discov. 2017;16:79.
Geenen JW, Baranova EV, Asselbergs FW, de Boer A, Vreman RA, Palmer CN, et al. Early health technology assessments in pharmacogenomics: a case example in cardiovascular drugs. Pharmacogenomics. 2017;18:1143–53.
Husereau D, Drummond M, Petrou S, Carswell C, Moher D, Greenberg D, et al. Consolidated Health Economic Evaluation Reporting Standards (CHEERS) statement. BMC Med. 2013;11:80.
National Institute for Health and Care Excellence. Blood and bone marrow cancers pathway. 2017. https://pathways.nice.org.uk/pathways/blood-and-bone-marrow-cancers/leukaemia#path=view%3A/pathways/blood-and-bone-marrow-cancers/lymphoid-leukaemia.xml&content=view-node%3Anodes-treatment-for-relapsed-or-refractory-chronic-lymphocytic-leukaemia. Accessed 25 Jul 2017.
Hoyle MW, Henley W. Improved curve fits to summary survival data: application to economic evaluation of health technologies. BMC Med Res Methodol. 2011;11:139.
Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370:997–1007.
Office for National Statistics. National life tables. 2017. https://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/lifeexpectancies/datasets/nationallifetablesunitedkingdomreferencetables. [Internet]. [cited 2019 Apr 4]. https://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/lifeexpectancies/datasets/nationallifetablesunitedkingdomreferencetables. Accessed 25 Jul 2017.
British Medical Association and the Royal Pharmaceutical Society of Great Britain. The British National Formulary (BNF). London, UK. https://www.medicinescomplete.com/mc/login.htm. Accessed 25 Jul 2017.
Department of Health. Hospital and community health services (HCHS) pay and price index. London: Department of Health; 2016.
Department of Health. National Health Service. Reference cost collection: national schedule of reference costs—year 2015-16—NHS trust and NHS foundation trusts. National Health Service, London.
Bekelman JE, Halpern SD, Blankart CR, Bynum JP, Cohen J, Fowler R, et al. Comparison of site of death, health care utilization, and hospital expenditures for patients dying with cancer in 7 developed countries. JAMA. 2016;315:272–83.
Beusterien KM, Davies J, Leach M, Meiklejohn D, Grinspan JL, O’Toole A, et al. Population preference values for treatment outcomes in chronic lymphocytic leukaemia: a cross-sectional utility study. Health Qual Life Outcomes. 2010;8:50.
Davis S, Tappenden P, Cantrell A. A review of studies examining the relationship between progression-free survival and overall survival in advanced or metastatic cancer. [Internet]. London: National Institute for Health and Care Excellence (NICE); 2012 [cited 2019 Apr 4]. http://www.ncbi.nlm.nih.gov/books/NBK425826/. Accessed 4 Apr 2019
Davis C, Naci H, Gurpinar E, Poplavska E, Pinto A, Aggarwal A. Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency: retrospective cohort study of drug approvals 2009-13. BMJ. 2017;359:j4530.
Guyot P, Ades A, Ouwens MJ, Welton NJ. Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC Med Res Methodol. 2012;12:9.
Wei Y, Royston P. Reconstructing time-to-event data from published Kaplan-Meier curves. Stat J. 2017;17:786–802.
Wan X, Peng L, Li Y. A review and comparison of methods for recreating individual patient data from published Kaplan-Meier survival curves for economic evaluations: a simulation study. PLoS One. 2015;10(3):e0121353.
Zia MI, Siu LL, Pond GR, Chen EX. Comparison of outcomes of phase II studies and subsequent randomized control studies using identical chemotherapeutic regimens. J Clin Oncol. 2005;23:6982–91.
Author information
Authors and Affiliations
Contributions
RAV and MJA devised and planned the study. RAV, JWG, AMH and MJA built the model. RAV, WGG, HGML and MJA analysed the model results and devised the results to be presented. RAV wrote the first draft of the manuscript. All authors revised the manuscript in subsequent iterations. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Funding
No funding was received for this study.
Conflict of interest
JWG has received an unrestricted grant from GlaxoSmithKline, unrelated to this study. AMH has received unrestricted grants from GlaxoSmithKline, unrelated to this study. HGML is a past-chairman of the Dutch Medicines Evaluation Board and a past-member of the EMA CHMP. He is also a member of the Lygature Leadership Team. RAV, WGG, MJA have no conflicts of interest that are directly relevant to the content of this article.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Vreman, R.A., Geenen, J.W., Hövels, A.M. et al. Phase I/II Clinical Trial-Based Early Economic Evaluation of Acalabrutinib for Relapsed Chronic Lymphocytic Leukaemia. Appl Health Econ Health Policy 17, 883–893 (2019). https://doi.org/10.1007/s40258-019-00496-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40258-019-00496-1