
Abstract
Purpose of Review
Cystic fibrosis (CF) is a monogenic recessive disease with multisystem involvement. The cause is a mutation in the gene that encodes the cystic fibrosis transmembrane conductance regulator (CFTR) protein. The aim is to review the literature involving the CFTR I1234V mutation and to provide recommendations for future research activities.
Recent Findings
The prevalence rates of CFTR mutations vary across the globe. The CFTR I1234V mutation is the most common mutation in Qatar, and one of the most common in the Arabian Gulf region.
Summary
Areas for future research include testing of the CFTR transcript and activity levels in different samples including nasal cells and organoids. Another area is applying Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) technology as a tool for gene editing.
Similar content being viewed by others


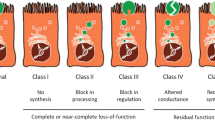
Introduction
Cystic fibrosis (CF) is a hereditary disease with multisystem involvement [1]. The cause is a mutation in the gene that encodes the cystic fibrosis transmembrane conductance regulator (CFTR) protein [1]. The number of CFTR gene mutations exceeds 2000 so far [2]. CF involves the presence of two disease-causing mutations on two separate chromosomes [3]. The CFTR gene was initially discovered in 1989, and is located on chromosome 7q31.2 and is comprised of 27 exons [4,5,6].
A comprehensive assessment of the CFTR mutations across the globe showed variations in prevalence rates for the various CFTR mutations [7]. Globally, the F508del is reported to be the most common mutation [8]. F508del was the commonest mutation among European patients in a study that described the spectrum of mutations among more than 25,000 patients [9]. The F508del mutation is also the most common in Latin America [10].
A range of mutations exist across Arab countries [11]. The spectrum of these mutations was recently described in a systematic review [12•]. The Middle East hosts several mutations, some of which are shared with other global regions, such as F508del, N1303K, W1282X, and 3120+1G>A, while others are less common globally, such as I1234V which is reported to be common among certain Arab tribes [10], 1548delG in Saudi Arabia [13], 2043delG in Bahrain [14], or S549R(T>G) in Oman [15].
CFTR I1234V Mutation
Scientists have identified the CFTR I1234V mutation (p.lle1234Val, c.3700A>G) [16] among patients from Qatar [17], Saudi Arabia [18, 19], France [20], and Israel [21]. In Qatar, 65% of the pediatric CF patients are homozygous for the CFTR I1234V mutation [22]. The I1234V mutation is a missense mutation in exon 19, at nucleotide position 3700, where adenine is replaced by guanine, resulting in the substitution of isoleucine by valine at codon 1234 in the mature CFTR protein [17].
The first report of a heterozygous I1234V mutation was in 1992, in a patient that was initially diagnosed at the age of 2 years. The patient carried the ∆F508 allele on the maternally inherited chromosome and presented with the following clinical features: severe pulmonary status, growth retardation, and presence of Pseudomonas aeruginosa [20]. The mutation was also described in 1997 as a homozygous I1234V mutation which was found in two sisters from Saudi Arabia, presenting with symptoms of recurrent diarrhea as well as failure to thrive [18]. More than a decade later, a study found a truncated exon 19 (r.3700_3717del; p.Ile1234_Arg1239del) which was similar to p.Phe508del, causing a primary defect in folding and processing [23].
Saudi patients with the I1234V mutation were described in another study to have nasal polyps, pansinusitis, and lung disease [13]. Subsequent studies reported on two Yemenite Jewish patients with the I1234V mutation, one had ΔF508 as the second mutation, while the second patient had W1282X as the second mutation [21]. Another study reported a 1:130 carrier frequency for the I1234V among Yemenite Jews [24].
The Clinical and Functional TRanslation of CFTR Database
The Clinical and Functional TRanslation of CFTR (CFTR2) database contains information on CF patients worldwide; this includes 28 patients with the I1234V variant [16]. The CFTR2 website indicates that those with the I1234V variant and a CF causing variant have a slightly lower average sweat chloride (94 vs. 96 mEq/L), but higher average age (22 vs. 20 years) when compared to the average for all patients with two CF causing variants [16]. As for lung function among patients with the I1234V variant, the website reports a forced expiratory volume test (FEV1%) predicted values of (68–118%, 32–103%,) for ages 10–20 and more than 20 years, respectively, while the FEV1% values for those with two CF causing variants are (42–118%) and (25–104%) for the same age groups [16]. Additionally, 40% of those with the variant I1234V were pancreatic insufficient, compared to 85% of all other patients in the database [16]. The rate of infection with Pseudomonas aeruginosa was 35% for those with the I1234V variant compared to 55% among all other patients in the database [16].
CFTR I1234V Mutation Milestones in Qatar
In 2000, a study conducted on 45 CF patients showed that Qatari’s have mild to moderate CF when compared to non-Qatari’s. The majority of patients suffered respiratory symptoms including cough (91%), crackles (64%), and recurrent chest infections (58%). Other characteristics of the sample included failure to thrive (51%), chronic diarrhea (20%), metabolic alkalosis (38%), and vomiting (20%). Radiographic findings included diffuse pulmonary infiltrates (36%), hyperinflation with peribronchial thickening (31%), normal chest X-ray (18%), atelectasis (9%), and bronchiectasis (7%) [25].
Later on, the homozygous I1234V mutation in exon 19 was discovered in 29 patients from 17 families from the same Arab tribe [17]. The rate of consanguinity among participants of the study was reported to be 96.6%, as parents were reported to be first degree cousins in 16 families. The majority of the sample presented with respiratory symptoms including cough (89.7%) and recurrent chest infections (55.2%). Other characteristics included metabolic alkalosis (44.8%), recurrent wheezy chest in early life (37.9%), and failure to thrive (34.5%) [17].
In 2003, a case report of late diagnosed CF in a multiparous Qatari lady at the age of 35 years, in whom the presenting symptom was chronic lung disease with bronchiectasis, suggested that the I1234V mutation has a variable expression of clinical severity and long survival [26]. Moreover, patients with the CFTR I1234V mutation are characterized by pancreatic sufficiency, as one study showed no statistical difference between fecal elastase-1 (FE1) levels of 40 CF patients from a large Arab kindred family compared to 25 controls [27]. Other research involved measuring sweat chloride levels, as one study reported a significantly higher mean among 41 CF patients homozygous for the I1234V mutation compared to 18 controls (99.2 ± 8.3 mmol/L vs. 15.4 ± 6.7 mmol/L) [28].
Several studies examined the microbiology of patients with the I1234V mutation, one of which reported that the most predominant pathogens among 36 CF patients were Pseudomonas aeruginosa, Staphylococcus aureus, and Haemophilus influenzae [29]. Alcaligenes xylosoxidans, Stenotrophomonas maltophilia, and Mycobacterium abscessus were among the unusual pathogens found in another study [30]. A third article studied the antimicrobial resistance patterns of multidrug-resistant Pseudomonas aeruginosa in 30 patients (83.3% were homozygous for the I1234V mutation), and found the following resistance rates: colistin (0%), piperacillin-tazobactam (50%), meropenem (53%), tobramycin (75%), ciprofloxacin (91.7%), gentamycin, amikacin, cefepime (100%) [31].
A fourth study showed a high prevalence rate of Candida dubliniensis in secretions of the lower respiratory tract of CF patients [32]. A fifth study showed that patients with persistent Candida dubliniensis had a higher BMI and lower FEV1 when compared to patients with intermittent Candida dubliniensis [33]. A sixth study conducted on 26 patients with the I1234V mutations showed a Pseudomonas aeruginosa infection rate of 61.5% and an FEV1 mean of 82.9 ± 14.7% [34].
Other research conducted on patients with the I1234V mutation involved assessing serum zinc levels. A study conducted on 45 CF patients homozygous for the CFTR I1234V mutation reported a mean zinc level of 0.78 ± 0.15 mcg/mL. The study also reported that patients diagnosed with hypozincemia (n = 7) had more respiratory exacerbations and more frequent colonization by Pseudomonas aeruginosa [35]. Another study with 40 CF patients with the I1234V mutation found them to be characterized by normal linear growth, as well as normal body mass index (BMI). However, the study found them to have a high prevalence rate of vitamin D deficiency [36].
Furthermore, a study that was conducted to assess fractional exhaled nitric oxide (FENO) levels among patients homozygous for the CFTR I1234V mutation and controls found the former to have significantly lower levels when compared to the latter, with females having significantly higher levels when compared to males [37]. Another study found a positive correlation between adiponectin levels in sputum and plasma, and negative correlation between BMI and adiponectin levels in sputum and plasma [38].
Conclusions
Areas for future research include testing of the CFTR transcript and activity levels in different samples including nasal cells and organoids [39•] among patients with the CFTR I1234V mutation, as studies have shown that organoids may be used for the development of personalized treatment for patients with CF [40•, 41•]. Applying Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) technology as a tool for gene editing along with its promising results as shown elsewhere on other mutations [2] is yet to take place for the CFTR I1234V mutation. In conclusion, a better future for CF patients worldwide and locally in Qatar is contingent upon intensifying research efforts and expanding on current collaborations.
Abbreviations
- BMI:
-
body mass index
- CF:
-
cystic fibrosis
- CFTR:
-
cystic fibrosis transmembrane conductance regulator,
- CFTR2:
-
Clinical and Functional TRanslation of CFTR
- CRISPR:
-
clustered regularly interspaced short palindromic repeats
- FE1:
-
fecal elastase-1
- FEV1:
-
forced expiratory volume test
- FENO:
-
fractional exhaled nitric oxide.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Elborn JS. Cystic fibrosis. Lancet. 2016;388(10059):2519–31.
Marangi M, Pistritto G. Innovative therapeutic strategies for cystic fibrosis: moving forward to CRISPR technique. Front Pharmacol. 2018;9:396.
Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration. 2000;67(2):117–33.
Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245(4922):1073–80.
Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–73.
Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245(4922):1059–65.
Bobadilla JL, Macek M Jr, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations—correlation with incidence data and application to screening. Hum Mutat. 2002;19(6):575–606.
CFTR Science. CFTR Mutations: 242 are known to be CF-causing. 2017; Available from: https://www.cftrscience.com/?q=cftr-mutations.
De Boeck K, et al. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J Cyst Fibros. 2014;13(4):403–9.
Human Genetics Programme WHO. The molecular genetic epidemiology of cystic fibrosis: report of a joint meeting of WHO/IECFTN/ICF(M)A/ECFS, Genoa. Italy. June 2002;19:2004.
Dawson K, Al Whadneh A. Cystic fibrosis and the gulf states. Qatar Med J. 2000;9(2):3–4.
• Al-Sadeq D, et al. Spectrum of mutations of cystic fibrosis in the 22 Arab countries: a systematic review. Respirology. 2018;24(2):127–36. A systematic review that describes the spectrum of cystic fibrosis mutations across Arab countries.
Banjar H, Kambouris M, Meyer BF, al-Mehaidib A, Mogarri I. Geographic distribution of cystic fibrosis transmembrane regulator gene mutations in Saudi Arabia. Ann Trop Paediatr. 1999;19(1):69–73.
Eskandarani HA. Cystic fibrosis transmembrane regulator gene mutations in Bahrain. J Trop Pediatr. 2002;48(6):348–50.
Fass UW, al-Salmani M, Bendahhou S, Shivalingam G, Norrish C, Hebal K, et al. Defining a mutational panel and predicting the prevalence of cystic fibrosis in oman. Sultan Qaboos Univ Med J. 2014;14(3):e323–9.
The Clinical and Functional TRanslation of CFTR (CFTR2). Available from: http://cftr2.org.
Abdul Wahab A, al Thani G, Dawod ST, Kambouris M, al Hamed M. Heterogeneity of the cystic fibrosis phenotype in a large kindred family in Qatar with cystic fibrosis mutation (I1234V). J Trop Pediatr. 2001;47(2):110–2.
el-Harith EA, et al. Novel and characteristic CFTR mutations in Saudi Arab children with severe cystic fibrosis. J Med Genet. 1997;34(12):996–9.
Kambouris M, Banjar H, Moggari I, Nazer H, al-Hamed M, Meyer BF. Identification of novel mutations in Arabs with cystic fibrosis and their impact on the cystic fibrosis transmembrane regulator mutation detection rate in Arab populations. Eur J Pediatr. 2000;159(5):303–9.
Claustres M, Gerrard B, Kjellberg P, Desgeorges M, Demaille J, Dean M. Screening for cystic fibrosis mutations in southern France: identification of a frameshift mutation and two missense variations. Hum Mutat. 1992;1(4):310–3.
Quint A, et al. Mutation spectrum in Jewish cystic fibrosis patients in Israel: Implication to carrier screening. Am J Med Genet A. 2005;136A(3):246–8.
Janahi IA. Assessing the variable molecular & clinical consequences of the rare CFTR I1234V mutation., in Regional CF Educational Meeting: Advancing cystic fibrosis total management. 2018: Muscat, Oman.
Molinski SV, et al. Genetic, cell biological, and clinical interrogation of the CFTR mutation c.3700 A>G (p.Ile1234Val) informs strategies for future medical intervention. Genet Med. 2014;16(8):625–32.
Reish O, et al. Dynamic modification strategy of the Israeli carrier screening protocol: inclusion of the Oriental Jewish Group to the cystic fibrosis panel. Genet Med. 2009;11(2):101–3.
Abdul Wahab A, Dawod ST, al Thani G. Cystic fibrosis in a large kindred family in Qatar. Ann Trop Paediatr. 2000;20(3):203–7.
Wahab AA. Cystic fibrosis mutation I1234V in a Qatari lady. J Trop Pediatr. 2003;49(1):54–5.
Abdel Rahman H, Abdul Wahab A, Abdel Rahman MO, Mostafa OA. Faecal elastase-1 concentration in cystic fibrosis patients with CFTR I1234V mutation. Acta Paediatr. 2006;95(9):1066–9.
Abdul-Wahab A, Janahi IA, Abdel-Rahman MO. Sweat chloride concentration in cystic fibrosis patients with cystic fibrosis trans-membrane conductance regulator I1234V mutation. Saudi Med J. 2009;30(8):1101–2.
Elshafie SS, Wahab AA, Janahi IA. Antimicrobial resistance of bacterial strains isolated from respiratory tract of cystic fibrosis patients with CFTR I1234V mutation. J Pediatr Infect Dis. 2007;02(01):039–43.
Wahab AA, Janahi IA, Marafia MM, el-Shafie S. Microbiological identification in cystic fibrosis patients with CFTR I1234V mutation. J Trop Pediatr. 2004;50(4):229–33.
AbdulWahab A, Zahraldin K, Sid Ahmed MA, Jarir SA, Muneer M, Mohamed SF, et al. The emergence of multidrug-resistant Pseudomonas aeruginosa in cystic fibrosis patients on inhaled antibiotics. Lung India. 2017;34(6):527–31.
Wahab AA, et al. High prevalence of Candida dubliniensis in lower respiratory tract secretions from cystic fibrosis patients may be related to increased adherence properties. Int J Infect Dis. 2014;24:14–9.
AbdulWahab A, Salah H, Chandra P, Taj-Aldeen SJ. Persistence of Candida dubliniensis and lung function in patients with cystic fibrosis. BMC Res Notes. 2017;10(1):326.
Abdul Wahab A, et al. Bone mineral density in cystic fibrosis patients with the CFTR I1234V mutation in a large kindred family is associated with pancreatic sufficiency. Int J Rheumatol. 2014;2014:465395.
AbdulWahab A, Abushahin A, Allangawi M, Chandra P, Abdel Rahman MO, Soliman A. Serum zinc concentration in cystic fibrosis patients with CFTR I1234V mutation associated with pancreatic sufficiency. Clin Respir J. 2017;11(3):305–10.
Wahab AA, Soliman A, Rahman MOA. Growth parameters and calcium homeostasis in cystic fibrosis patients with CFTR I1234V mutation. Ann of Saudi Med. 2009;29(6):487–8.
Saadoon A, et al. Baseline exhaled nitric oxide level in cystic fibrosis patients with I1234V mutation is lower than normal and affected by gender and airway microbiome, in D25. Pediatric Cystic Fibrosis. 2014: San Diego, CA. p. A5518-A5518.
Abdul Wahab A, et al. Sputum and plasma adiponectin levels in clinically stable adult cystic fibrosis patients with CFTR I1234V mutation. Eur Respir J. 2018;52(suppl 62).
• Iwona P, et al. Correction of CFTR function in nasal epithelial cells from cystic fibrosis patients predicts improvement of respiratory function by CFTR modulators. Vol. 7. 2017. Provides evidence that correction of CFTR function in human nasal epithelial cultures can predict respiratory improvement by CFTR modulaters.
• Berkers G, et al. Rectal organoids enable personalized treatment of cystic fibrosis. Cell Rep. 2019;26(7):1701–1708.e3. Shows that organoids may be used for the development of personalized treatment for patients with CF.
• Noordhoek J, et al. Intestinal organoids and personalized medicine in cystic fibrosis: a successful patient-oriented research collaboration. Curr Opin Pulm Med. 2016;22(6):610–6. Shows that organoids may be used for the development of personalized treatment for patients with CF.
Acknowledgements
Open Access funding provided by the Qatar National Library.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Samer Hammoudeh, Wessam Gadelhak, Atqah AbdulWahab, Mona Al-Langawi, and Ibrahim A. Janahi each declare no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Clinical Genetics
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hammoudeh, S., Gadelhak, W., AbdulWahab, A. et al. An Overview of the Homozygous Cystic Fibrosis Transmembrane Conductance Regulator Mutation c.3700 A>G (p.Ile1234Val) in Qatar. Curr Genet Med Rep 7, 187–190 (2019). https://doi.org/10.1007/s40142-019-00174-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40142-019-00174-7