
Abstract
Introduction
There is no approved effective drug for diabetic peripheral neuropathic pain (DPNP) in China. Gabapentinoids including mirogabalin have shown promise, although data in Chinese patients are scarce.
Methods
This phase 3, multicenter, randomized, double-blind, placebo-controlled trial investigated the efficacy and safety of mirogabalin for treating DPNP in China. Mirogabalin was administered at 5 mg twice daily for the first week and uptitrated to 15 mg twice daily for a total duration of 14 weeks. The primary efficacy endpoint was the change from baseline in weekly average daily pain score (ADPS) at week 14; secondary endpoints included the ADPS responder rate, Short-Form McGill Pain Questionnaire visual analogue scale score, patient global impression of change (PGIC), average daily sleep interference score (ADSIS), EuroQol 5-dimensions 5-levels (EQ-5D-5L), and incidence of treatment-emergent adverse events (TEAEs).
Results
Of 393 patients (mirogabalin, n = 196; placebo n = 197), the mean age was 58.2 years (mirogabalin, 58.7 years; placebo, 57.7 years) and 54.2% were male (mirogabalin, 56.1%; placebo, 52.3%). Mirogabalin elicited a greater change from baseline in the weekly ADPS vs. placebo at week 14: least-squares mean difference (95% confidence interval) vs. placebo − 0.39 (− 0.74, − 0.04), p = 0.0301. PGIC, ADSIS, and EQ-5D-5L data reflected significantly better improvements for patients receiving mirogabalin vs. placebo. The incidence of TEAEs was 75.0% and 75.1% in the mirogabalin and placebo groups, respectively. Most TEAEs were mild or moderate, and the incidence of TEAEs leading to treatment discontinuation was 2.6% in the mirogabalin group and 1.5% in the placebo group.
Conclusions
Although the effect size of mirogabalin was reduced due to the placebo effect, mirogabalin is a safe and effective treatment option for Chinese patients with DPNP.
Trial Registration
ClinicalTrials.gov identifier, NCT04094662.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Diabetic peripheral neuropathic pain (DPNP) is a common and intractable symptom of diabetes mellitus, but gabapentinoids such as mirogabalin have shown promise in treating DPNP. |
However, previous studies have not successfully confirmed efficacy in the Chinese population. |
We hypothesized that mirogabalin, titrated up to 15 mg twice daily, would elicit improvement in the weekly average daily pain score (ADPS) from baseline after 14 weeks of treatment of DPNP in Chinese patients. |
What was learned from this study? |
Mirogabalin elicited a greater change from baseline in the weekly ADPS vs. placebo after 14 weeks of treatment (primary endpoint). |
Mixed results (i.e., both significant and non-significant changes) were observed in various secondary outcomes such as the patient global impression of change, average daily sleep interference score, EuroQol 5-dimensions 5-levels, ADPS responder rate, and Short-Form McGill Pain Questionnaire; the rates of treatment-emergent adverse events were similar between mirogabalin and placebo groups and most events were mild or moderate. |
Mirogabalin is considered a safe and potentially effective treatment option for Chinese patients with DPNP. |
Introduction
Diabetic peripheral neuropathic pain (DPNP) is a common symptom of diabetic peripheral neuropathy presenting as pain in both legs, usually below the knee [1]. DPNP is often accompanied by difficulty sleeping, anxiety, or depression, resulting in reduced quality of life [2,3,4]. Neuropathic pain is generally classified as either central or peripheral; DPNP is one of the major forms of peripheral neuropathic pain and affects a considerable number of patients with diabetes mellitus [5,6,7]. The prevalence of DPNP among patients with diabetes in the US is estimated to be approximately 28% [8, 9], between 6 and 34% in Europe [10], and between 28 and 37% in Japan [11, 12]. The prevalence in China is currently unknown, but DPNP is a growing public health concern, as it is thought to be underreported in China owing to low screening rates; it likely represents a considerable disease burden, as the incidence of type 2 diabetes continues to increase [13].
Patients with bilateral foot pain/pins and needles/tingling sensations who are suspected to have DPNP often report a lack of satisfaction with their treatment [14]. Although a variety of drugs may be used to treat DPNP as off-label use in China [15], there is a lack of evidence for their safety and efficacy in Chinese populations. Duloxetine demonstrated a significantly greater pain relief effect vs. placebo [16]; however, due to Good Clinical Practice violations, duloxetine has not been approved for DPNP in China. Thus, there is currently no approved medicine for treatment of DPNP on the market in China, which is a serious clinical concern.
Mirogabalin is an oral gabapentinoid that has recently been approved in several Asian countries excluding China for treatment of neuropathic pain including peripheral and central neuropathic pain [17]. Phase 2 and 3 studies of mirogabalin conducted in Asia (Japan, 72.3%; Korea, 15.6%; Taiwan region, 8.8%; and Malaysia, 3.4%) and the US have demonstrated its efficacy in treatment of DPNP [18, 19] while being generally well tolerated. A recent meta-analysis has further confirmed that mirogabalin has favorable safety and efficacy profiles for the treatment of DPNP [20]. Although previous studies have been conducted throughout Asia, there is a lack of evidence regarding the safety and efficacy of mirogabalin specifically in Chinese populations. Based on the above-mentioned background and rationale, we hypothesized that mirogabalin, titrated up to 15 mg twice daily, would elicit improvement in the weekly average daily pain score (ADPS) from baseline after 14 weeks of treatment of DPNP in Chinese patients.
The present study objectives were as follows: to evaluate the efficacy of mirogabalin in the treatment of DPNP in Chinese patients, as measured by the change from baseline in weekly ADPS at 14 weeks of treatment and other secondary endpoints; and to examine the safety of mirogabalin.
Methods
Study Design/Interventions
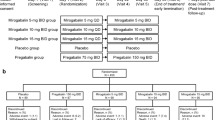
This was a phase 3, randomized, double-blind, placebo-controlled, parallel-group study conducted from June 2019 to January 2022 in 44 centers in China. A full list of participating centers and investigators is provided in Table S1 in the electronic supplementary material and the overall study design is illustrated in Fig. 1. The study and its risks for participation were explained to patients, after which informed consent was obtained, and patients who were receiving prohibited concomitant medications underwent a 1-week washout phase. Patients then underwent screening (visit 1), followed by a 2-week observation period, and then were randomly allocated to receive mirogabalin or placebo for a 2-week dose titration period, a 12-week fixed-dose period, and finally a 1-week follow-up period. For the first week of the titration period, patients received 5 mg of mirogabalin twice daily (BID), followed by 10 mg BID throughout the second week. The dose was increased to 15 mg BID for the fixed-dose period. Patients were instructed to take one tablet of mirogabalin or matching placebo in the morning and at bedtime. All concomitant medications/therapies administered to patients throughout the study were recorded. Prohibited and restricted concomitant medications are listed in Text S1 in the electronic supplementary material.

Study design. Patients underwent screening at visit 1, at which demographics and baseline data were collected and physical exams were performed. Patients were also instructed on the use of electronic diaries and completed SF-MPQ, HADS, and C-SSRS. At visit 2 (observation), patient diaries were reviewed. Patients were randomly allocated to treatment groups at visit 3 and underwent minimal physical exams, laboratory tests, and completed SF-MPQ, HADS, EQ-5D-5L, C-SSRS, and MOS-Sleep evaluations. Patients attended the study site every week for visits 3–5 and every 2 weeks for visits 6–10 during the treatment period. At visit 11 (end of treatment), patients underwent full physical exams, laboratory tests, and completed SF-MPQ, PGIC, HADS, EQ-5L-5D, C-SSRS, and MOS-Sleep evaluations. Post-treatment follow-up was conducted at visit 12 and included the HADS and C-SSRS evaluations, as well as physical exams and laboratory tests. BID twice daily, C-SSRS Colombia-Suicide Severity Rating Scale, HADS Hospital Anxiety and Depression Scale, IC informed consent, MOS-Sleep Medical Outcomes Study Sleep Scale, PGIC patient global impression of change, SF-MPQ Short-Form McGill Pain Questionnaire
Patients
To be eligible for the study, patients had to meet all of the following criteria: ≥ 18 years of age at the time of informed consent; able to provide written informed consent for study participation, understand procedures of this study, and complete patient-reported questionnaires adequately; diagnosed with either type 1 or type 2 diabetes mellitus, diagnosed with painful distal symmetric polyneuropathy at least 6 months prior to screening, and a score of ≥ 40 mm on the visual analogue scale (VAS) of the Short-form McGill Pain Questionnaire (SF-MPQ) at screening and at randomization. With respect to the diagnosis of painful distal symmetric polyneuropathy, patients were required to meet the “confirmed” or “probable” criteria for typical diabetic peripheral neuropathy recommended by the American Diabetes Association. Specifically, for “confirmed diabetic sensorimotor peripheral neuropathy”, this was defined as “the presence of an abnormality of nerve conduction and a symptom or symptoms or a sign or signs of neuropathy”. If the results of a nerve conduction study were normal, patients could meet the “probable” criterion if they met at least two of the following criteria: (1) neuropathic symptoms, (2) decreased distal sensation, and (3) unequivocally decreased or absent ankle reflexes [21]. Neuropathic symptoms were distal, symmetrical, often associated with nocturnal exacerbations, and commonly described by patients as prickling, deep aching, sharp, like an electric shock, or burning with hyperalgesia and frequently allodynia upon examination. Furthermore, at randomization, patients had to have completed at least 4 days of a daily pain diary with an ADPS of ≥ 4 over the previous 7 days on an 11-point numerical rating scale.
Patients were excluded if they met any of the following criteria: a VAS pain score of ≥ 90 mm (SF-MPQ) at screening or randomization; a daily pain score of 10 recorded at any time during the observation period; glycated hemoglobin (National Glycohemoglobin Standardization Program) > 10% at screening; uncontrolled blood glucose within 1 month prior to screening/at screening/randomization; other severe pain, neurological disorders, or skin conditions unrelated to DPN that could confound the assessment of mirogabalin; major psychiatric disorder; lower limb amputation (other than toes); previous use of pregabalin ≥ 300 mg/day or gabapentin ≥ 1200 mg/day with lack of efficacy; impaired renal function (creatinine clearance [CrCL] using the Cockcroft–Gault equation < 60 ml/min at screening), drug/alcohol abuse within 1 year prior to screening; and pregnancy/breastfeeding or unwilling to use reliable contraceptive measures during the study and for 4 weeks after study completion. A comprehensive list of all eligibility criteria is provided in Text S2 in the electronic supplementary material. Patients attended the study centers (national/university hospitals) on an outpatient basis.
Outcomes
The primary efficacy endpoint was the change from baseline in weekly ADPS (defined as the mean daily pain score over a 7-day period based on entries in patient daily pain diaries) at the end of the fixed-dose period (week 14). The secondary efficacy endpoints included ADPS responder rate, SF-MPQ VAS score, patient global impression of change (PGIC), average daily sleep interference score (ADSIS), EuroQol 5-dimensions 5-levels (EQ-5D-5L) VAS score and index values, and the Medical Outcomes Study Sleep Scale (MOS-Sleep).
The safety endpoints were as follows: incidence of treatment-emergent adverse events (TEAEs), incidence of adverse drug reactions (ADRs), laboratory test results, vital signs, body weight, 12-lead electrocardiography, physical and neurological examinations, Colombia-Suicide Severity Rating Scale (C-SSRS), Hospital Anxiety and Depression Scale (HADS), and incidence of edema. ADRs were defined as any AE that was considered by the investigator to be related to the study drug, based on clinical judgement and the timing of drug administration and onset of the AE. AEs defined as “significant” included dizziness, somnolence, edema, and weight gain. Definitions of AE seriousness and severity are provided in Text S3 in the electronic supplementary material. All AEs were coded using the Medical Dictionary for Regulatory Activities version 24.1.
Randomization and Blinding
Patients were randomly allocated in a 1:1 ratio to receive mirogabalin or placebo, with stratification by baseline ADPS (< 6.0 or ≥ 6.0). Further details are provided in Text S4 in the electronic supplementary material.
Ethical Considerations
The study protocol, amendments, informed consent forms, and information sheets were approved by the relevant Independent Ethics Committee or Institutional Review Board of each study center. The study was conducted in compliance with the Declaration of Helsinki and the International Council for Harmonisation consolidated Guideline E6 for Good Clinical Practice. All participants provided written informed consent prior to study participation. The study was registered in ClinicalTrials.gov under the identifier NCT04094662.
Statistical Analysis
The modified intention-to-treat analysis set was used for all efficacy analyses and included all randomly allocated patients who received at least one dose of the study drug. The safety analysis set similarly included all patients who received at least one dose of the study drug.
Regarding the primary endpoint analysis, the change from baseline in the weekly ADPS at week 14 was compared between the mirogabalin and placebo groups using a multiple imputation method and mixed effects model for repeated measurements (Text S5 in the electronic supplementary material). Summary statistics or frequency tables were used to describe secondary efficacy endpoints, with no adjustment for multiple comparisons. All hypothesis testing was performed at a two-sided significance level of 0.05.
The sample size was calculated by applying Student’s t test with a one-sided significance level of 0.025. A total of 356 patients (178 per group) was necessary to provide 85% statistical power under the assumption of a difference of 0.6 (vs. placebo) and common standard deviation (SD) of 1.88 for the change from baseline in weekly ADPS at week 14. The treatment difference is assumed to represent a minimum clinically important difference [22], and the common SD represents an expected value that we conservatively estimated based on the results of a previous phase 3 study of mirogabalin for Asian patients with DPNP [18] and that of a phase 3 Chinese study of duloxetine [16].
The sample size calculation was designed using nQuery Advisor® version 7.0 software (Statsols, San Diego, CA, USA), and all statistical analyses were performed using SAS software version 9.2 (SAS Institute Inc., Cary, NC, USA).
Results
Patients
The patient disposition is illustrated in Fig. 2. A total of 393 patients were included in the study (196 in the mirogabalin group and 197 in the placebo group), and 359 patients (91.3%) completed the study. The completion rate was similar between treatment groups, and the most common reason for study discontinuation was withdrawal by patient decision, which included reasons such as AEs and lack of efficacy.

Patient disposition. aIncludes discontinuations due to the COVID-19 pandemic. mITT modified intention-to-treat
In the overall study population, the mean age at informed consent was 58.2 years, 54.2% of patients were male, the mean CrCL was 105.0 ml/min, and 97.5% of patients were diagnosed with type 2 diabetes mellitus (Table 1). There were no notable differences in patient background characteristics between treatment groups, with the exception of the percentage of patients with reduced renal function. Because of an error in the calculation of CrCL at randomization, three patients with CrCL between 30 and < 60 ml/min failed to be excluded from the study. All three patients were allocated to placebo, and no patients with CrCL < 60 ml/min were included in the mirogabalin group.
Weekly Average Daily Pain Score
The least-squares (LS) mean weekly ADPS (standard error) gradually decreased from week 1 to week 14 in both treatment groups (Fig. 3), and the change from baseline at week 14 (primary endpoint) was significantly greater in the mirogabalin group compared with placebo: LS mean difference (95% confidence interval [CI]) vs. placebo was − 0.39 (− 0.74, − 0.04); p = 0.0301 (Table 2). The results of the sensitivity analyses were consistent with those of the primary analysis.

Time-course of least-squares mean weekly average daily pain score (modified intention-to-treat analysis set). Data were imputed using a multiple imputation method using a pattern mixture model with shifting parameters of [adverse event, lack of efficacy, any other reason] = [1.0, 1.0, 0.5] and a mixed-effects model for repeated measures. Data are least-squares mean with standard error. 0 = no pain; 10 = worst pain imaginable
There were missing data for the weekly ADPS starting from week 3 in the mirogabalin group and from week 2 in the placebo group. The most common reason for the missing weekly ADPS data up to week 14 was “any other reason” (6.1% [12/196] in the mirogabalin group and 4.6% [9/197] in the placebo group). Missing data rates in the mirogabalin and placebo groups for the weekly ADPS (up to week 14) due to AE-related discontinuations were 2.6% (5/196) and 2.0% (4/197) and for lack of efficacy, 0.0% (0/196) and 2.0% (4/197), respectively.
Regarding the results of the ADPS-related secondary efficacy endpoints, at week 14, the percentage of patients with a ≥ 30% or ≥ 50% reduction in weekly ADPS from baseline was numerically greater in the mirogabalin group than the placebo group (Table 2).
Other Secondary Endpoints
The LS mean improvement in week 14 SF-MPQ VAS score vs. baseline was higher in patients who received mirogabalin vs. placebo, but the difference did not reach statistical significance (Table 3). The results of the PGIC are described in Table 3 and Table S3 in the electronic supplementary material. The percentage of patients with PGIC scores of ≤ 2 (“much improved” or “very much improved”) and of ≤ 3 (“minimally improved” or better) were significantly higher in the mirogabalin group vs. placebo.
Regarding the weekly ADSIS, a statistically significantly greater improvement in the mirogabalin group than the placebo group was observed. Regarding the EQ-5D-5L analysis, treatment with mirogabalin resulted in statistically significantly greater improvements in both index value and VAS score at week 14 vs. placebo (Table 3). Regarding the MOS-Sleep scale, no consistent trends were found in any sleep parameters after 14 weeks of treatment (Table S4 in the electronic supplementary material).
Safety Analysis
The incidence of TEAEs was similar in the mirogabalin (75.0%) and placebo (75.1%) groups (Table 4). The incidence of ADRs was higher in the mirogabalin group than in the placebo group: 34.2 vs. 21.8%, respectively.
The most common TEAEs (reported in ≥ 5% of patients in the mirogabalin group) for mirogabalin and placebo, respectively, were: hyperuricemia (12.8 vs. 6.6%), urinary tract infection (10.7 vs. 12.7%), hyperlipidemia (10.7 vs. 11.2%), upper respiratory tract infection (6.6 vs. 5.1%), dizziness (6.6 vs. 3.6%), somnolence (6.1 vs. 3.0%), weight gain (5.6 vs. 2.0%), increased blood creatinine phosphokinase (5.1 vs. 3.0%), and peripheral edema (5.1 vs. 1.5%). Most TEAEs were mild or moderate and the incidence of severe TEAEs was 2.6% in the mirogabalin group and 1.0% in the placebo group. No severe TEAEs were reported in ≥ 2 patients in either group, and no TEAEs leading to death were reported.
The incidence of serious TEAEs in the mirogabalin and placebo groups was 4.1% and 4.6%, respectively. Details of all serious TEAEs and their outcomes and causality are described in Table S5 in the electronic supplementary material. No serious TEAEs occurred at a higher incidence in the mirogabalin group. Only one serious TEAE in the mirogabalin group was judged to be an ADR (blood glucose fluctuation); most serious TEAEs were resolved or resolving with or without treatment by the end of the study. The incidence of TEAEs leading to treatment discontinuation was 2.6% in the mirogabalin group and 1.5% in the placebo group. No notable abnormal findings were reported in clinical laboratory tests, vital signs, 12-lead electrocardiography, C-SSRS, or the HADS.
Discussion
This phase 3, multicenter, randomized, double-blind, placebo-controlled trial assessed the efficacy and safety of 14 weeks of treatment with mirogabalin 15 mg BID for Chinese patients with DPNP. Mirogabalin elicited significant improvement vs. placebo in the primary endpoint (change from baseline in weekly ADPS at week 14). Results from the secondary endpoints generally supported the efficacy of mirogabalin, although not all improvements met the threshold of statistical significance. Mirogabalin was generally well tolerated, and no novel safety concerns were identified. The majority of TEAEs that occurred more frequently in the mirogabalin group were related to the central nervous system, weight gain, or edema, and are expected class effects of gabapentinoids [18, 23, 24]. TEAEs were mostly mild or moderate and few patients discontinued treatment because of a TEAE.
The ADPS, which was the primary efficacy endpoint in this study, was recommended to be used in clinical trials of neuropathic pain by the Japanese [25], European [26], and the mission of the Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) [27] guidelines. Mirogabalin elicited a statistically significant improvement vs. placebo in the ADPS, but this was not reflected in some of the secondary efficacy endpoints, such as the ADPS responder rates and SF-MPQ VAS scores. These inconsistencies may be due to a high placebo response in the present Chinese study compared with a previous phase 3 trial of mirogabalin conducted elsewhere in Asia (primarily in Japan and Korea) [18], which had a similar study design and patient characteristics to the present trial. Both studies found that both placebo and mirogabalin elicited reductions in the weekly ADPS from baseline, but the baseline scores in the placebo group were higher in the present study (6.09 vs. 5.60). The numerically greater reduction in the weekly ADPS (placebo group) from baseline at week 14 in the present study (LS mean − 1.81 vs. − 1.31) may therefore suggest a strong placebo effect in the present study, although the 14-week ADPS values of the placebo groups were similar between studies: 4.30 (present study) vs. 4.26. These abovementioned inconsistencies between Chinese and other Asian populations regarding the placebo effect were also supported by previous studies of pregabalin [28, 29], with respect to both ADPS and SF-MPQ VAS scores. Additionally, both weekly ADPS and VAS scores continued to decrease throughout the treatment period. Both scores remained at consistently lower levels in the mirogabalin group compared with the placebo group soon after treatment initiation and throughout the treatment period. This suggests that mirogabalin has a stronger analgesic effect than placebo, and the difference is not random but corresponds to a pharmacological effect. It is important to note that in our study, the weekly ADPS was assessed using daily pain evaluations, whereas the VAS was evaluated at each study visit, looking back over the past week. Therefore, the ADPS is a more sensitive measure that is affected by daily pain changes, and the favorable ADPS results of our study support the effect of mirogabalin even if there was a contributing placebo effect, although it should also be noted that the ADPS relies on patients’ subjective interpretation of their symptoms. Importantly, in our study, the week 14 ADPS in the mirogabalin group decreased to < 4 (a change of − 2.19), whereas the score in the placebo group did not. Generally, ADPS ≥ 4 is defined as moderate pain, and it is stated that if the severity changes, changing the treatment is clinically meaningful [30]; thus, the reduction of the mean to the lower limit of moderate, ADPS = 4, is clinically meaningful although the difference in the effect size of mirogabalin vs. placebo was reduced due to the placebo effect.
Because there was a slight imbalance in baseline CrCL between treatment groups, with the placebo group having a higher percentage of patients with normal renal function than the mirogabalin group, we performed an additional analysis with adjustment by baseline CrCL as covariate, and a similar tendency was observed in ADPS change from baseline: LS mean difference (95% CI) vs. placebo was − 0.39 (− 0.74, − 0.04); p = 0.0276 (Table S2 in the electronic supplementary material). We found that the ADPS significantly improved with mirogabalin vs. placebo as in the primary analysis. This indicates that the imbalance in baseline CrCL had no influence on the overall greater improvements observed in the ADPS for mirogabalin vs. placebo.
The change from baseline in weekly ADPS was broadly comparable with that of previous studies of mirogabalin and pregabalin (600 mg/day). The LS mean weekly ADPS change from baseline at week 14 (mirogabalin group) in the present study was similar to those of a phase 3 study [18], a study of mirogabalin for postherpetic neuralgia (primarily in Japanese and Korean patients) [31], an 11-week Chinese trial of pregabalin for DPNP [28], and a 14-week Japanese trial of pregabalin for DPNP [29]. The SF-MPQ VAS score data from the present study were consistent with those of the two previous phase 3 Asian (primarily Japanese and Korean) studies of mirogabalin [18, 31]. Regarding the ADSIS, our results were also comparable with those reported for the two previous mirogabalin phase 3 studies for neuropathic pain [18, 31]. Taken together, these data indicate that mirogabalin is effective for treatment of DPNP in the Chinese patient population.
Neuropathic pain is intractable, and the goal of its treatment is not necessarily to eliminate the pain itself, but to reduce it as much as possible and improve activities of daily living and quality of life for patients [32]. Therefore, the EQ-5D-5L and the PGIC are important tools in the evaluation of treatments for neuropathic pain. The present study demonstrated that mirogabalin elicited tangible improvements in quality of life based on the favorable EQ-5D-5L and PGIC results, and such improvements were both statistically and clinically meaningful. With respect to the EQ-5D-5L results, the LS mean difference (95% CI) vs. placebo in EQ-5D-5L index value in a previous study of mirogabalin for central neuropathic pain secondary to spinal cord injury was 0.0287 (− 0.0009, 0.0583) [33] vs. 0.0291 (0.0068, 0.0514) in the present study. Regarding the EQ-5D-5L VAS score, the LS mean difference vs. placebo (95% CI) was 6.2 (2.0, 10.4) in the previous study of mirogabalin for central neuropathic pain [33] vs. 2.8 (0.1, 5.6) in the present study. Quality of life based on the EQ-5D-5L was not assessed in previous phase 3 studies for neuropathic pain [18, 31]; thus, this historical comparison was made with neuropathic pain of different etiologies, which should be noted in interpreting this comparison.
The present study’s PGIC results indicated that the percentage of patients who received mirogabalin and achieved a score of ≤ 3 (“minimally improved” or better) was numerically higher than in previous similar studies: 87.2 vs. 70.3% [18] and 69.0% [31]. Taken together, the above results suggest that mirogabalin not only effectively reduces pain, but also improves quality of life.
The safety results were broadly comparable with those of the previous phase 3 study for DPNP [18], with similar rates of somnolence, dizziness, peripheral edema, and weight gain in patients who received mirogabalin, which are expected class effects of gabapentinoids [24]. However, hyperuricemia as a TEAE was higher in both treatment groups, which was a different trend from the previous research [18]. A recent study reported a relatively high prevalence of hyperuricemia in China [34]. Risk factors for hyperuricemia reported in Chinese studies include high body mass index [35, 35, 36], hypertension [34], and dyslipidemia (high triglycerides, high total cholesterol, high low-density lipoprotein cholesterol, and low high-density lipoprotein cholesterol) [34]. Of the patients who developed hyperuricemia, 38.6% had a history of hyperuricemia or high baseline uric acid, which may explain the relatively high incidence of hyperuricemia in the present study. Notably, patients who developed hyperuricemia did not require any specific treatment or discontinuation/dose interruption of the study drug, most cases were mild and transient, and no severe or serious hyperuricemia TEAEs occurred. Furthermore, the events of hyperuricemia as ADRs were similar between the mirogabalin (n = 2, 1.0%) and placebo groups (n = 1, 0.5%). Thus, mirogabalin treatment raised no new safety concerns in our study, consistent with results from the previous Asian (primarily Japanese and Korean) phase 3 DPNP study [18]. Nevertheless, because hyperuricemia was more common in the mirogabalin group, further study is warranted to confirm whether there is an increased risk of hyperuricemia in Chinese patients with DPNP treated with mirogabalin.
The present study was limited to Chinese patients, so generalization to non-Asian populations may not be valid. Patients with impaired renal function (< 60 ml/min) were excluded, so the safety and efficacy of mirogabalin in Chinese patients with renal disease remains to be determined. The weekly ADPS decreased gradually throughout the study, and it is unclear whether the steady decrease would have continued past 14 weeks of treatment. The lack of efficacy in the secondary outcome measures may not be able to be fully explained by the placebo effect. Neurological examinations such as ankle jerk, vibratory sensation, pain sensation including hyperalgesia, allodynia, muscle strength (ankle dorsiflexion), and gait/station (observation of regular walking, heel-to-toe [tandem] walking, and the Romberg test, each assessed as normal or abnormal) were performed in this study during screening, but these were not performed to evaluate efficacy after study drug administration. Finally, long-term safety and efficacy data of mirogabalin remain to be collected in the Chinese DPNP patient population.
Conclusions
Although the effect size of mirogabalin was reduced due to the placebo effect, mirogabalin, uptitrated to 15 mg BID, was found to be safe and effective in the treatment of DPNP in Chinese patients, suggesting that mirogabalin may be a new treatment option for DPNP in China.
Data Availability
The datasets generated and/or analyzed during the current study are not publicly available, but are available from the corresponding author on reasonable request at https://vivli.org/ourmember/daiichi-sankyo/.
References
Gylfadottir SS, Weeracharoenkul D, Andersen ST, Niruthisard S, Suwanwalaikorn S, Jensen TS. Painful and non-painful diabetic polyneuropathy: Clinical characteristics and diagnostic issues. J Diabetes Investig. 2019;10:1148–57.
Gore M, Brandenburg NA, Dukes E, Hoffman DL, Tai KS, Stacey B. Pain severity in diabetic peripheral neuropathy is associated with patient functioning, symptom levels of anxiety and depression, and sleep. J Pain Symptom Manage. 2005;30:374–85.
Tesfaye S, Vileikyte L, Rayman G, et al. Painful diabetic peripheral neuropathy: consensus recommendations on diagnosis, assessment and management. Diabetes Metab Res Rev. 2011;27:629–38.
Girach A, Julian TH, Varrassi G, Paladini A, Vadalouka A, Zis P. Quality of life in painful peripheral neuropathies: a systematic review. Pain Res Manag. 2019;2019:2091960.
Treede RD, Jensen TS, Campbell JN, et al. Neuropathic pain: Redefinition and a grading system for clinical and research purposes. Neurology. 2008;70:1630–5.
Loeser JD, Treede RD. The Kyoto protocol of IASP basic pain terminology. Pain. 2008;137:473–7.
Nicholson B. Differential diagnosis: Nociceptive and neuropathic pain. Am J Manag Care. 2006;12:S256–62.
Pop-Busui R, Boulton AJ, Feldman EL, et al. Diabetic neuropathy: a position statement by the American Diabetes Association. Diabetes Care. 2017;40:136–54.
Gregg EW, Gu Q, Williams D, et al. Prevalence of lower extremity diseases associated with normal glucose levels, impaired fasting glucose, and diabetes among U.S. adults aged 40 or older. Diabetes Res Clin Pract. 2007;77:485–8.
Alleman CJ, Westerhout KY, Hensen M, et al. Humanistic and economic burden of painful diabetic peripheral neuropathy in Europe: A review of the literature. Diabetes Res Clin Pract. 2015;109:215–25.
Satoh J, Baba M, Yagihashi S, et al. Frequency of diabetic polyneuropathy (DPN) and clinical significance of Achilles tendon reflex in diagnosis of DPN. J Japan Diabetes Soc. 2007;50:799–806. (Japanese).
Yokoyama H, Tsuji T, Hayashi S, Kabata D, Shintani A. Factors associated with diabetic polyneuropathy-related sensory symptoms and signs in patients with polyneuropathy: A cross-sectional Japanese study (JDDM 52) using a non-linear model. J Diabetes Investig. 2020;11:450–7.
Zhang Y, Li N, Zhao Y, et al. Painful Diabetic Peripheral Neuropathy Study of Chinese OutPatiEnts (PDN-SCOPE): Protocol for a multicentre cross-sectional registry study of clinical characteristics and treatment in China. BMJ Open. 2019;9: e025722.
Deguchi T, Takatsuna H, Yokoyama M, et al. A Cross-sectional web survey of satisfaction with treatment for pain in participants with suspected diabetic peripheral neuropathic pain in both feet. Adv Ther. 2021;38:4304–20.
Lian J, Wang H, Cui R, Zhang C, Fu J. Status of analgesic drugs and quality of life results for diabetic peripheral neuropathy in China. Front Endocrinol (Lausanne). 2022;12: 813210.
Gao Y, Guo X, Han P, et al. Treatment of patients with diabetic peripheral neuropathic pain in China: a double-blind randomised trial of duloxetine vs. placebo. Int J Clin Pract. 2015;69:957–66.
Daiichi Sankyo Press Release. Tarlige® Tablets Approved in Japan for Treatment of Patients with Neuropathic Pain. March 18, 2022. https://www.daiichisankyo.com/files/news/pressrelease/pdf/202203/20220328_E.pdf. Accessed 28 May 2024.
Baba M, Matsui N, Kuroha M, Wasaki Y, Ohwada S. Mirogabalin for the treatment of diabetic peripheral neuropathic pain: A randomized, double-blind, placebo-controlled phase III study in Asian patients. J Diabetes Investig. 2019;10:1299–306.
Vinik A, Rosenstock J, Sharma U, et al. Efficacy and safety of mirogabalin (DS-5565) for the treatment of diabetic peripheral neuropathic pain: a randomized, double-blind, placebo- and active comparator-controlled, adaptive proof-of-concept phase 2 study. Diabetes Care. 2014;37:3253–61.
Alyoubi RA, Alshareef AA, Aldughaither SM, et al. Efficacy and safety of mirogabalin treatment in patients with diabetic peripheral neuropathic pain: a systematic review and meta-analysis of randomised controlled trials. Int J Clin Pract. 2021;75: e13744.
Tesfaye S, Boulton AJM, Dyck PJ, et al. Diabetic neuropathies: Update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care. 2010;33:2285–93.
Kirby S, Chuang-Stein C, Morris M. Determining a minimum clinically important difference between treatments for a patient-reported outcome. J Biopharm Stat. 2010;20:1043–54.
Satoh J, Yagihashi S, Baba M, Suzuki M, Arakawa A, Yoshiyama T. Efficacy and safety evaluation of pregabalin treatment over 52 weeks in patients with diabetic neuropathic pain extended after a double-blind placebo-controlled trial. J Diabetes Investig. 2011;2:457–63.
Rose MA, Kam PCA. Gabapentin: pharmacology and its use in pain management. Anaesthesia. 2002;57:451–62.
Ministry of Health, Labour and Welfare. Guidelines for the Clinical Evaluation of Neuropathic Pain Medications. 2020. Available at: https://www.pmda.go.jp/files/000238736.pdf. Accessed 1 Dec 2023. (in Japanese)
European Medicines Agency. Guideline on the clinical development of medicinal products intended for the treatment of pain. 2016. Available at: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-development-medicinal-products-intended-treatment-pain-first-version_en.pdf. Accessed 1 Dec 2023.
Dworkin RH, Turk DC, Peirce-Sandner S, et al. Considerations for improving assay sensitivity in chronic pain clinical trials: IMMPACT recommendations. Pain. 2012;153:1148–58.
Mu Y, Liu X, Li Q, et al. Efficacy and safety of pregabalin for painful diabetic peripheral neuropathy in a population of Chinese patients: a randomized placebo-controlled trial. J Diabetes. 2018;10:256–65.
Satoh J, Yagihashi S, Baba M, et al. Efficacy and safety of pregabalin for treating neuropathic pain associated with diabetic peripheral neuropathy: a 14-week, randomized, double-blind, placebo-controlled trial. Diabet Med. 2011;28:109–16.
Woo A, Lechner B, Fu T, et al. Cut points for mild, moderate, and severe pain among cancer and non-cancer patients: a literature review. Ann Palliat Med. 2015;4:176–83.
Kato J, Matsui N, Kakehi Y, Murayama E, Ohwada S, Sugihara M. Mirogabalin for the management of postherpetic neuralgia: a randomized, double-blind, placebo-controlled phase 3 study in Asian patients. Pain. 2019;160:1175–85.
Nikaido T, Takatsuna H, Tabata S, Shiosakai K, Nakatani T, Konno S. Efficacy and safety of add-on mirogabalin to NSAIDs in lumbar spinal stenosis with peripheral neuropathic pain: a randomized, open-label study. Pain Ther. 2022;11:1195–214.
Ushida T, Katayama Y, Hiasa Y, et al. Mirogabalin for central neuropathic pain after spinal cord injury: a randomized, double-blind, placebo-controlled, phase 3 study in Asia. Neurology. 2023;100:e1193–206.
Song J, Jin C, Shan Z, Teng W, Li J. Prevalence and risk factors of hyperuricemia and gout: a cross-sectional survey from 31 provinces in mainland China. J Transl Int Med. 2022;10:134–45.
Piao W, Zhao L, Yang Y, et al. The prevalence of hyperuricemia and its correlates among adults in China: Results from CNHS 2015–2017. Nutrients. 2022;14:4095.
Li Y, Fan X, Li C, et al. The relationships among hyperuricemia, body mass index and impaired renal function in type 2 diabetic patients. Endocr J. 2018;65:281–90.
Acknowledgements
We thank the participants of the study.
Medical Writing/Editorial Assistance
The authors wish to thank all of the institutions and investigators that participated in this study (Table S1 in the electronic supplementary material). The authors thank Mary Richardson, MSc, of Edanz (www.edanz.com), for providing medical writing support, which was funded by Daiichi Sankyo Co., Ltd., in accordance with Good Publication Practice Guidelines (https://www.ismpp.org/gpp-2022).
Funding
This study was funded by Daiichi Sankyo Co., Ltd., which also funded the Rapid Service Fee for publication of this manuscript. The authors thank the patients and staff of all participating institutions for their support of this research.
Author information
Authors and Affiliations
Contributions
All authors contributed to the conceptualization, methodology, and writing – review & editing of the manuscript. Xiaohui Guo, Yang Yu, Yongbo Zhang, Li Sun, Yufeng Li, and Bing Song conducted the clinical investigation. Masayuki Baba provided oversight for the safe conduct of the study. Kunika Kikumori conducted the formal analysis. Yosuke Wasaki, Kunika Kikumori, Li Hang, and Emiko Murayama contributed to validation of the data. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Corresponding author
Ethics declarations
Conflict of Interest
Xiaohui Guo, Yang Yu, Yongbo Zhang, Li Sun, Yufeng Li, and Bing Song received investigator fees from Daiichi Sankyo Co., Ltd. Masayuki Baba received lecture and consultancy fees from Daiichi Sankyo Co., Ltd. Yosuke Wasaki, Kunika Kikumori, and Emiko Murayama are employees of the study sponsor, Daiichi Sankyo Co., Ltd., and Li Hang is an employee of Daiichi Sankyo (China) Holdings Co., Ltd.
Ethical Approval
The protocol for this research project has been approved by a suitably constituted Ethics Committee of the participating institutions and it conforms to the provisions of the Declaration of Helsinki. All patients provided written informed consent prior to participation. This study was registered in ClinicalTrials.gov on September 19, 2019, under the identifier NCT04094662.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Guo, X., Yu, Y., Zhang, Y. et al. A Phase 3, Multicenter, Randomized, Double-Blind, Placebo-Controlled 14-Week Study of Mirogabalin in Chinese Patients with Diabetic Peripheral Neuropathic Pain. Pain Ther 13, 937–952 (2024). https://doi.org/10.1007/s40122-024-00617-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40122-024-00617-2