
Abstract
Introduction
Recent cross-sectional research has demonstrated a substantial link between tuberculosis (TB) and gut microbiota. Nevertheless, the causal impact of the gut microbiota on TB susceptibility in humans remains unknown.
Methods
The Mendelian randomization (MR) method was utilized for investigating the causality between them. The main method used for MR analysis was the inverse variance weighted (IVW) test, with the MR-Egger, weighted median, weighted mode, and simple median methods serving as supplements. And several sensitivity tests were carried out to validate the MR findings.
Results
The IVW outcomes suggested that three bacterial traits exhibited associations with susceptibility to respiratory TB after Bonferroni correction, namely Lachnospiraceae UCG010 (odds ratio [OR] 1.73, 95% confidence interval [CI] 1.17–2.55, P = 0.005), Eubacterium (brachy group) (OR 1.33, 95% CI 1.07–1.65, P = 0.009), and Ruminococcaceae UCG005 (OR 0.71, 95% CI 0.52–0.98, P = 0.034). Sensitivity analyses demonstrated that horizontal pleiotropy and heterogeneity were absent, thereby guaranteeing the reliability of the results.
Conclusion
This research sheds light on the causal impact of gut microbiota on respiratory tuberculosis susceptibility, improving our knowledge of therapeutic strategies for managing TB.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
The second-most common infectious illness worldwide, tuberculosis (TB) poses a huge burden on global health. |
Although some studies have proposed that gut microbiota may affect TB susceptibility, a causal relationship has not yet been demonstrated. |
What was learned from this study? |
This study is the first to use the Mendelian randomization (MR) method to prove the causality between gut microbiota and TB in humans. |
Three bacterial traits—Ruminococcaceae UCG005, Lachnospiraceae UCG010, and Eubacterium (brachy group)—are associated with TB susceptibility, which provides a new perspective on TB prevention. |
Introduction
Tuberculosis (TB), the second most deadly infectious illness globally, is caused by Mycobacterium tuberculosis (Mtb) and kills approximately 1.4 million human immunodeficiency virus-negative individuals each year [1]. In many TB cases, the respiratory system serves as the primary entrance and is the system most frequently affected during active TB infections [2].
Emerging research has demonstrated that a variety of lung diseases, including cystic fibrosis, asthma, and COVID-19, are linked with dysregulation of the gut microbiota, acting as a potential contributory factor that impacts their onset and progression [3,4,5,6]. For TB, some animal experiments have indicated that the gut microbiota could affect the susceptibility to TB [7,8,9,10,11,12]. TB infection can be promoted by broad-spectrum antibiotics through disrupting the gut microbiota [7,8,9,10], and direct oral administration of gut bacteria or fecal transplantation could reduce TB infection in mice [7, 11, 12]. Only a few observational studies have explored the association between patients with TB and healthy individuals [13,14,15,16,17,18,19,20]. Besides, those studies were based on small samples (fewer than 100 cases), do not account for significant confounders, and have conflicting findings with minimal overlap [13,14,15,16,17,18,19,20]. For instance, investigations by Wang et al. and Naidoo et al. found that Bifidobacterium was abundant in healthy individuals [9, 19], whereas research by Wipperman et al. and Khaliq et al. showed the opposite [18, 20]. The causal associations have not been completely investigated in observational studies in humans [13,14,15,16,17,18,19,20].
A method called Mendelian randomization (MR) could infer causal relationships between exposures and outcomes. This method utilizes genes as instrumental variables (IVs) which are less susceptible to confounding factors as they depend on the random distribution of genetic variation during conception [21, 22]. Several studies have shown a causal relationship between the gut microbiota and various diseases (e.g., COVID-19, major depressive disorder, and cancer) by MR method [23,24,25]. But no research has looked into the causality between gut microbiota and TB; therefore, this study employed the MR approach to explore this relationship.
Methods
Exposure and Outcome Samples
We selected the human gut microbiota as the exposure variable from a genome-wide association study (GWAS) meta-analysis comprising 24 cohorts with a total of 18,340 participants [26]. Regarding the human gut microbiota, the lowest level of classification is the genus. Initially, there were 131 genera, of which 12 were unknown. After these 12 unknown genera were excluded, 119 bacterial traits were further studied. The outcome variable respiratory TB was obtained from the FinnGen Consortium and included 341,987 participants. The diagnosis of TB relied on the International Classification of Diseases (ICD) codes, encompassing ICD-8, ICD-9, and ICD-10, post hospital discharge or following mortality. Additionally, factors such as sex, age, the first ten principal components, and genotyping batch were adjusted for all samples. In a two-sample MR study, the two groups need to come from different populations, and the two populations have less overlap to ensure higher statistical power [27, 28].
The data used as part of this study was from publicly available sources and thus no ethical approval was needed. All data was de-identified before use. Ethics committee approval was not required.
Study Design
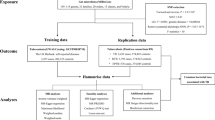
The specific MR methodology is illustrated in Fig. 1. On the basis of three core assumptions, IVs were chosen: (1) Relevance assumption: the IVs needed to show a strong association with gut microbiota (P < 1 × 10−5) [23, 25]. (2) Independence assumption: the IVs were not affected by TB-related confounding factors (r2 < 0.001; clumping distance = 10,000 kb) [29]. (3) Exclusion restriction: the IVs impacted the TB solely via the gut microbiota without any alternative routes [30]. For TB, diabetes [31], alcohol consumption [32], smoking [33], low body mass index [34], and acquired immunodeficiency syndrome [35] were considered to be common risk factors. So, single nucleotide polymorphisms (SNPs) associated with those risk factors should be screened via the Phenoscanner website and excluded prior to MR analysis. Besides, alleles on the forward strand were ascertained using allele frequency data for palindromic SNPs.

Flowchart of Mendelian randomization. IVs instrumental variables, SNPs single-nucleotide polymorphisms, MR Mendelian randomization, MR-PRESSO MR-pleiotropy residual sum and outlier
The F statistic is an important index of the statistical strength of IVs. An F statistic > 10 was assumed to indicate no weak IV bias [36]. The formula F = R2 (N − K − 1)/K (1 − R2) was used to obtain the F statistic [37].
Five MR methods were employed: the inverse variance weighted (IVW) test, MR-Egger, weighted median, weighted mode, and simple median. The IVW test served as the primary technique, which would be impartial if horizontal pleiotropy did not exist, with other methods serving as supplements [38, 39].
Sensitivity analyses included heterogeneity, horizontal pleiotropy, and leave-one-out test. For heterogeneity, Cochrane’s Q test was taken, and P < 0.05 was regarded as heterogeneous. The MR-pleiotropy residual sum and outlier (MR-PRESSO) and MR-Egger methods were employed to evaluate for horizontal pleiotropy, with P < 0.05 as evidence of horizontal pleiotropy. Furthermore, visual assessment of the scatter plots and MR-PRESSO can both identify potential outliers. MR-PRESSO can identify potential outliers and reassess causal effects. Lastly, to ensure robustness, the leave-one-out test was applied.
Taking multiple comparisons into account, the Bonferroni correction was used to prevent false-positive results. If P was less than 0.05 but higher than 4.20 × 10−4 (0.05/119), it was deemed suggestive evidence of causality between gut microbiota and respiratory TB [40].
All statistical analyses were performed by the R software. MR analyses were conducted using the TwoSampleMR and MRPRESSO packages within R.
Results
IVs Selection
Of the initial 131 bacterial traits, 1379 SNPs were selected for IVs. After 12 unknown genera were excluded, 119 bacterial traits and 1235 SNPs remained. Furthermore, our exploration using the Phenoscanner website did not reveal any potential IVs associated with the identified risk factors. The F statistics for SNPs ranged from 14.58 to 88.42, indicating no weak IV bias. Details of the IVs are given in Supplementary Table S1.
Causal Effects Between Gut Microbiota and Respiratory TB
As shown in Fig. 2 and Supplementary Table S2, the IVW test identified three bacterial traits significantly associated with respiratory TB. Among these, two bacterial traits were potentially linked with a higher risk of respiratory TB, namely genus Lachnospiraceae UCG010 (odds ratio [OR] 1.73, 95% confidence interval [CI] 1.17–2.55, P = 0.005) and genus Eubacterium (brachy group) (OR 1.33, 95% CI 1.07–1.65, P = 0.009). In contrast, genus Ruminococcaceae UCG005 (OR 0.71, 95% CI 0.52–0.98, P = 0.034) was linked with a lower risk of respiratory TB. These causal relationships remain significant even after Bonferroni correction.

Causal effects between gut microbiota and respiratory tuberculosis. MR Mendelian randomization, OR odds ratio, CI confidence interval
Moreover, neither the Cochran Q test nor the intercept test of the MR-Egger analysis indicated heterogeneity or horizontal pleiotropy for these suggestive significant causal associations (P > 0.05) (Table 1 and Supplementary Tables S3, S4). Visual assessment of the scatter plots (Fig. 3) identified potential outliers for Ruminococcaceae UCG005 (Fig. 3c). Subsequently, MR-PRESSO was conducted, and the results (Table 1 and Supplementary Table S5) suggested that there was inadequate evidence of outliers and horizontal pleiotropy (P > 0.05). Finally, we conducted a leave-one-out test to evaluate the robustness of the results (Fig. S4). The causal impact of Eubacterium (brachy group) (Fig. S4B) in respiratory TB was not driven by single SNPs, confirming its robustness. A single SNP had minimal impact on the overall estimates of the remaining bacterial traits, with no discernible shift in the significance levels (Figs. S4A, C).

Scatter plots from gut microbiota on respiratory tuberculosis susceptibility. MR Mendelian randomization
Discussion
This study utilized the MR method to examine the causal impact of gut microbiota on respiratory TB susceptibility. This finding revealed significant associations between three bacterial traits and susceptibility to respiratory TB, and all associations were significant after Bonferroni correction. Among the three bacterial traits, Ruminococcaceae UCG005 exhibited protective effects against respiratory TB, whereas Lachnospiraceae UCG010 and Eubacterium (brachy group) were linked with an increased risk of respiratory TB.
In children with pulmonary TB, Ruminococcaceae has been found to be reduced compared with levels in healthy children, which is consistent with our finding [41]. For Ruminococcaceae UCG005.
Chen et al. demonstrated that it could reduce insulin resistance and the incidence of type 2 diabetes [42, 43]. It is interesting to note that diabetes is one of the key risk factors for developing TB [44]. In addition, it has been shown that Ruminococcus UCG005 is positively associated with HDL cholesterol but negatively associated with triglyceride levels [45]. In the development of TB, host lipids are crucial. For instance, Mtb can use triglycerides in macrophages to help it survive inside the cell [46], and decreased infection-related mortality risks were associated with greater baseline cholesterol levels in patients with TB [47].
Lachnospiraceae UCG010 belongs to the Lachnospiraceae family. The role of Lachnospiraceae in TB remains controversial. In a rhesus macaque model, Lachnospiraceae were abundant in monkeys that were more prone to infection Mtb [48]. This result supports the adverse effect of Lachnospiraceae on TB; however, observational studies found that Lachnospiraceae were enriched in healthy individuals [17, 49]. In contrast to Ruminococcaceae UCG005, Lachnospiraceae impair glucose metabolism and promote the onset of diabetes [50, 51].
Maji et al. found higher levels of Eubacterium in patients with TB than in healthy individuals. This might be because Eubacterium could produce a lot of propionate and butyrate, which affects the immune systems in humans [52]. And Segain et al. demonstrated that butyrate could reduce the expression of proinflammatory cytokine mRNA and the generation of tumor necrosis factor (TNF) [53]. TNF, especially TNFα, is crucial for the control of TB infection, including granuloma formation [54, 55], and thus their reduction could raise the risk of TB.
This is the first investigation to establish the causality between gut microbiota and TB in humans, excluding confounding factors. Additionally, several sensitivity studies were conducted to validate the MR results [28]. However, this study has several limitations. First, the sample sizes for gut microbiota and respiratory TB, despite being the largest in GWAS to date, were relatively small. A limited sample size may lack the sufficient statistical power to detect variants with low frequency or effect sizes, resulting in increased false negative results [56]. Besides, gut microbiota was investigated at the genus level, causing the results to be restricted. Second, the study participants were predominantly of European heritage; therefore, it might be difficult to generalize those findings to other ethnic groups. Third, our study did not further explore the specific mechanism of gut microbiota affecting TB susceptibility and this has to be done in future research. Lastly, factors such as diet and medication can influence gut microbiome abundance. Consequently, the proportion of variance attributable to genetics might diminish.
Conclusion
This comprehensive exploration of the potential causality between gut microbiota and respiratory TB suggests that it might represent a diagnostic marker and a possible therapeutic target for respiratory TB. Future studies should validate these findings in humans again and investigate the underlying mechanisms in greater detail.
Data Availability
The MiBioGen consortium (www.mibiogen.org) and FinnGen consortium (https://www.r8.finngen.fi/) provided the data used in this work. For detailed MR methodology, readers can refer to “Reading Mendelian Randomisation Studies: A Guide, Glossary, and Checklist for Clinicians” [22] by Davies et al. to understand.
References
World Health Organization. Geneva: Global Tuberculosis Report; 2021. https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2022. Accessed 2023 Jun 23.
Lyon SM, Rossman MD. Pulmonary tuberculosis. Microbiol Spectrum. 2017. https://doi.org/10.1128/microbiolspec.tnmi7-0032-2016.
Budden KF, Gellatly SL, Wood DL, et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol. 2017;15(1):55–63. https://doi.org/10.1038/nrmicro.2016.142.
Barcik W, Boutin RCT, Sokolowska M, Finlay BB. The role of lung and gut microbiota in the pathology of asthma. Immunity. 2020;52(2):241–55. https://doi.org/10.1016/j.immuni.2020.01.007.
Dayama G, Priya S, Niccum DE, Khoruts A, Blekhman R. Interactions between the gut microbiome and host gene regulation in cystic fibrosis. Genome Med. 2020;12(1):12. https://doi.org/10.1186/s13073-020-0710-2.
Zhang F, Lau RI, Liu Q, Su Q, Chan FKL, Ng SC. Gut microbiota in COVID-19: key microbial changes, potential mechanisms and clinical applications. Nat Rev Gastroenterol Hepatol. 2023;20(5):323–37. https://doi.org/10.1038/s41575-022-00698-4.
Yang F, Yang Y, Chen L, et al. The gut microbiota mediates protective immunity against tuberculosis via modulation of lncRNA. Gut Microbes. 2022;14(1):2029997. https://doi.org/10.1080/19490976.2022.2029997.
Khan N, Vidyarthi A, Nadeem S, Negi S, Nair G, Agrewala JN. Alteration in the gut microbiota provokes susceptibility to tuberculosis. Front Immunol. 2016;7:529. https://doi.org/10.3389/fimmu.2016.00529.
Wang H, Yao J, Chen Y, et al. Gut dysbacteriosis attenuates resistance to Mycobacterium bovis infection by decreasing cyclooxygenase 2 to inhibit endoplasmic reticulum stress. Emerg Microbes Infect. 2022;11(1):1806–18. https://doi.org/10.1080/22221751.2022.2096486.
Dumas A, Corral D, Colom A, et al. The host microbiota contributes to early protection against lung colonization by Mycobacterium tuberculosis. Front Immunol. 2018;9:2656. https://doi.org/10.3389/fimmu.2018.02656.
Eribo OA, Naidoo CC, Theron G, Walzl G, du Plessis N, Chegou NN. An archetypical model for engrafting Bacteroides fragilis into conventional mice following reproducible antibiotic conditioning of the gut microbiota. Microorganisms. 2023. https://doi.org/10.3390/microorganisms11020451.
Chen L, Zhang G, Li G, et al. Ifnar gene variants influence gut microbial production of palmitoleic acid and host immune responses to tuberculosis. Nat Metab. 2022;4(3):359–73. https://doi.org/10.1038/s42255-022-00547-3.
Ding X, Zhou J, Chai Y, et al. A metagenomic study of the gut microbiome in PTB’S disease. Microbes Infect. 2022;24(2): 104893. https://doi.org/10.1016/j.micinf.2021.104893.
Hu Y, Feng Y, Wu J, et al. The gut microbiome signatures discriminate healthy from pulmonary tuberculosis patients. Front Cell Infect Microbiol. 2019;9:90. https://doi.org/10.3389/fcimb.2019.00090.
Luo M, Liu Y, Wu P, et al. Alternation of gut microbiota in patients with pulmonary tuberculosis. Front Physiol. 2017;8:822. https://doi.org/10.3389/fphys.2017.00822.
Hu Y, Yang Q, Liu B, et al. Gut microbiota associated with pulmonary tuberculosis and dysbiosis caused by anti-tuberculosis drugs. J Infect. 2019;78(4):317–22. https://doi.org/10.1016/j.jinf.2018.08.006.
Shi W, Hu Y, Ning Z, et al. Alterations of gut microbiota in patients with active pulmonary tuberculosis in China: a pilot study. Int J Infect Dis. 2021;111:313–21. https://doi.org/10.1016/j.ijid.2021.08.064.
Khaliq A, Ravindran R, Afzal S, et al. Gut microbiome dysbiosis and correlation with blood biomarkers in active-tuberculosis in endemic setting. PLoS ONE. 2021;16(1):e0245534. https://doi.org/10.1371/journal.pone.0245534.
Naidoo CC, Nyawo GR, Sulaiman I, et al. Anaerobe-enriched gut microbiota predicts pro-inflammatory responses in pulmonary tuberculosis. EBioMedicine. 2021;67:103374. https://doi.org/10.1016/j.ebiom.2021.103374.
Wipperman MF, Fitzgerald DW, Juste MAJ, et al. Antibiotic treatment for tuberculosis induces a profound dysbiosis of the microbiome that persists long after therapy is completed. Sci Rep. 2017;7(1):10767. https://doi.org/10.1038/s41598-017-10346-6.
Zuccolo L, Holmes MV. Commentary: Mendelian randomization-inspired causal inference in the absence of genetic data. Int J Epidemiol. 2017;46(3):962–5. https://doi.org/10.1093/ije/dyw327.
Davies NM, Holmes MV, Davey SG. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ. 2018;362: k601. https://doi.org/10.1136/bmj.k601.
Song J, Wu Y, Yin X, Ma H, Zhang J. The causal links between gut microbiota and COVID-19: a Mendelian randomization study. J Med Virol. 2023;95(5):e28784. https://doi.org/10.1002/jmv.28784.
Long Y, Tang L, Zhou Y, Zhao S, Zhu H. Causal relationship between gut microbiota and cancers: a two-sample Mendelian randomisation study. BMC Med. 2023;21(1):66. https://doi.org/10.1186/s12916-023-02761-6.
Amin N, Liu J, Bonnechere B, et al. Interplay of metabolome and gut microbiome in individuals with major depressive disorder vs control individuals. JAMA Psychiat. 2023;80(6):597–609. https://doi.org/10.1001/jamapsychiatry.2023.0685.
Kurilshikov A, Medina-Gomez C, Bacigalupe R, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet. 2021;53(2):156–65. https://doi.org/10.1038/s41588-020-00763-1.
Pierce BL, Burgess S. Efficient design for Mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am J Epidemiol. 2013;178(7):1177–84. https://doi.org/10.1093/aje/kwt084.
Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two-sample Mendelian randomization. Genet Epidemiol. 2016;40(7):597–608. https://doi.org/10.1002/gepi.21998.
Bulik-Sullivan BK, Loh PR, Finucane HK, et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47(3):291–5. https://doi.org/10.1038/ng.3211.
Boef AG, Dekkers OM, le Cessie S. Mendelian randomization studies: a review of the approaches used and the quality of reporting. Int J Epidemiol. 2015;44(2):496–511. https://doi.org/10.1093/ije/dyv071.
Restrepo BI. Diabetes and tuberculosis. Microbiol Spectr. 2016. https://doi.org/10.1128/microbiolspec.TNMI7-0023-2016.
Simou E, Britton J, Leonardi-Bee J. Alcohol consumption and risk of tuberculosis: a systematic review and meta-analysis. Int J Tuberc Lung Dis. 2018;22(11):1277–85. https://doi.org/10.5588/ijtld.18.0092.
Amere GA, Nayak P, Salindri AD, Narayan KMV, Magee MJ. Contribution of smoking to tuberculosis incidence and mortality in high-tuberculosis-burden countries. Am J Epidemiol. 2018;187(9):1846–55. https://doi.org/10.1093/aje/kwy081.
Lönnroth K, Williams BG, Cegielski P, Dye C. A consistent log-linear relationship between tuberculosis incidence and body mass index. Int J Epidemiol. 2010;39(1):149–55. https://doi.org/10.1093/ije/dyp308.
Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388(10053):1545–602. https://doi.org/10.1016/s0140-6736(16)31678-6.
Mukamal KJ, Stampfer MJ, Rimm EB. Genetic instrumental variable analysis: time to call mendelian randomization what it is. The example of alcohol and cardiovascular disease. Eur J Epidemiol. 2020;35(2):93–7. https://doi.org/10.1007/s10654-019-00578-3.
Dan YL, Wang P, Cheng Z, et al. Circulating adiponectin levels and systemic lupus erythematosus: a two-sample Mendelian randomization study. Rheumatology (Oxf). 2021;60(2):940–6. https://doi.org/10.1093/rheumatology/keaa506.
Boehm FJ, Zhou X. Statistical methods for Mendelian randomization in genome-wide association studies: a review. Comput Struct Biotechnol J. 2022;20:2338–51. https://doi.org/10.1016/j.csbj.2022.05.015.
Walker VM, Davies NM, Hemani G, et al. Using the MR-Base platform to investigate risk factors and drug targets for thousands of phenotypes. Wellcome Open Res. 2019;4:113. https://doi.org/10.12688/wellcomeopenres.15334.2.
Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent Estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–14. https://doi.org/10.1002/gepi.21965.
Li W, Zhu Y, Liao Q, Wang Z, Wan C. Characterization of gut microbiota in children with pulmonary tuberculosis. BMC Pediatr. 2019;19(1):445. https://doi.org/10.1186/s12887-019-1782-2.
Chen Z, Radjabzadeh D, Chen L, et al. Association of insulin resistance and type 2 diabetes with gut microbial diversity: a microbiome-wide analysis from population studies. JAMA Netw Open. 2021;4(7):e2118811. https://doi.org/10.1001/jamanetworkopen.2021.18811.
Esquivel-Hernández DA, Martínez-López YE, Sánchez-Castañeda JP, et al. A network perspective on the ecology of gut microbiota and progression of type 2 diabetes: linkages to keystone taxa in a Mexican cohort. Front Endocrinol (Lausanne). 2023;14:1128767. https://doi.org/10.3389/fendo.2023.1128767.
Lönnroth K, Roglic G, Harries AD. Improving tuberculosis prevention and care through addressing the global diabetes epidemic: from evidence to policy and practice. Lancet Diabetes Endocrinol. 2014;2(9):730–9. https://doi.org/10.1016/s2213-8587(14)70109-3.
Sommer AJ, Peters A, Rommel M, et al. A randomization-based causal inference framework for uncovering environmental exposure effects on human gut microbiota. PLoS Comput Biol. 2022;18(5): e1010044. https://doi.org/10.1371/journal.pcbi.1010044.
Brandenburg J, Marwitz S, Tazoll SC, et al. WNT6/ACC2-induced storage of triacylglycerols in macrophages is exploited by Mycobacterium tuberculosis. J Clin Invest. 2021. https://doi.org/10.1172/jci141833.
Chidambaram V, Zhou L, Ruelas Castillo J, et al. Higher serum cholesterol levels are associated with reduced systemic inflammation and mortality during tuberculosis treatment independent of body mass index. Front Cardiovasc Med. 2021;8:696517. https://doi.org/10.3389/fcvm.2021.696517.
Namasivayam S, Kauffman KD, McCulloch JA, et al. Correlation between disease severity and the intestinal microbiome in Mycobacterium tuberculosis-infected Rhesus macaques. mBio. 2019. https://doi.org/10.1128/mBio.01018-19.
Huang Y, Tang JH, Cai Z, et al. Alterations in the nasopharyngeal microbiota associated with active and latent tuberculosis. Tuberculosis (Edinb). 2022;136:102231. https://doi.org/10.1016/j.tube.2022.102231.
Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55–60. https://doi.org/10.1038/nature11450.
Kameyama K, Itoh K. Intestinal colonization by a Lachnospiraceae bacterium contributes to the development of diabetes in obese mice. Microbes Environ. 2014;29(4):427–30. https://doi.org/10.1264/jsme2.ME14054.
Maji A, Misra R, Dhakan DB, et al. Gut microbiome contributes to impairment of immunity in pulmonary tuberculosis patients by alteration of butyrate and propionate producers. Environ Microbiol. 2018;20(1):402–19. https://doi.org/10.1111/1462-2920.14015.
Segain JP, Raingeard de la Blétière D, Bourreille A, et al. Butyrate inhibits inflammatory responses through NFkappaB inhibition: implications for Crohn's disease. Gut. 2000;47(3):397–403. https://doi.org/10.1136/gut.47.3.397.
Casanova JL, Abel L. Genetic dissection of immunity to mycobacteria: the human model. Annu Rev Immunol. 2002;20:581–620. https://doi.org/10.1146/annurev.immunol.20.081501.125851.
McCaffrey EF, Donato M, Keren L, et al. The immunoregulatory landscape of human tuberculosis granulomas. Nat Immunol. 2022;23(2):318–29. https://doi.org/10.1038/s41590-021-01121-x.
Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90(1):7–24. https://doi.org/10.1016/j.ajhg.2011.11.029.
Funding
No funding or sponsorship was received for this study or publication of this article. The Rapid Service Fee was funded by the authors.
Author information
Authors and Affiliations
Contributions
The study was created and designed by Jian-Qing He and Jiayu Wen. The statistical analysis, figures, and supplementary material were encoded by Jiayu Wen. Jian-Qing He evaluated the data and drafted the initial version of the paper.
Corresponding author
Ethics declarations
Conflict of Interest
Jiayu Wen and Jian-Qing He declare that they have no competing interests.
Ethical Approval
The data used as part of this study was from publicly available sources and thus no ethical approval was needed. All data was de-identified before use. Ethics committee approval was not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Wen, J., He, JQ. The Causal Impact of the Gut Microbiota on Respiratory Tuberculosis Susceptibility. Infect Dis Ther 12, 2535–2544 (2023). https://doi.org/10.1007/s40121-023-00880-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-023-00880-4