
Abstract
Background
The causal association between gut microbiome and HIV infection remains to be elucidated. We conducted a two-sample mendelian randomization analysis to estimate the causality between gut microbiome and HIV infection.
Methods
Publicly released genome-wide association studies summary data were collected to perform the mendelian analysis. The GWAS summary data of gut microbiome was retrieved from the MiBioGen consortium, which contains 18 340 samples from 24 cohorts. GWAS summary data of HIV infection was collected from the R5 release of FinnGen consortium, including 357 HIV infected cases and 218 435 controls. The SNPs were selected as instrumental variables according to our selection rules. And SNPs with a F-statistics less than ten were regarded as weak instrumental variables and excluded. Mendelian randomization analysis was conducted by five methods, including inverse variance weighted (IVW), MR-Egger, weighted median, weighted mode, and simple mode. The Cochran’s Q test and MR-Egger intercept test were performed to identify heterogeneity and pleiotropy. Leave-one-out analysis were used to test the sensitivity of the results.
Results
Fifteen gut microbiota taxa showed causal effects on HIV infection according to the MR methods. Four taxa were observed to increase the risk of HIV infection, including Ruminococcaceae (OR: 2.468[1.043, 5.842], P: 0.039), Ruminococcaceae UCG005 (OR: 2.051[1.048, 4.011], P: 0.036), Subdoligranulum (OR: 3.957[1.762, 8.887], P < 0.001) and Victivallis (OR: 1.605[1.012, 2.547], P=0.044). Erysipelotrichaceae was protective factor of HIV infection (OR: 0.278[0.106, 0.731], P < 0.001) and Methanobrevibacter was also found to be associated with reduced risk of HIV infection (OR: 0.509[0.265, 0.980], P=0.043). Horizontal pleiotropy was found for Fusicatenibacter (P<0.05) according to the MR-Egger regression intercept analysis. No heterogeneity was detected.
Conclusion
Our results demonstrate significant causal effects of gut microbiome on HIV infection. These findings facilitate future studies to develop better strategies for HIV prophylaxis through gut microbiome regulation. Further explorations are also warranted to dissect the mechanism of how gut microbiome affects HIV susceptibility.
Similar content being viewed by others


Background
Human immunodeficiency virus (HIV) was firstly isolated from lymph node in 1983, which caused acquired immune deficiency syndrome [1]. At present, HIV infection has been globally pandemic, with near 39 million people living with HIV [2]. In China, the prevalence of new HIV infection was increasing [3], especially among men who have sex with men (MSM) [4]. Mechanically, HIV interacts with receptor CD4 and coreceptor CCR5 to entry T cells, allowing the virus to replicate [5]. Exposure to the semen or mucosal surface were the most common route of HIV transmission. However, as reported, the probability of HIV transmission was only 0.01%-0.4% per sexual contact [6], suggesting that HIV infection also depends on the interaction between the virus and host. Host immune activation might facilitate HIV transmission and infection. Research showed that higher levels of inflammatory factors in male penile, including IL-6 and IL-10, contributed to HIV virus shedding [7]. Besides, a nested case-control study indicated that the rate of HIV seroconversion was significantly higher in women with higher genital inflammatory cytokines [8]. In MSM, HIV mainly transmitted through anal intercourse, thus the immune barrier of rectal mucosa plays important role in the early process of HIV infection.
Gut microbiome takes part in constituting gut immune barrier and could regulate functions of small intestinal innate lymphoid cells [9]. Recent studies found that HIV infection was associated with gut microbiome dysbiosis [10,11,12], with decreased α-diversity, enrichment of genus Prevotella and depletion of Bacteroides in HIV patients. Furthermore, in a prospective study gut microbiome dysbiosis was demonstrated to contribute to increased HIV susceptibility in MSM [13]. The relative abundance of family Succinivibrionaceae, Erysipelotrichaceae and Coriobacteriaceae were significantly higher while Bacteroidaceae and Rikenellaceae were significantly depleted in HIV sero-converters. The gut microbiome dysbiosis in HIV patients were associated with immunologic response to antiretroviral therapy [14], activation of T cells [15] and increased inflammatory factors [16]. However, the association between HIV infection and gut microbiome remains controversial. After stratifying sexual preference, HIV status was reported to have subtle effects on gut microbiome [17, 18]. Meanwhile it’s also unclear whether gut microbiome dysbiosis was induced by HIV infection or prompting to the virus infection [19]. The causal relationship between gut microbiome and HIV infection was remained to be elucidated.
Mendelian randomization (MR) is an analytic approach that utilizes genetic variants as instrumental variations to assess the causality between exposure and outcome [20]. With the help of genome-wide association studies (GWAS), MR analysis is widely used to explain the causal effects of an interested exposure on outcomes, which overcomes the potential influence of confounding factors in observational studies [21]. To our knowledge, no studies have documented the causal effects of gut microbiome on HIV infection before. Hence, this study aimed to explore the causal associations between gut microbiome and HIV infection through a two-sample mendelian randomization analysis.
Methods
GWAS summary data source
The GWAS summary data of gut microbiome was retrieved from the MiBioGen consortium. MiBioGen is a large-scale genome-wide meta-analysis to study the association between gut microbiome and human genetic variants, which contains 18 340 samples from 24 cohorts from the USA, Canada, Israel, South Korea, Germany, Denmark, the Netherlands, Belgium, Sweden, Finland and the UK [22]. The microbiome data were generated by 16S rRNA sequencing using an Illumina sequencing platform. Multiple hyper-variable regions of the 16S rRNA gene was sequenced, including V1-V2, V3-V4 and V4 region. After adjustment for age, sex, technical covariates and genetic principal components, 211 gut microbiome taxa had GWAS summary statistics, including 9 phyla, 16 classes, 20 orders, 35 families and 131 genera. Fifteen unknown bacteria were excluded, and 196 gut microbiotas were enrolled for the final analysis. The GWAS summary data for HIV infection were collected from the 5th release of the FinnGen consortium in May 2021, composed of 357 HIV infected cases and 218 435 controls [23]. We selected the gut microbiome as the exposure and the HIV infection as the outcome.
Instrumental variables selection
To indicate causal association between gut microbiome and HIV infection, we selected SNPs that were associated with gut microbiome at the level of P < 1× 10 -5 as instrument variables (IVs). This value was identified as the optimal threshold in many gut microbiota-related MR research to increase the amount of genetic variance explained by the genetic predictors [24].The linkage disequilibrium (LD) across SNPs were calculated in a 10,000-kb window and the LD threshold for R2 was 0.001. In the condition of no SNPs shared between gut microbiome and HIV infection, proxy SNPs with r2 > 0.8 were selected to replace the original SNPs. Then F-statistics was calculated for each SNP with the following formula: F = β2/Se2, whereas β and Se are the coefficient and stand error of exposure respectively. SNPs with F-statistics < 10 were regarded as weak IVs and were discarded in the following analysis. MR pleiotropy residual sum and outlier (MR‐PRESSO) tests was also conducted to remove outlier SNPs.
Mendelian randomization analysis
Five methods were used to perform the MR analysis, including inverse variance weighted (IVW), weighted mode, MR-egger, weighted median, and simple mode. IVW uses a meta-analysis approach combined with the Wald estimates for each SNP to obtain an overall estimate, which assumes that all SNPs are valid instruments [25]. The weighted mode produces a robust causal effects estimate when the horizontal pleiotropy existed [26]. ME-egger could provide a valid effect on the condition that all SNPs are invalid [27]. The estimates based on weighted median are consistent even when more than 50% of the information comes from invalid instrumental variables [28]. Simple mode is an unweighted causal estimation [29]. The primary method was IVW, and only when the result of IVW analysis was significant, the causal effects of exposure on outcome presented. Other four MR methods were used as supplement. Additionally, false discovery rate (FDR) was calculated based on the Benjamini-Hochberg (BH) method to adjust the multiple tests. FDR < 0.1 were considered significant [30].
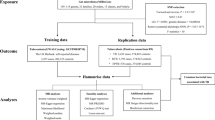
Sensitivity analysis was conducted to evaluate the stability of the results. Cochran’s Q statistic was used to test the heterogeneity. MR egger intercept was used to assess the horizontal pleiotropy. P value < 0.05 indicated no significant heterogeneity or horizontal pleiotropy. Besides, leave-one out analysis was conducted to identify whether the overall estimates were influenced by one single SNP. To further exclude the impact of confounding factors, such as behavior and environment, we searched the Phenoscanner database (http://www.phenoscanner.medschl.cam.ac.uk/) to explore the reported traits that were associated with our selected IVs. The study flowchart was stated in Fig. 1.

The flowchart of this mendelian randomization study
All the analysis were performed by R software (version: R 4.3.0). R packages “TwosampleMR” was used to conduct the two-sample mendelian randomization analysis. And R package “MRPRESSO” was used to identify the outlier SNPs.
Results
Causal effects of gut microbiome on HIV infection
At the level of p < 1 x 10-5, 2 774 SNPs were associated with gut microbiota at phylum, class, order, family, and genus levels. Fifty-three SNPs were excluded due to the F-statistic < 10. Thus, a total of 2 721 SNPs were enrolled for MR analysis. Fifteen bacteria were found to have causal effects on HIV, including 1 class, 2 orders, 3 families and 9 genera (Table 1). One genus of phylum Actinobacteria was negatively related to HIV infection. One class, 2 orders, 3 families and 6 genera of phylum Firmicutes were significantly associated with HIV infection. One genus of phylum Lentisphaerae and 1 genus of phylum Methanomada were also showed causal effects on HIV infection.
At the class level, 13 SNPs were selected for genetically predicting Erysipelotrichia. Based on IVW analysis and weighted median analysis, Erysipelotrichia was protective factor of HIV infection (IVW: OR: 0.278[0.106, 0.731], P < 0.01; weighted median: OR: 0.161[0.045, 0.575], P<0.01). Order Bacillales and Erysipelotrichales, both belonged to Firmicutes, were significantly associated with HIV. The results of IVW analysis indicated that both Bacillales and Erysipelotrichales were protective factors of HIV (P<0.05). Three families of Firmicutes, Defluviitaleaceae, Erysipelotrichaceae and Ruminococcaceae had causal effects on HIV infection. Defluviitaleaceae and Erysipelotrichaceae were significantly associated with decreased risk of HIV infection, while Ruminococcaceae was identified as risk factor of HIV infection in IVW and weighted median analysis (IVW: OR:2.468[1.043,5.842], P=0.04; Weighted median: OR:3.365[1.045, 10.836], P=0.04.). At the genus level, Eggerthella, Anaerofilum, Anaerotruncus, Clostridium, Coprococcus2, Fusicatenibacter and Methanobrevibacter were protective factors. Hungatella, Ruminococcaceae UCG005, Subdoligranulum and Victivallis were risk factors of HIV infection. After adjusting for the multiple testing, genus Eggerthella and genus Subdoligranulum were found to significantly decrease the risk of HIV acquirement (FDR < 0.1). All these results indicated that gut microbiome had significant causal effects on HIV infection.
Among all these fifteen gut microbiotas, the results of Cochran’s Q test indicated no significant heterogeneity existed. However, horizontal pleiotropy was found for genus Fusicatenibacter (P<0.05) according to the MR-Egger regression intercept analysis. Leave-one out sensitivity analysis suggested that the causal association between gut microbiome and HIV infection were not driven by one single SNP (Fig. 2, Figure S1). Besides, we searched for the Phenoscanner database to explore whether potential confounding factors existed. The results (Table S1) showed that our selected IVs were associated with a few physical indicators, such as impedance of arms, body mass index, arm fat mass, weight, height and so on. These factors were known as influencing factors of gut microbiome composition, but were not reported to affect HIV infection, which demonstrated that the causal association between gut microbiome and HIV was robust.

Leave-one-out analysis of the significant causal effects of gut microbiome on HIV infection. The X-axis indicates the estimated β value. In each panel, the red line stands for the overall estimates, and each blank line indicates the overall estimate after excluding the left SNP. A: class Erysipelotrichia, B: family Erysipelotrichaceae, C: family Ruminococcaceae, D: family Defluviitaleaceae, E: order Erysipelotrichales, F: order Bacillales, G: genus Ruminococcaceae UCG005, H: genus Methanobrevibacter, I: genus Eggerthella
Discussion
The present study was the first MR analysis to evaluate the causal relationships between gut microbiome and HIV infection. We demonstrated causal effects of 15 gut microbiotas on HIV infection. In our study, we found that family Ruminococcaceae, and genus Hungatella, Ruminococcaceae UCG005, Subdoligranulum and Victivallis were significantly associated with HIV infection, which might facilitate HIV infection. Genus Clostridium, Coprococcus2 and Fusicatenibacter were identified as protective factors against HIV infection. Typically, most of our reported gut microbiotas were belonged to Firmicutes phylum. Importantly our results stated causality between HIV infection and family Erysipelotrichaceae and genus Methanobrevibacter, which was consistent with one prior study [13], further demonstrating that gut microbiome dysbiosis had major impact on HIV incidence. After adjusting for the multiple testing, genus Eggerthella and genus Subdoligranulum were found to significantly decrease the risk of HIV acquirement, indicating robust causal effects of gut microbiome on HIV infection.
In the field of HIV infection, gut microbiome has aroused researchers’ interests. Researchers found that the composition of gut microbiome significantly changed in HIV-infected patients [18]. To explain the mechanism how gut microbiome affect HIV infection, researches have focused on exploring the association between gut microbiome and immune. In MSM, the high-risk population of HIV infection, gut microbiome can influence the CCR5 expression and influx of HIV targeted cells to colon through modulation of integrin and chemokines expression on T cells [31]. Using gnotobiotic mice model, researchers found that gut microbiome from high-risk MSM activated T cells, with higher frequency of CD69+ and CD103+ T cells [14]. In vitro assays, gut microbiome from HIV-infected patients could induce significantly higher levels of activated monocytes and T cells, which were mediated by TNF-α and TLR (toll like receptor) [16]. Further study revealed that Holdemanella biformis was the main signature that was responsible for elevated frequency of CCR5+ CD4+ T cells in the vitro stimulation assay of Lamina Propria Mononuclear Cells with fecal bacterial communities [32]. All these evidences demonstrated gut microbiome had the potential of increasing HIV transmission through activating immune reaction.
In the present study, family Erysipelotrichaceae decreased risk of HIV infection (OR: 0.28, P < 0.05), which means that higher the relative abundance of Erysipelotrichaceae protects patients against HIV invasion. As reported members of this bacterial were highly immunogenic, for example Erysipelotrichi was reported to correlate with TNF in chronic HIV infection patients [33]. Besides, Erysipelotrichaceae has been found to associate with inflammatory bowel diseases (IBD) and colon cancers [34]. Actually, Erysipelotrichaceae is a kind of butyrate-producing bacteria [35]. Butyrate is the primary energy source of colon epithelial cells and plays important role on ameliorating mucosal inflammation and oxidative status, reinforcing the epithelial defense barrier to maintain intestinal health [36]. Moreover, butyrate is a well-known histone deacetylase inhibitor and could regulate gene expression through modulating chromatin structure, indicating its potential therapeutic implication for clinical use [37]. Studies documented that oral butyrate supplementation in patients with IBD activated epithelial peroxisome proliferator-activated receptor-γ (PPAR-γ) signaling and promoted generation of Treg cells in colonic lamina propria [38]. Animal experiment also demonstrated that oral butyrate downregulated inflammation in mice implanted with polyether-polyurethane sponge, presented with decreased neutrophil infiltration and reduced levels of TNF-α, IL-10 and TGF-β1 [39]. All these evidences shed light on the hypothesis that Erysipelotrichaceae decreased the risk of HIV infection through enhancing the production of butyrate which inhibited excessive activation of immune system. Meanwhile, Methanobrevibacter was also showed protective effects on HIV infection in our analysis. Methanobrevibacter belongs to methanogens and plays important physiological role in human health by producing methane from hydrogen [40]. Study indicated that Methanobrevibacter smithii colonized in the human gastric mucosa just early after birth [41]. Methanobrevibacter was documented to be associated with irritable bowel diseases and possibly reversed the susceptibility of irritable bowel diseases [42, 43]. However, further research is needed on the specific relationship between Methanobrevibacter and host health.
Moreover, we also found Subdoligranulum and Victivallis were positively related to HIV infection. Consistent with previous studies, Subdoligranulum and Victivallis were enriched in HIV infected patients, which further supporting our results [11, 44]. Interestingly, contrary to Erysipelotrichaceae, butyrate-producing bacteria Ruminococcaceae was demonstrated to increase the risk of HIV infection in our study. This conclusion seems to be contradictory, but in fact it implies the complex interaction between gut microbiome and suggests that the effects of gut microbiome on HIV infection are not simply caused by single microbiota. This also indicates that the structure of gut microbiome is in dynamic balance, and only when the balance is broken will it affects health of the host.
What is worth to note is that genus Prevotella was not statistically correlated with HIV infection in this MR analysis. However, Prevotella was the most frequently reported HIV associated genus and might promote to virus infection. For example, researchers found that the abundance of Prevotella was higher in women with high-risk HPV infection and associated with NF-KB signaling, suggesting its role in promoting virus infection by altering immune regulators [45]. Besides, the abundance of Prevotella was associated with immune response, which could drive Th17-mediated mucosal inflammation and stimulate epithelial cells to produce inflammatory factors, such as IL-8 and IL-6 [46]. The impact of genus Prevotella on HIV infection remains to be further elucidated.
To be honest, there are limitations in this study. Firstly, the results of reverse MR analysis exploring causal effects of HIV infection on gut microbiome was not stated here. In fact, we indeed conducted the reverse MR analysis and attempted to explore the causal effects of HIV infection on gut microbiome. However, there are no SNPs available as instrumental variables in the currently used summary GWAS data of HIV infection. On the other hand, we also tried using the latest release HIV GWAS summary data of FinnGen consortium in May 2023, but still lack of available instrumental variables. Secondly, we recommend to treat the significant results carefully. We adjusted the P values based on BH method to avoid false discovery and only two taxa passed the BH correction (FDR < 0.1). More studies are expected to elucidate the role of gut microbiome in HIV infection.
Conclusions
In conclusion, this study analyzed the causal association between gut microbiome and HIV infection by a two-sample MR analysis. Our results demonstrate that several gut taxa increase the risk of HIV acquirement, such as Ruminococcaceae, Subdoligranulum and Victivallis. While Erysipelotrichaceae and Methanobrevibacter significantly reduce the risk of HIV infection. We hold the concept that the risk of HIV infection may not be significantly increased by one single microbiome, it is the results of complex interaction of variable gut microbiome. These findings facilitate future studies to develop better strategies for HIV prophylaxis through gut microbiome regulation. Further explorations are also warranted to dissect the mechanism of how gut microbiome affects HIV susceptibility.
Availability of data and materials
The datasets analyzed during the current study are available in the Finngen database (https://www.finngen.fi/en) and MiBioGen database (https://mibiogen.gcc.rug.nl/), which are publicly available.
Abbreviations
- GWAS:
-
Genome-wide association studies
- IVW:
-
Inverse variance weighted
- HIV:
-
Human immunodeficiency virus
- MSM:
-
Men who have sex with men
- MR:
-
Mendelian randomization
- IVs:
-
Instrument variables
- LD:
-
Linkage disequilibrium
- MR‐PRESSO:
-
MR pleiotropy residual sum and outlier
- FDR:
-
False discovery rate
References
Moir S, Chun TW, Fauci AS. Pathogenic mechanisms of HIV disease. Ann Rev Pathol. 2011;6:223–48.
UNAIDS_Global HIV & AIDS Statistics FactSheet(2023 updated). https://www.unaids.org/en/resources/fact-sheet.
Qiao YC, Xu Y, Jiang DX, Wang X, Wang F, Yang J, Wei YS. Epidemiological analyses of regional and age differences of HIV/AIDS prevalence in China, 2004–2016. Int J Infect Dis. 2019;81:215–20.
Dong MJ, Peng B, Liu ZF, Ye QN, Liu H, Lu XL, Zhang B, Chen JJ. The prevalence of HIV among MSM in China: a large-scale systematic analysis. BMC Infect Dis. 2019;19(1):1000.
Chen B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019;27(10):878–91.
Gray RH, Wawer MJ, Brookmeyer R, Sewankambo NK, Serwadda D, Wabwire-Mangen F, Lutalo T, Li X, vanCott T, Quinn TC. Probability of HIV-1 transmission per coital act in monogamous, heterosexual, HIV-1-discordant couples in Rakai Uganda. Lancet. 2001;357(9263):1149–53.
Patel EU, Kirkpatrick AR, Grabowski MK, Kigozi G, Gray RH, Prodger JL, Redd AD, Nalugoda F, Serwadda D, Wawer MJ, et al. Penile immune activation and risk of HIV shedding: a prospective cohort study. Clin Infect Dis. 2017;64(6):776–84.
Masson L, Passmore JA, Liebenberg LJ, Werner L, Baxter C, Arnold KB, Williamson C, Little F, Mansoor LE, Naranbhai V, et al. Genital inflammation and the risk of HIV acquisition in women. Clin Infect Dis. 2015;61(2):260–9.
Gury-BenAri M, Thaiss CA, Serafini N, Winter DR, Giladi A, Lara-Astiaso D, Levy M, Salame TM, Weiner A, David E, et al. The spectrum and regulatory landscape of intestinal innate lymphoid cells are shaped by the microbiome. Cell. 2016;166(5):1231-1246.e1213.
Sortino O, Phanuphak N, Schuetz A, Ortiz AM, Chomchey N, Belkaid Y, Davis J, Mystakelis HA, Quinones M, Deleage C, et al. Impact of acute HIV infection and early antiretroviral therapy on the human gut microbiome. Open Forum Infect Dis. 2020;7(12):ofz367.
Cook RR, Fulcher JA, Tobin NH, Li F, Lee D, Javanbakht M, Brookmeyer R, Shoptaw S, Bolan R, Aldrovandi GM, et al. Effects of HIV viremia on the gastrointestinal microbiome of young MSM. AIDS. 2019;33(5):793–804.
Tuddenham SA, Koay WLA, Zhao N, White JR, Ghanem KG, Sears CL. Consortium HIVMR-a: the impact of human immunodeficiency virus infection on gut microbiota alpha-diversity: an individual-level meta-analysis. Clin Infect Dis. 2020;70(4):615–27.
Chen Y, Lin H, Cole M, Morris A, Martinson J, McKay H, Mimiaga M, Margolick J, Fitch A, Methe B, et al. Signature changes in gut microbiome are associated with increased susceptibility to HIV-1 infection in MSM. Microbiome. 2021;9(1):237.
Li SX, Sen S, Schneider JM, Xiong KN, Nusbacher NM, Moreno-Huizar N, Shaffer M, Armstrong AJS, Severs E, Kuhn K, et al. Gut microbiota from high-risk men who have sex with men drive immune activation in gnotobiotic mice and in vitro HIV infection. PLoS Pathog. 2019;15(4):e1007611.
Zhao H, Feng A, Luo D, Wu H, Zhang G, Zhang L, Yuan J, Lin YF, Li L, Zou H. Altered gut microbiota is associated with different immunologic responses to antiretroviral therapy in HIV-infected men who have sex with men. J Med Virol. 2023;95(3):e28674.
Neff CP, Krueger O, Xiong K, Arif S, Nusbacher N, Schneider JM, Cunningham AW, Armstrong A, Li S, McCarter MD, et al. Fecal microbiota composition drives immune activation in HIV-infected individuals. EBioMedicine. 2018;30:192–202.
Noguera-Julian M, Rocafort M, Guillen Y, Rivera J, Casadella M, Nowak P, Hildebrand F, Zeller G, Parera M, Bellido R, et al. Gut microbiota linked to sexual preference and HIV infection. EBioMedicine. 2016;5:135–46.
Vujkovic-Cvijin I, Sortino O, Verheij E, Sklar J, Wit FW, Kootstra NA, Sellers B, Brenchley JM, Ananworanich J, Loeff MSV, et al. HIV-associated gut dysbiosis is independent of sexual practice and correlates with noncommunicable diseases. Nat Commun. 2020;11(1):2448.
Lopera TJ, Lujan JA, Zurek E, Zapata W, Hernandez JC, Toro MA, Alzate JF, Taborda NA, Rugeles MT, Aguilar-Jimenez W. A specific structure and high richness characterize intestinal microbiota of HIV-exposed seronegative individuals. PloS one. 2021;16(12):e0260729.
Sekula P, Del Greco MF, Pattaro C, Köttgen A. Mendelian randomization as an approach to assess causality using observational data. J Am Soc Nephrol. 2016;27(11):3253–65.
Zuccolo L, Holmes MV. Commentary: Mendelian randomization-inspired causal inference in the absence of genetic data. Int J Epidemiol. 2017;46(3):962–5.
Kurilshikov A, Medina-Gomez C, Bacigalupe R, Radjabzadeh D, Wang J, Demirkan A, Le Roy CI, Raygoza Garay JA, Finnicum CT, Liu X, et al. Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat Genet. 2021;53(2):156–65.
Kurki MI, Karjalainen J, Palta P, Sipilä TP, Kristiansson K, Donner KM, Reeve MP, Laivuori H, Aavikko M, Kaunisto MA, et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023;613(7944):508–18.
Sanna S, van Zuydam NR, Mahajan A, Kurilshikov A, Vich Vila A, Võsa U, Mujagic Z, Masclee AAM, Jonkers D, Oosting M, et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat Genet. 2019;51(4):600–5.
Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658–65.
Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46(6):1985–98.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–25.
Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet Epidemiol. 2016;40(4):304–14.
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, Tan VY, Yarmolinsky J, Shihab HA, Timpson NJ, Evans DM, Relton C, Martin RM, Smith GD, Gaunt TR, Haycock PC, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7:e34408.
Li P, Wang H, Guo L, Gou X, Chen G, Lin D, Fan D, Guo X, Liu Z. Association between gut microbiota and preeclampsia-eclampsia: a two-sample Mendelian randomization study. BMC Med. 2022;20:443. https://doi.org/10.1186/s12916-022-02657-x.
Coleman SL, Neff CP, Li SX, Armstrong AJS, Schneider JM, Sen S, Fennimore B, Campbell TB, Lozupone CA, Palmer BE. Can gut microbiota of men who have sex with men influence HIV transmission? Gut Microbes. 2020;11(3):610–9.
Yamada E, Martin CG, Moreno-Huizar N, Fouquier J, Neff CP, Coleman SL, Schneider JM, Huber J, Nusbacher NM, McCarter M, et al. Intestinal microbial communities and Holdemanella isolated from HIV+/- men who have sex with men increase frequencies of lamina propria CCR5(+) CD4(+) T cells. Gut Microbes. 2021;13(1):1997292.
Dinh DM, Volpe GE, Duffalo C, Bhalchandra S, Tai AK, Kane AV, Wanke CA, Ward HD. Intestinal microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J Infect Dis. 2015;211(1):19–27.
Kaakoush NO. Insights into the Role of Erysipelotrichaceae in the Human Host. Front Cell Infect Microbiol. 2015;5:84.
Loman BR, Jordan KR, Haynes B, Bailey MT, Pyter LM. Chemotherapy-induced neuroinflammation is associated with disrupted colonic and bacterial homeostasis in female mice. Sci Rep. 2019;9(1):16490.
Canani RB, Costanzo MD, Leone L, Pedata M, Meli R, Calignano A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World J Gastroenterol. 2011;17(12):1519–28.
Berni Canani R, Di Costanzo M, Leone L. The epigenetic effects of butyrate: potential therapeutic implications for clinical practice. Clin Epigenet. 2012;4(1):4.
Sitkin S, Vakhitov T, Pokrotnieks J. Oral butyrate modulates the gut microbiota in patients with inflammatory bowel disease, most likely by reversing proinflammatory metabolic reprogramming of colonocytes. Neurogastroenterol Motil. 2021;33(1):e14038.
de Lazari MGT, Pereira LX, Orellano LAA, Scheuermann K, Machado CT, Vasconcelos AC, Andrade SP, Campos PP. Sodium butyrate downregulates implant-induced inflammation in mice. Inflammation. 2020;43(4):1259–68.
Sogodogo E, Drancourt M, Grine G. Methanogens as emerging pathogens in anaerobic abscesses. Eur J Clin Microbiol Infect Dis. 2019;38(5):811–8.
Grine G, Boualam MA, Drancourt M. Methanobrevibacter smithii, a methanogen consistently colonising the newborn stomach. Eur J Clin Microbiol Infect Dis. 2017;36(12):2449–55.
Villanueva-Millan MJ, Leite G, Wang J, Morales W, Parodi G, Pimentel ML, Barlow GM, Mathur R, Rezaie A, Sanchez M, et al. Methanogens and hydrogen sulfide producing bacteria guide distinct gut microbe profiles and irritable bowel syndrome subtypes. Am J Gastroenterol. 2022;117(12):2055–66.
Ghavami SB, Rostami E, Sephay AA, Shahrokh S, Balaii H, Aghdaei HA, Zali MR. Alterations of the human gut Methanobrevibacter smithii as a biomarker for inflammatory bowel diseases. Microb Pathog. 2018;117:285–9.
Parbie PK, Mizutani T, Ishizaka A, Kawana-Tachikawa A, Runtuwene LR, Seki S, Abana CZ, Kushitor D, Bonney EY, Ofori SB, et al. Dysbiotic fecal microbiome in HIV-1 infected individuals in Ghana. Front Cell Infect Microbiol. 2021;11:646467.
Dong B, Huang Y, Cai H, Chen Y, Li Y, Zou H, Lin W, Xue H, Feng A, Zhao H, et al. Prevotella as the hub of the cervicovaginal microbiota affects the occurrence of persistent human papillomavirus infection and cervical lesions in women of childbearing age via host NF-κB/C-myc. J Med Virol. 2022;94(11):5519–34.
Larsen JM. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology. 2017;151(4):363–74.
Acknowledgements
Not applicable.
Funding
This research was funded by National Youth Science Foundation Project (grant number [82204159]) and Science and Technology Research Program of Chongqing Municipal Education Commission (Grant No. KJQN202300423).
Author information
Authors and Affiliations
Contributions
Conceptualization, Xiaoni Zhong and Biao Xie; Methodology, Kangjie Li; Formal Analysis, Kangjie Li, Jielian Deng; Data Curation, Cong Zhang, Yuan Zhang, Haijiao Zeng, Guichuan Lai; Writing – Original Draft Preparation, Kangjie Li; Writing – Review & Editing, Xiaoni Zhong; Funding Acquisition, Biao Xie.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, K., Zhang, C., Deng, J. et al. Causal effects of gut microbiome on HIV infection: a two-sample mendelian randomization analysis. BMC Infect Dis 24, 280 (2024). https://doi.org/10.1186/s12879-024-09176-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-024-09176-5