
Abstract
Introduction
This phase I clinical trial was conducted to evaluate the safety of RP22 as a skin test reagent for tuberculosis (TB) diagnosis and to explore the appropriate dosage.
Methods
We used a randomized, double-blind, placebo-controlled identification allergen (IA) skin test. A total of 72 healthy adult volunteers with negative chest X-ray results were randomized into six groups and given a QuantiFERON-TB Gold (QFT) test. Of the 12 participants in each group, eight received RP22 and four received placebo. The doses of RP22 in the six experimental groups ranged from 0.1 to 4.0 μg in a single intradermal injection of 0.1 ml. Skin reactions and adverse events were recorded at intervals.
Results
All doses of RP22 except the highest were well tolerated and safe. No serious adverse events associated with the injection were observed in all groups. There were 11 participants who had positive QFT results, eight had a skin reaction with a redness or induration area diameter of greater than 10 mm at 48–72 h, one had no skin reaction. Among the 60 negative-QFT participants, none had a reaction area diameter of greater than 10 mm.
Conclusion
The RP22 skin test was well tolerated and safe, it could play a key role in screening for latent tuberculosis infection (LTBI) by providing a much-wanted alternative to the tuberculin skin test (TST) and interferon-γ release assays (IGRAs).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
RP22 was well tolerated and safe in healthy normal participants and those with LTBI. |
The maximum response was obtained 48–72 h after antigen injection; the suitable response induration diameter could be defined as 10 mm and the suitable dose could be defined as 0.5 μg or 1 μg. |
As a skin test reagent for Mycobacterium tuberculosis infection, RP22 could play an important role in the screening of LTBI. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.14208284.
Introduction
Tuberculosis (TB) is still a leading infectious disease with high morbidity and mortality worldwide, and delayed detection of TB is a serious problem [1]. The existing diagnostic tests are not ideal. The tuberculin skin test (TST) has been widely used to diagnose Mycobacterium tuberculosis (Mtb) infection for more than a century, and it is simple to operate and has a low cost. However, the TST has some limitations including cross-reaction against the Bacille Calmette-Guerin (BCG) vaccine strain and some non-tuberculosis mycobacteria (NTM). In some contexts, it lacks the required specificity and sensitivity, especially in those with human immunodeficiency virus (HIV) infection, severe organ dysfunction, organ transplants, malnutrition, and in young children [2, 3]. ESAT6 and CFP10 are specific antigens of Mtb, both of which are coded by the region of difference 1 (RD1) which only exists in the genome of Mtb and a few other pathogenic mycobacteria; all BCG strains and most environmental mycobacteria do not have this genome region [4]. Interferon-γ release assays (IGRAs) based on these two antigens have been used to diagnose Mtb infection, providing an attractive alternative to the TST. There are two main commercial IGRAs: the QuantiFERON-TB Gold in Tube (QFT-GIT) (Cellestis, Carnegie, Australia) and the T-SPOT.TB (T-SPOT) (Oxford Immunotec, Abingdon, UK) assay [5]. However, the price of IGRAs is high, the requirements for laboratories and supervision are high, and the results are variable, which is considered highly dynamic, high rates of conversions and reversions when the IGRAs were tested repeatedly on the same subjects [6,7,8]. Therefore, a new point-of-care test method with high specificity and lower costs is urgently required.
An identification allergen (IA) skin test procedure, which retains the characteristics of simple operation of the traditional PPD skin test method and exploits the specificity of IGRA technology, has been extensively studied in many countries. Two examples, the C-Tb skin test (Statens Serum Institute, Copenhagen, Denmark) from the Danish National Serum Institute [9] and the Diaskintest (DST) skin test from Russia [10], have both shown high safety and efficacy in humans. In China, an improved skin test reagent, RP22, a recombinant fusion protein CFP10–ESAT6 (HS625) with excipient, had shown safety and high specificity in our unpublished preliminary animal studies. So we conducted this phase I clinical trial to find safe doses of RP22 for the diagnosis of Mtb infection in humans.
Methods
RP22
RP22 reagent is a freeze-dried powder of recombinant fusion protein ESAT6/CFP10 mixed with excipient manufactured by Zhejiang Hisun Pharmaceutical Co. Ltd, China. The ratio of ESAT6 to CFP10 is 1:1. The excipient comprises sodium citrate, citric acid, trehalose dihydrate, polysorbate, and purified water. The protein in RP22 is slightly different from the existing ESAT6/CFP10 fusion protein with an optimized coding sequence to increase the production efficiency. China Food and Drug Administration (CFDA) approved the phase I clinical trials of RP22 in 2016 (batch number 2016L10792, test number HS625-I); the registration number is CTR20170520 (http://www.chinadrugtrials.org.cn/). The drug specification is 10 µg/vial, we add 10 ml, 4 ml, 2 ml, 1 ml, 0.5 ml, 0.25 ml saline to different vials respectively, then prepare 0.1 μg/0.1 ml, 0.25, 0.5, 1, 2, and 4 μg/0.1 ml experiment drug, respectively. Each participant received experiment drug with a total volume of 0.1 ml; the remaining volume was disposed of.
In unpublished preliminary studies, RP22 was tested in animal models. A valence study in a guinea pig sensitivity model showed that 24 h after intradermal injection was an ideal observation time for skin reaction. In mice, a safety evaluation study of RP22 showed that the maximum dose tested by intradermal injection (250 μg/kg; equivalent to 15,000 times that of human clinical dose) was well tolerated. No obvious dose–effect relationship was found within the range of 0.6–1.2 μg. On the basis of these animal studies, the appropriate dose for human use was assumed to be about 1.0 μg, and therefore five other dose groups above or below 1.0 μg were used here to assess the safety of RP22.
QFT-GIT
The QuantiFERON-TB Gold In-Tube (QFT-GIT) assay uses three tubes: the negative control (nil) tube that measures the background interferon-γ (IFNγ) response, the antigen tube that measures the antigen-specific response, and the positive control (mitogen) tube that measures the nonspecific T cell response. The qualitative result (negative, positive, or indeterminate) is interpreted from the quantification of IFNγ in international units (IU) per milliliter. An IFNγ response above 0.35 IU/mL at screening is regarded as showing possible Mtb infection. The QFT-GIT assay was done at the screening visit [11].
Study Design
We designed a randomized, double-blind, placebo-controlled phase I clinical trial. It was performed at the phase I Clinical Trial Department of the Shanghai Public Health Clinical Center affiliated with Fudan University in China from June 24, 2017, to January 18, 2018. Seventy-two eligible trial participants were divided into six groups of 12. Eight participants of each group received RP22, and four participants received the placebo (excipient; ratios of male to female were 1:1). Escalating doses of RP22 were tested sequentially in the six groups, A to F, that received 0.1, 0.25, 0.5, 1, 2, and 4 μg, respectively. Members of a group who received placebo only were given the dose of excipient corresponding to the dose present in RP22 in that group. All of the participants were randomized according to the sequence of screening numbers, and we use random number table to do this job and a clinical coordinator will supervise the whole procedure to guarantee allocation concealment. Each participant received only one dose. Every two participants in a same dose group received the skin test at the same time in different rooms; another two participants in the same dose group received the skin test 1 hour later. If adverse events (AEs) were observed in more than half of the participants in any dose group or a serious adverse event (SAEs) occurred, the trial at that dose would be terminated. Otherwise, the dose escalation continued.
Study Participants
The trial population was mainly recruited by advertising on the Internet. Persons aged from 18 to 45 years were primarily screened. All gave signed informed consent, and received a physical examination, electrocardiogram (ECG), chest radiograph, sputum acid-fast bacilli smear, QFT-GIT, and tests for liver and kidney function, virus detection, nicotine, alcohol allergy, and routine blood, urine, and stool tests, etc.
The inclusion criteria were (1) Each participant signed an informed consent and complied with the requirements of the clinical trial program. (2) Age was between 18 and 45 years old, the age span in each dose group was no more than 10 years; female body weight greater than 45 kg, male body weight greater than 50 kg, and body mass index (BMI) between 18 and 28 kg/m2 (BMI = body weight /height2). (3) No TB history, no family history of tuberculosis, no close contact history of TB. (4) No intrapulmonary or extrapulmonary tuberculosis (EPTB), respiratory symptoms, or systemic discomfort. (5) Chest X-ray and sputum smear confirmed that there was no TB infection. (6) Negative pregnancy blood test.
The exclusion criteria were (1) Allergic to two or more medicines or foods in the past. (2) Had malignancy, organ function failure, HIV, immunosuppressive disease, undergone major surgery within 6 months, or disease that could significantly affect the judgment of a skin test reaction. (3) Had severe scar formation, burns, rashes, eczema, psoriasis, or any other skin disease around the injection site that could affect the judgment of a skin test reaction. (4) Had participated in other clinical trials within 3 months. (5) Had been infected within 4 weeks with bacteria, viruses, fungi, parasites, etc. requiring anti-infective treatment. (6) Failed to meet the health standards in general physical examination such as abnormal vital signs, abnormal laboratory examinations, abnormal clinical significance in electrocardiogram examination. (7) Planned to conceive or donate sperm within 6 months. (8) Had other reasons for non-enrollment.
Skin Test Procedure
RP22 or placebo was injected by the Mantoux technique with a short-beveled sterile needle, sized 0.51 mm (21 gauge), in the anterior 1/3 of either the left or the right volar forearm. Each participant received one dose with a total volume of 0.1 ml. The needles were pierced into the dermal surface, with the bevel of the needle upward on a 5–10° angle. Digital photographs of the injection sites were taken at 15 min, 30 min, 1, 2, 4, 8, 24, 48, and 72 h after the injection. A vernier caliper was used to measure the longitudinal and transverse diameters of the skin induration and redness around the injection site.
Safety Assessment
The participants were monitored closely for local skin reactions and systemic reactions at 15 min, 30 min, 1, 2, 4, 8, 24, 48, and 72 h after the skin test. Local skin reactions included redness, swelling, induration, and blister reactions; all these reactions were graded according to the criteria listed in Table 1, which are based on the “principle of quantitative criterion and grading system for adverse events from vaccine for clinical trials” released by the CFDA in 2005. When a local skin reaction reached a severe grade (grade 3), it was recorded as an adverse event. Systemic reactions included systemic allergic rash, anaphylactic shock, generalized urticaria, lymphangitis, allergic purpura, fever, and other adverse events. The participants’ blood pressure, respiratory rate, heart rate, body temperature, and electrocardiogram at every time point were recorded. One day before and the third and seventh day after the skin test, blood, urine, liver function, and kidney function were examined. All the severe local skin reactions, systemic symptoms and signs, and abnormal laboratory examination results were recorded as adverse events. The causality of adverse events was assessed as certainly related, probably related, possibly related, possibly unrelated, and unrelated to the injection [12]. The grade of adverse events was assessed as mild, moderate, severe, life-threatening, or death according to the Common Terminology Criteria for Adverse Events (CTCAE) v5.0. Emergency plans were prepared to respond to severe adverse events.
Statistical Analysis
Data entry was completed by data editors using EpiData 3.0 software (EpiData Association, Odense, Denmark). Statistical analysis was performed with SAS 9.3 software (SAS Institute, Cary, NC, USA). Continuous variables were summarized by descriptive statistics, including numbers, average, median, standard deviation, maximum and minimum. Classified variables were described by the number and percentage of cases. Data were presented as mean ± standard deviation (SD). Significant differences between the means using Student’s t test and Wilcoxon test. P < 0.05 was considered significant. All adverse events were evaluated descriptively.
Compliance with Ethics Guidelines
This study was strictly in compliance with the Good Clinical Practice (GCP) principle according to the CFDA and approved by the Shanghai Public Health Clinical Center Medical Ethics Committee (2017-E028-01). All participants have provided informed consent to participate in the study. Our study was performed in accordance with the declaration of Helsinki 1964 and its later amendments.
Results
Participants
A total of 230 healthy adult participants between 18 and 45 years of age were assessed for eligibility; all these participants were screened according to the study protocol including assessing vital signs, electrocardiogram, chest radiograph, sputum acid-fast bacilli smear, virus detection, nicotine, alcohol allergy, liver and kidney function, and routine blood, urine, and stool tests. As a pre-defined standard, any abnormal qualitative indicator would lead to exclusion; abnormal was defined as any quantitative indicator with an excess of 20% of the reference value (lower more than 20% of lower limit or higher more than 20% of upper limit). Finally, 158 were excluded through not meeting the inclusion criteria or for personal reasons. The remaining 72 participants were recruited and randomized into six groups (group A to F). Within a group, the recipients of RP22 or recipients of the placebo did not differ significantly in age, weight, BMI index, and the distribution was balanced between the groups. All participants completed the skin test and follow-up, except for one participant in group A who had an abnormal ECG on the day of receiving intradermal injection and quitted before the measurement of skin test response, so there were seven participants in the 0.1 μg RP22 group and eight participants who received RP22 in each of the other groups. In total, there were 24 participants who received the placebo; all 71 participants completed the procedure. No more than half of them had an AE in each group and the highest dose level group, group F, was completed (Fig. 1).

Flowchart of the enrollment. *One participant had abnormal ECG on the day of receiving the intradermal injection
Safety Results
The incidence of AEs associated with RP22 injection ranged from 12.5% (1/8, 0.25 μg group) to 50% (4/8, 2 μg and 4 μg groups). The major AEs were in the local injection area. The incidence of local injection area AEs was 4.2% (1/24) in the placebo group and ranged from 0% (0/7) to 50.0% (4/8) in the RP22 participants. Systemic AEs included dizziness, sweating after the skin test, and abnormal ECG or laboratory examination result. The incidence was 12.5% (3/24) in the placebo group and ranged from 0% (0/8) to 37.5% (3/8) in the RP22 participants and was not associated with dose size. The two abnormal ECGs were reported as transient sinus bradycardia, with a heart rate of 50–60 bpm, without any clinical symptoms, and were restored in the following test within 2 h. Six subjects had mild elevated liver function and recovered within 1 week without taking any hepatoprotective drugs. One subject caught a cold so some of the blood routine indexes were elevated mildly. No serious AEs were observed in any of the groups, except for one participant in the 0.5 μg group who was involved in a minor car accident 72 h after the injection, which had no bearing on the skin test reading (Table 2).
Secondary Evaluation Indicators
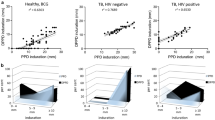
In the 1, 2, and 4 μg groups, there was transient redness within 15–30 min after injection; seven of the eight participants who received 4 μg RP22 had transient redness, a higher incidence rate than other dose groups (Fig. 2). Four participants in the placebo group had transient redness. After unblinding, retrospective analysis showed that three of these placebo recipients were in the 4 μg RP22 placebo group and one in the 1 μg RP22 placebo group, which was consistent with the higher redness response rates in the RP22 recipients in these groups. All the redness disappeared within 2 h after injection. There was no transient redness in the three lower dose groups (0.1 μg, 0.25 μg, and 0.5 μg) after intradermal injection.

Skin reaction of RP22 and QFT result of all participants. *Participants who received placebo,  QFT positive,
QFT positive,  redness,
redness,  induration, number in the
induration, number in the  blank is average redness diameter, number in the
blank is average redness diameter, number in the  blank is average induration diameter, the unit is millimeter (mm), average diameter = (longitudinal + transverse diameter)/2
blank is average induration diameter, the unit is millimeter (mm), average diameter = (longitudinal + transverse diameter)/2
Comparison Between RP22 and QFT-IT Assay in All Participants
Eleven participants (15.5%) had positive QFT results, and 60 participants (84.5%) had negative QFT results. Of the 11 positive-QFT participants, five had induration diameter of greater than 10 mm at 48–72 h. Of the 60 negative-QFT participants, five participants had induration but no diameter was larger than 10 mm (Fig. 2, Table 3). On the basis of this, if the cutoff value of induration was set as 10 mm, the agreement between the RP22 and QFT was 0.92, and the kappa value was 0.59 (Table 4).
Placebo Group
The placebo group included 12 women and 12 men with a mean age of 24 ± 3 years. Two of them had positive QFT-GIT results, and none of them had induration within 72 h.
0.1 μg RP22 Group
This group included three women and four men, mean age 25 years (SD 4 years). One participant in this group had a positive QFT-GIT result but an unresponsive skin test result within 72 h.
0.25 μg RP22 Group
This group included four women and four men, mean age 25 years (SD 2 years). One participant in this group had a positive QFT-GIT result but an unresponsive skin test result within 72 h.
0.5 μg RP22 Group
This group included four women and four men, mean age 26 years (SD 3 years). Two participants in this group had positive QFT-GIT results and responsive skin test results, one had a maximum induration diameter (27.5 mm) at 72 h, the other one had a maximum induration diameter (40.1 mm) at 72 h.
1.0 μg RP22 Group
This group included four women and four men, mean age 25 years (SD 3 years). Three participants showed positive QFT-GIT results and one of them showed a responsive skin test result with maximum induration diameter (27.3 mm) at 72 h, the other two participants had unresponsive skin test results within 72 h. Two participants in this group showed negative QFT-GIT results but responsive skin test results, one had a maximum induration diameter (8.6 mm) at 72 h, the other one had the maximum induration diameter (7.5 mm) at 72 h.
2.0 μg RP22 Group
This group included four women and four men, mean age 23 years (SD 2 years). One had a positive QFT-GIT result and a responsive skin test result, which had a maximum induration diameter (66.5 mm) at 72 h. Two participants in this group showed negative QFT-GIT results but responsive skin test results, one had a maximum induration diameter (6.3 mm) at 72 h, the other one had the maximum induration diameter (6.2 mm) at 72 h.
4.0 μg RP22 Group
This group included four women and four men, mean age 25 years (SD 2 years). One had a positive QFT-GIT result and a responsive skin test result, which had the maximum induration diameter (45.4 mm) at 72 h.
Discussion
The goals of the World Health Organization’s End TB Strategy have led to a renewed focus on screening for LTBI in individuals at risk [13] and the IA skin test approach has become a new focus. It has simplicity in that it requires no laboratory processing of clinical samples. This new test uses MTB antigens that are not present in the BCG vaccine or most environmental mycobacteria and have well-established specificity [14].
Through this phase I clinical trial, we confirmed that all doses RP22 except 0.4 μg as a skin test reagent for the diagnosis of Mtb infection was well tolerated and safe. No serious adverse events associated with the injection were observed in any of the groups, all the adverse reactions are mild. The major adverse reactions after injection included systemic AEs and local injection area AEs. Systemic AEs included dizziness, sweating, and abnormal ECG or laboratory examination results. There were two participants who had dizziness and a sweating reaction within 5 min after the skin test in groups A and B, respectively. Two participants had transient sinus bradycardia and returned to normal rhythm (more than 60 beats per minute) within 2 h; we think they did not have organic heart disease. The seven other systemic AEs were mild, and all the abnormal indicators had returned to normal within 1 week.
In 83.3% (10/12) participants, a transient redness was observed within 15–30 min after injection in the highest doses that had disappeared within 2 hours after injection. We speculated that the phenomenon of transient redness occurring in the higher dose groups was related to a higher osmotic pressure of the solution of RP22 and placebo in the higher dose groups. The later redness that occurred at 48–72 h after antigen injection was ascribed to a delayed type hypersensitivity (DTH) reaction to the antigen by participants with LTBI. We recorded the early redness as a local adverse event, but the later redness was not considered as a local adverse event unless the diameter was larger than 30 mm. As the performance of redness was unstable, we agreed that the effect measure of this test was induration, just like the widely accepted measure for skin test. There were some differences in the time and diameter of DTH reaction between the animal model and humans. Although the maximum induration in the guinea pig model was at 24 h after antigen injection, in humans this occurred at 48–72 h after antigen injection; this difference is consistent with other studies of antigen skin tests in humans and animals [15, 16].
In animal studies, intradermal tests with ESAT6 and a combination of ESAT6 with CFP10 protein had shown safety and sensitivity to inducing specific skin test responses [17, 18]. Several large and ongoing follow-up studies in humans have also shown specificity and sensitivity, proof of safety, feasibility, and dosage tolerability of a recombinant dimeric version of ESAT6 (rdESAT6) [19, 20]. However, the sensitivity with rdESAT6 was not ideal, and combined use of CFP10 and ESAT6 showed higher sensitivity in the diagnosis of TB than the use of either antigen alone and the specificity was not lower [21]. Further studies showed that combined rdESAT6/rCFP10 (C-Tb) could discriminate patients with TB from BCG-vaccinated healthy individuals with excellent sensitivity in phase I/II clinical trials [22, 23]. Ruhwald [9] reported an assessment of C-Tb in a phase III clinical trial. A total of 979 participants comprising negative controls, close contacts, occasional contacts, and patients with active TB were enrolled at 13 centers in Spain. C-Tb and QFT results were concordant in 785 (94%) of 834 participants aged 5 years and older, and results did not differ significantly between exposure groups.
As expected, there was a clear association between a positive QFT-IT test result (11 participants) and positive RP22 result (5 of the 11 participants had induration larger than 10 mm at 48–72 h) and a dose–response relationship was evident in the RP22 results. It was notable that the middle doses of RP22 (0.5 μg and 1 μg) gave higher concordance with TST and IGRA than the lower doses and fewer adverse reactions than the higher doses. Induration diameter larger than 10 mm could be used as a positive cutoff to diagnose LTBI. Our study suggested that RP22 could play a key role in screening for LTBI and provide a much-wanted alternative to the existing TST and IGRA methods. A limitation of our study was the inclusion of only adult healthy individuals. In further studies, safety evaluation and diagnostic performance in a wider range of population including patients with TB, those with recent TB infection, close contacts, children, immunocompromised, and high/low TB burden context citizens should be conducted as phase II and III clinical trials. Furthermore, we will focus on assessing the consistency of RP22 with IGRAs, evaluate the mechanism of false positive or false negative cases, and determine whether this approach can be used as an independent diagnostic index or just a complementary test for TST.
Conclusions
RP22 was well tolerated and safe in healthy normal participants and those with LTBI. The maximum response was obtained 48–72 h after antigen injection; the suitable response induration diameter could be defined as 10 mm and the suitable dose could be defined as 0.5 μg or 1 μg. As a skin test reagent for Mtb infection, RP22 could play an important role in the screening of LTBI.
References
WHO. Global tuberculosis report 2019. Geneva: World Health Organization; 2019.
Getahun H, Matteelli A, Chaisson RE, Raviglione M. Latent Mycobacterium tuberculosis infection. N Engl J Med. 2015;372:2127–35.
Wang L, Turner MO, Elwood RK, Schulzer M, FitzGerald JM. A meta-analysis of the effect of Bacille Calmette Guerin vaccination on tuberculin skin test measurements. Thorax. 2002;57:804–9.
Kahwati LC, Feltner C, Halpern M, et al. Primary care screening and treatment for latent tuberculosis infection in adults: evidence report and systematic review for the US preventive services task force. JAMA. 2016;316:970–83.
Mori T. Usefulness of interferon-gamma release assays for diagnosing TB infection and problems with these assays. J Infect Chemother. 2009;15:143–55.
Pai M. Spectrum of latent tuberculosis - existing tests cannot resolve the underlying phenotypes. Nat Rev Microbiol. 2010;8:242.
Pai M, Joshi R, Dogra S, et al. Serial testing of health care workers for tuberculosis using interferon-gamma assay. Am J Respir Crit Care Med. 2006;174:349–55.
Pai M, Joshi R, Dogra S, et al. T-cell assay conversions and reversions among household contacts of tuberculosis patients in rural India. Int J Tuberc Lung Dis. 2009;13:84–92.
Ruhwald M, Aggerbeck H, Gallardo RV, et al. Safety and efficacy of the C-Tb skin test to diagnose Mycobacterium tuberculosis infection, compared with an interferon gamma release assay and the tuberculin skin test: a phase 3, double-blind, randomised, controlled trial. Lancet Respir Med. 2017;5:259–68.
Slogotskaya L, Bogorodskaya E, Ivanova D, Sevostyanova T. Comparative sensitivity of the test with tuberculosis recombinant allergen, containing ESAT6-CFP10 protein, and Mantoux test with 2 TU PPD-L in newly diagnosed tuberculosis children and adolescents in Moscow. PLoS One. 2018;13:e0208705.
Andrews JR, et al. Serial QuantiFERON testing and tuberculosis disease risk among young children: an observational cohort study. Lancet Respir Med. 2017;5(4):282–90.
Wei X, Xie YM. [Principle of adverse drug reaction causality judgement and interpretation of causality assessment method both in China and abroad]. Zhongguo Zhong Yao Za Zhi. 2012;37:2744–7.
World Health Organization. Guidance for national tuberculosis programmes on the management of tuberculosis in children. WHO: Geneva: 2014.
Abubakar I, Jackson C, Rangaka MX. C-Tb: a latent tuberculosis skin test for the 21st century? Lancet Respir Med. 2017;5:236–7.
Wu X, Zhang L, Zhang J, Zhang C, Zhu L, Shi Y. Recombinant early secreted antigen target 6 protein as a skin test antigen for the specific detection of Mycobacterium tuberculosis infection. Clin Exp Immunol. 2008;152:81–7.
Desem N, Jones SL. Development of a human gamma interferon enzyme immunoassay and comparison with tuberculin skin testing for detection of Mycobacterium tuberculosis infection. Clin Diagn Lab Immunol. 1998;5:531–6.
Du WX, Chen BW, Lu JB, et al. Preclinical study and phase I clinical safety evaluation of recombinant Mycobacterium tuberculosis ESAT6 protein. Med Sci Monit Basic Res. 2013;19:146–52.
van Pinxteren LA, Ravn P, Agger EM, Pollock J, Andersen P. Diagnosis of tuberculosis based on the two specific antigens ESAT-6 and CFP10. Clin Diagn Lab Immunol. 2000;7:155–60.
Arend SM, Franken WP, Aggerbeck H, et al. Double-blind randomized phase I study comparing rdESAT-6 to tuberculin as skin test reagent in the diagnosis of tuberculosis infection. Tuberculosis (Edinb). 2008;88:249–61.
Lillebaek T, Bergstedt W, Tingskov PN, et al. Risk of sensitization in healthy adults following repeated administration of rdESAT-6 skin test reagent by the Mantoux injection technique. Tuberculosis (Edinb). 2009;89:158–62.
Skinner MA, Buddle BM, Wedlock DN, et al. A DNA prime-Mycobacterium bovis BCG boost vaccination strategy for cattle induces protection against bovine tuberculosis. Infect Immun. 2003;71:4901–7.
Bergstedt W, Tingskov PN, Thierry-Carstensen B, et al. First-in-man open clinical trial of a combined rdESAT-6 and rCFP-10 tuberculosis specific skin test reagent. PLoS One. 2010;5:e11277.
Aggerbeck H, Giemza R, Joshi P, et al. Randomised clinical trial investigating the specificity of a novel skin test (C-Tb) for diagnosis of M. tuberculosis infection. PLoS One. 2013;8:e64215.
Acknowledgments
We thank all the participants of the study.
Funding
This work was supported by grants from the National Science Foundation of China (81770011, 31771004), and technique supports from the Chinese National Mega Science and Technology Program on Infectious Diseases (2018ZX10302301, 2018ZX10731301), the National Key R&D Program of China (2018YFD0500900), and the Clinical Research Plan of SHDC (16CR2041B). The supporting grants are also funding the journals rapid service fee.
Authorship
All authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Lu Xia, Xu-hui Liu, Zhang-yan Zhao, Tao Li, Xiu-hong Xi, Ping Liu, Wei Huang, Xiao-yong Fan, Xue-qiong Wu, and Shui-hua Lu declare that they have no conflict of interest.
Compliance with Ethics Guidelines
This study was strictly in compliance with the Good Clinical Practice (GCP) principle according to the CFDA and approved by the Shanghai Public Health Clinical Center Medical Ethics Committee (2017-E028-01). All participants have provided informed consent to participate in the study. Our study was performed in accordance with the declaration of Helsinki 1964 and its later amendments.
Data Availability
The datasets generated during and analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Xia, L., Liu, Xh., Zhao, Zy. et al. Safety Evaluation of Recombinant Fusion Protein RP22 as a Skin Test Reagent for Tuberculosis Diagnosis: A Phase I Clinical Trial. Infect Dis Ther 10, 925–937 (2021). https://doi.org/10.1007/s40121-021-00435-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-021-00435-5