
Abstract
Introduction
To assess the safety, tolerability and pharmacokinetics of a single dose of SYHA1402 in healthy Chinese subjects.
Methods
This was a randomized, double-blind, placebo-controlled, single-ascending dose study in healthy subjects. Subjects received a single dose of SYHA1402 25 mg, 50 mg, 100 mg, 200 mg, 400 mg or 800 mg, or matching placebo. Safety and tolerability were assessed throughout the study. The pharmacokinetic (PK) parameters of SYHA1402 were estimated using non-compartmental analysis.
Results
In all, 54 subjects were enrolled and completed the study. Specifically, there were no deaths, serious adverse events or withdrawals from study due to adverse events. All treatment-emergent adverse events were mild. The most common drug-related adverse event was sinus bradycardia. The time to maximum concentration ranged from 1.13 to 2.25 h, and the terminal elimination half-life range was 1.51–4.70 h. SYHA1402 exhibited nonlinear PK parameters with less than dose-proportional increases in exposure after single oral doses of 25 to 800 mg.
Conclusion
SYHA1402 administered as a single dose was well tolerated and safe over the dose range of 25–800 mg. More than 50% of the unchanged SYHA1402 was excreted in urine within the dose range of 25–100 mg.
Trial registration
NCT03988413 (https://www.clinicaltrials.gov/; registration date: 17 June 2019).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Our aim was to evaluate the safety, tolerability and pharmacokinetic (PK) characteristics of a single dose of SYHA1402 in healthy Chinese subjects. |
What was learned from this study? |
SYHA1402 was tolerable in healthy Chinese subjects in the dose range of 25–800 mg, and the safety was good. |
The incidence of drug-related adverse events did not increase with dose, and there was no correlation between severity and dose. |
After the administration of a single oral dose of SYHA1402 in the range of 25–800 mg, the drug was absorbed rapidly in healthy volunteers, and the median Tmax was 1.13–2.25 h. |
SYHA1402 showed nonlinear PK parameters in the dose range of 25–800 mg, but linear PK characteristics in the dose ranges of 25–100 mg and 200–800 mg. |
Introduction
The SYHA1402 tablet is a highly selective aldose reductase inhibitor (ARI) that has been developed to treat diabetic peripheral neuropathy (DPN). Aldose reductase (AR) is a key rate-limiting enzyme in the polyol pathway of glucose metabolism [1]. Under hyperglycemic conditions, excess glucose is converted to sorbitol by AR activation through the polyol pathway. Increased sorbitol accumulation in cells results in injury to both cells and organs. Additionally, the accumulation of sorbitol in cells results in the loss of myoinositol, an essential component of sodium/potassium (Na/K) ATPase, thereby impairing normal nerve physiology [2,3,4,5]. ARI inhibits the activity of AR in the polyol pathway, reduces the accumulation of sorbitol in cells and has the potential to play an important role in the treatment of DPN [6]. Several ARIs have been reported in the literature, but epalrestat is the only ARI currently marketed for use in the treatment of DPN in Japan and China [7]. In that study, the radioligand binding assay and enzyme test in vitro showed that SYHA1402 could effectively inhibit AR activity (50% inhibitory concentration [IC50] = 22.8 nM), with an IC50 that was 5.86-fold higher that of epalrestat (133.7 nM). The pharmacodynamic studies in vivo confirmed that SYHA1402 significantly inhibited the deceleration of motor nerve conduction velocity of the sciatic nerves and tail nerves, increased the activity of (Na/K) ATPase in sciatic nerves and inhibited the accumulation of sorbitol in sciatic nerve cells. In the safety pharmacology study, the NOAEL (no-observed-adverse-effect level) in SD rats and Beagle dogs was 500 and 150 mg/kg, respectively.
The purposes of the present study were to evaluate the safety, tolerability and pharmacokinetic (PK) characteristics of a single-dose of SYHA1402 in healthy Chinese subjects.
Methods
Compliance with Ethics Guidelines
This clinical trial was conducted in accordance with the Helsinki Declaration of 1964 and its later amendments and with Good Clinical Practice (GCP) requirements [8]. The pilot scheme/protocol was approved by the Ethics Committee of the Chinese PLA General Hospital before implementation (No. C2019-009–01), with the researchers guaranteeing that the clinical trial would be conducted in accordance with the laws, regulations, scientific and ethical standards of the People’s Republic of China concerning medical trials. Subjects were required to sign a written informed consent form before entering the trial (screening or any other trial-related activities). The trial is registered on ClinicalTrials.gov (https://clinicaltrials.gov/; Identifier: NCT03988413).
Subjects
Healthy male or female subjects aged 18–45 years, with a body mass index (BMI) of 19–26 kg m−2 and a total body weight ≥ 45.0 kg for female subjects and ≥ 50.0 kg for male subjects were eligible for inclusion in the study. Health was defined as no clinically related abnormalities that were identifiable by medical history, physical examinations, vital signs, electrocardiogram (ECG) or clinical laboratory tests. Female subjects who were pregnant, lactating or planning a pregnancy within 3 months after the end of the trial were not eligible to participate. Also, people who were taking drugs and/or had a history of alcohol abuse, substance abuse or other conditions that researchers believed were not suitable for inclusion in the trial were excluded. In accordance with the National Cancer Institute-Common Terminology Criteria for Adverse Events Version 5.0 (NCI-CTCAE), the researcher needed to discuss with the sponsor and decide whether to continue the trial when the following circumstances occurred during the trial: (1) if ≥ 50% of the subjects in each group had adverse events determined by the researchers to be related to the experimental drug, and the subjects in each group had grade ≥ 3 adverse events related to the experimental drug; (2) serious adverse events (SAEs) occurred during the course of the trial, and the researchers determined that the SAEs were related to the experimental drugs.
Trial Design
This phase I randomized, double-blind, single-dose increment study in healthy Chinese subjects was conducted at Chinese PLA General Hospital, Beijing, China, from August 2019 to January 2020. The subjects were randomly assigned to SYHA1402 tablet dose groups: 25, 50, 100, 200, 400 and 800 mg, respectively. In the 25 mg group, six subjects were randomly assigned at a 2:1 distribution, with four subjects receiving SYHA1402 tablet and two subjects receiving placebo. Ten subjects were assigned to each of the 50, 100, 200 and 400 mg groups, with subjects randomly assigned to receive SYHA1402 and placebo at a 4:1 distribution. In the 800 mg group, eight subjects were randomly assigned at a 3:1 distribution, with six subjects receiving SYHA1402 tablet and two receiving placebo.
The study included a screening period (up to 14 days) and an 8-day period of treatment and evaluation. From day 1 to day 4, the subjects were hospitalized. On day 1, the subjects received a dose of SYHA1402 or a matching placebo. Study visits and safety assessments were planned from day 1 to day 4. On day 8, follow-up assessments were conducted by telephone to identify any adverse effects (AEs).
Study Drug
SYHA1402 tablets, formulated as a tablet in two dose strengths of 25 mg (lot number: HA1403181201) and 100 mg (lot number: HA1403181203), and placebo tablets, formulated at the two dose strengths of 25 mg (lot number: HA1403181201k) and 100 mg (lot number: HA1403181203k) were manufactured by CSPC Pharmaceutical Group Co., Ltd. (Shijiazhuang City, Hebei Province, China). Both the study drug and placebo were taken orally under the fasting condition.
Pharmacokinetic Assessment
Blood samples were collected pre-dose (within 3 h before dosing), and at 10, 20, 30 and 45 min and 1, 1.25, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 24, 36, 48 and 72 h post-dose. Urine samples were also collected prior to dosing and at 0–4, 4–8, 8–12, 12–24, 24–48 and 48–72 h post-dose. The feces samples were collected only in the 400 mg dose group at 0–72 h. Collected blood samples were centrifuged at 4 °C, 6200 g for 10 min, and the plasma was collected and stored in a freezer at − 80 °C for future analysis.
The concentrations of SYHA1402 in plasma and urine were determined on a validated liquid chromatography–tandem mass spectrometry (LC–MS/MS) system using a Kinetex C18 column (2.6 µm, 50 mm × 2.1 mm; Phenomenex, Torrance, CA, USA) and mobile phases (solvent A, water containing 0.1% formic acid; solvent B, 50% methanol and 50% acetonitrile) for gradient elution. The concentrations of SYHA1402 in feces were determined on a validated LC–MS/MS system using a Zorbax SB-C18 column (3.5 µm, 100 mm × 2.1 mm; Agilent Technologies, Inc., Santa Clara, CA, USA) and mobile phases (solvent A, water containing 0.5% formic acid; solvent B, 50% methanol and 50% acetonitrile) for gradient elution. The compound was detected by MS/MS with electrospray ionization operated with multiple reaction monitoring in the positive ionization mode, and d9-tolbutamide was used as the internal standard. Focus was on the following ion transitions: m/z 422.0 → 216.0 for SYHA1402, and m/z 280.1 → 155.0 for d9-tolbutamide. The quantitative range of SYHA1402 in plasma, urine and feces was 1.00–1000, 100–200,000 and 0.500–500 ng mL−1, respectively.
Safety Assessment
Safety evaluation included monitoring for AEs and analysis of laboratory test results (hematology, clinical chemistry and routine urinalysis), vital signs, 12-lead ECG and physical examinations. The severity of AEs was assessed using the criteria of Common Terminology Criteria for Adverse Events version 5.0 (https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcae_v5_quick_reference_5x7.pdf).
Pharmacokinetics Parameters
The PK parameters of SYHA1402 in plasma, urine and feces were calculated with a non-compartmental analysis model based on Phoenix WinNonlin 8.1 software (Certara, Princeton, NJ, USA). The following PK parameters were calculated: time to maximum plasma concentration (Tmax), maximum plasma concentration (Cmax), terminal elimination half-life (t½), apparent volume of distribution (Vz/F), apparent clearance rate (CLz/F) and area under the concentration–time curve from time zero to the last measurable concentration (AUC0–t) or infinity (AUC0–∞). The unchanged SYHA1402 recovered in urine was calculated based on the urine data as follows: cumulative urinary drug excretion from t1 to t2 (Aet1–t2), cumulative urinary drug excretion from 0 to 72 h (Ae0–t), cumulative urinary drug excretion percentage from 0 to 72 h (Fe0–t). The unchanged SYHA1402 recovered in feces were calculated as follows: cumulative fecal drug excretion from 0 to 72 h (Af0–t) and cumulative fecal drug excretion percentage from 0 to 72 h (Ff0–t).
Statistical Analysis
Descriptive statistics were carried out on the demographic parameters. There were no formal comparisons between different dose groups. The drug concentration and PK parameters were summarized and compared in the drug administration cohort by descriptive statistical methods. The power model was used to test the relationship between Cmax, AUC0–∞ and dose, i.e. dose proportionality, natural logarithm (ln) (AUC0–t, AUC0–∞ or Cmax) = α + β × ln (dose). A β value of approximately 1 was considered to indicate that the PK parameters were linear.
Results
Baseline Characteristics
A total of 54 subjects were randomized to one of six dose groups. The demographics of the subjects in the six SYHA1402 dose groups (including those subjects on matching placebo) are summarized in Table 1. Overall, the demographic characteristics were similar across the six treatment groups. All 54 subjects completed the trial.
Safety and Tolerability
The safety results are pooled in Table 2. A total of 54 treatment-emergent AEs (TEAEs) were reported by 33 subjects (61%) (25 in the study groups and 8 in the placebo groups). All TEAEs were classified as mild.
Overall, 14 AEs (26% of all events) reported by 12 subjects (13 events reported by 11 SYHA1402 recipients and 1 event reported by 1 placebo recipient) were considered to be related to the drug used in the study. Among these, eight subjects experienced sinus bradycardia, including two in each of the 50 and 800 mg dose groups, one in each of the 100, 200 and 400 mg dose groups and one in the placebo group. The other drug-related AE included one case of positive white blood cells in urine (100 mg dose group), one case of positive urine protein (200 mg dose group), one case of positive urine ketone body (400 mg dose group), one case of low blood pressure (25 mg dose group) and one case of diarrhea (25 mg dose group). No deaths or SAEs were reported. None of the subjects withdrew from the study because of AEs. There was no apparent relationship between the frequency and intensity of AEs and the increasing doses of SYHA1402.
Pharmacokinetic Evaluation
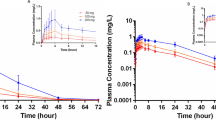
After each of the 42 healthy subjects had been administered a single dose of SYHA1402 tablet (6 dose groups: 25 mg group, N = 4; 50–400 mg group, N = 8; 800 mg group, N = 6), the mean plasma concentration–time curves of SYHA1402 were deteremined, as shown in Fig. 1.

a Mean SYHA1402 plasma concentration–time profiles after single-dose oral administration. b Mean SYHA1402 plasma concentration–time profiles from 0 to 8 h after single-dose oral administration. Results are shown as the mean ± standard deviation
After oral administration, SYHA1402 was absorbed quickly, and the median Tmax was 1.13–2.25 h. Of all the doses evaluated in the study, the mean t½ of SYHA1402 ranged from 1.51 to 4.70 h. The plasma Cmax increased from 865 ng mL−1 in the 25 mg dose group to 15,750 ng mL−1 in the 800 mg dose group. As shown in Table 3 and Fig. 1, the Cmax of SYHA1402 was significantly less than the dose proportional with the increase of dose, indicating that SYHA1402 has a nonlinear PK. The AUC0–∞ and the area AUC0–t of SYHA1402 also increased in a less than dose-proportional manner over the dose range from 25 to 800 mg. There were significant differences between the estimated values of the power model exponents and the expected value 1 under the dose proportionality. The estimated values and 95% confidence intervals (CIs) of Cmax, AUC0–t and AUC0–∞ were 0.782 (95% CI 0.686–0.877) ng/mL, 0.833 (95% CI 0.754–0.913) h ng/mL and 0.832 (95% CI 0.753–0.911) h ng/mL, respectively. In the dose groups 25, 50 and 100 mg and in the dose groups 200, 400 and 800 mg, Cmax, AUC0–t and AUC0–∞ increased in proportion to dose. The estimated values and 95% CIs were 0.960 (95% CI 0.661–1.259) ng/mL, 1.030 (95% CI 0.809–1.250) h ng/mL and 1.028 (95% CI 0.807–1.248) h ng/mL for dose groups 25, 50 and 100 mg, and 0.944 (95% CI 0.689–1.199) ng/mL, 0.840 (95% CI 0.603–1.077) h ng/mL and 0.840 (95% CI 0.603–1.077) h ng/mL for dose groups 200, 400 and 800 mg.
In addition to determining the PK of plasma SYHA1402, we also evaluated urine excretion of unchanged SYHA1402 in the six dose groups (Table 4). Figure 2 shows the time course of cumulative excretion of unchanged SYHA1402 in urine within 72 h post-dose. In the 25, 50, 100, 200, 400 and 800 mg groups, the mean Fe0–72 h of SYHA1402 was 64.08%, 57.97%, 59.28%, 24.64%, 8.49% and 7.15%, respectively, and the mean renal clearance rate (CLr) was 7266.52, 6014.60, 6489.26, 3918.37, 1270.18 and 1483.06 mL h−1, respectively. More than 50% of the unchanged SYHA1402 was excreted in urine within the dose range of 25–100 mg, while with the dose range from 200 to 800 mg, the Fe0–72 h and CLr of SYHA1402 decreased significantly. According to the data on Ae0–72 h in each group, the amount of SYHA1402 excreted in the urine gradually trended towards saturation.

a Cumulative urinary drug excretion-time curve after administration of a single oral dose of 25 mg to 800 mg SYHA1402 tablet to healthy subjects. b Histogram of cumulative urinary drug excretion percentage following oral administration of a single dose of 25 to 800 mg SYHA1402 tablet to healthy subjects
The unchanged SYHA1402 recovered in feces was also evaluated, but only in the 400 mg dose group. The mean Af0–72 h was 6803.08 μg, and only a small amount of unchanged SYHA1402 was recovered from feces, with a Ff0–72 h of 1.70% (Table 5).
Discussion
Diabetic peripheral neuropathy is one of the most common long-term complications of diabetes, affecting nearly two-thirds of all people with diabetes [9]. It is associated with a risk of foot ulcer, gangrene and subsequent amputations, which significantly decreases the quality of life of people with diabetes. AR is the key enzyme of the polyol pathway that reduces the metabolism of glucose to sorbitol in the presence of nicotinamide adenine dinucleotide phosphate, and the accumulation of sorbitol is the key underlying factor for the development of DPN [10, 11]. ARI inhibits reduces the accumulation of sorbitol and plays a protective role in DPN [6]. However, with the exception of epalrestat, which was the first ARI epalrestat to be developed, as early as the mid-1960s, and successfully listed in countries or regions such as Japan and China [7], there have been no new medicines [12]. SYHA1402 is a new highly selective ARI with an IC50 value of 22.8 nM and has been found to be effective in neuropathy by reducing sorbitol levels in sciatic nerves in rats with diabetes mellitus. In the present study, which is the first study on SYHA1402 to be conducted in humans, we show the PK parameters and safety results of single ascending doses in healthy Chinese subjects.
The starting dose for humans was calculated and justified by both the NOAEL [13] and minimal-anticipated-biological-effect level (MABEL) [14], and the starting dose of human body in this study was determined as 25 mg. At present, the only clinical application of ARI drugs is epalrestat at the application dose of 150 mg/day. The only ARI, namely ranirestat, currently under trial in the USA and Japan has a dose of 20, 40, 80 mg/day for phase II/III trials. Considering the results of SYHA1402 preclinical pharmacodynamics, PK parameters, toxicology and the safety data of clinical trials of similar mechanism products, it has been predicted that the compound was safe.
In the present study, we found that SYHA1402 administered at a single dose of up to 800 mg was well tolerated; there were no deaths or SAEs. All AEs were mild, and there was no apparent relationship between the frequency and intensity of AEs and the increasing doses of SYHA1402. Sinus bradycardia was the most common drug-related AE reported in the study (8 subjects: 8 events in 7 SYHA1402 recipients and 1 event in 1 placebo recipient). All of the sinus bradycardia events were mild and temporary, with most occurring at 1–2 h after administration. There have been no report of sinus bradycardia events with epalrestat treatment. More attention should be paid to this AE in the future clinical trials, and ECG monitoring should be emphasized at 1–2 h after administration. However, minor elevations in liver enzymes, which are the most frequently reported AEs in epalrestat package insert, were not observed in this study [15].
SYHA1402 exhibited rapid oral absorption. The absorption rate and t½ of 50 mg SYHA1402 were similar to those of 50 mg epalrestat. According to the epalrestat package insert [16], a plasma Cmax of 3.9 µg mL−1 is reached at approximately 1 h after a 50 mg oral dose and t½ is about 1.83 h. SYHA1402 exhibited significant nonlinear PK parameters with less than dose proportional increases in exposure after single doses ranging from 25 to 800 mg. The terminal half-lives and CLz/F were similar among groups receiving doses of 25, 50 and 100 mg but varied greatly among groups receiving 25 to 800 mg, while a single dose in the range of 25–100 mg exhibited linear PK parameters. Epalrestat was reported with linear PK parameters with a single dose in the range of 50 to 200 mg [17].
More than 50% of the unchanged SYHA1402 was excreted in urine within the dose range of 25–100 mg. However, with increases in the dose from 200 to 800 mg, both the Fe0–72 h and CLr of SYHA1402 appeared to fall significantly to a plateau and the amount of SYHA1402 excreted in the urine gradually trended towards saturation. In the 400 mg dose group, 10.19% of unchanged SYHA1402 was recovered from the feces and urine, of which 1.7% and 8.49% were detected in feces and urine, respectively.
A few study limitations should be mentioned. Although the study was designed as a blind placebo-controlled trial in accordance with the standard guidelines, it was a single, ascending dose study with a small sample size. Due to only four to eight subjects for each dose regimen, the data on treatment-related AEs should be carefully interpreted and individual differences need to be considered.
Conclusions
This study evaluated the PK parameters of a single oral dose of SYHA1402 in healthy Chinese adults under the fasting condition, and provided preliminary data on the dose, safety and tolerability of SYHA1402. The incidence of drug-related AEs did not increase with increasing dose, and there was no correlation between severity of AEs and dose. The findings clearly indicate that SYHA1402 was tolerable in our healthy Chinese subjects in the dose range of 25–800 mg and that the safety of this drug was good. SYHA1402 at the dose of 50 mg had similar absorption and elimination rates as epalrestat at a dose of 50 mg, which is the usual dose of epalrestat. A detailed human mass balance study should be performed to verify and explain the study results, and the safety and PK characteristics of SYHA1402 also need to be evaluated in further clinical trial studies.
References
Hodgkinson AD, Søndergaard KL, Yang B, et al. Aldose reductase expression is induced by hyperglycemia in diabetic nephropathy. Kidney Int. 2001;60(1):211–8. https://doi.org/10.1046/j.1523-1755.2001.00788.x.
Giugliano D, Ceriello A, Paolisso G. Oxidative stress and diabetic vascular complications. Diabetes Care. 1996;19(3):257–67. https://doi.org/10.2337/diacare.19.3.257.
Dyck PJ, Zimmerman BR, Vilen TH, et al. Nerve glucose, fructose, sorbitol, myo-inositol, and fiber degeneration and regeneration in diabetic neuropathy. N Engl J Med. 1988;319(9):542–8. https://doi.org/10.1056/nejm198809013190904.
Greene DA, Sima AA, Stevens MJ, Feldman EL, Lattimer SA. Complications: neuropathy, pathogenetic considerations. Diabetes Care. 1992;15(12):1902–25. https://doi.org/10.2337/diacare.15.12.1902.
Gabbay KH, Merola LO, Field RA. Sorbitol pathway: presence in nerve and cord with substrate accumulation in diabetes. Science. 1966;151(3707):209–10. https://doi.org/10.1126/science.151.3707.209.
Cameron NE, Cotter MA, Basso M, Hohman TC. Comparison of the effects of inhibitors of aldose reductase and sorbitol dehydrogenase on neurovascular function, nerve conduction and tissue polyol pathway metabolites in streptozotocin-diabetic rats. Diabetologia. 1997;40(3):271–81. https://doi.org/10.1007/s001250050674.
Hamada Y, Nakamura J, Naruse K, et al. Epalrestat, an aldose reductase ihibitor, reduces the levels of Nepsilon-(carboxymethyl)lysine protein adducts and their precursors in erythrocytes from diabetic patients. Diabetes Care. 2000;23(10):1539–44. https://doi.org/10.2337/diacare.23.10.1539.
State Food and Drug Administration of China. Good Clinical Practice. 2003. https://www.nmpa.gov.cn/yaopin/ypfgwj/ypfgbmgzh/20030806010101443.html. Accessed Aug 2003.
Tesfaye S, Boulton AJ, Dyck PJ, et al. Diabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care. 2010;33(10):2285–93. https://doi.org/10.2337/dc10-1303.
Yagihashi S. Glucotoxic mechanisms and related therapeutic approaches. Int Rev Neurobiol. 2016;127:121–49. https://doi.org/10.1016/bs.irn.2016.03.006.
Feldman EL, Nave KA, Jensen TS, Bennett DLH. New horizons in diabetic neuropathy: mechanisms, bioenergetics, and pain. Neuron. 2017;93(6):1296–313. https://doi.org/10.1016/j.neuron.2017.02.005.
Zhu C. Aldose reductase inhibitors as potential therapeutic drugs of diabetic complications. In: Oguntibeju O, editor. Diabetes mellitus-insights and perspectives. London: InTech; 2013. https://doi.org/10.5772/54642.
U.S. Food and Drug Administration. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. 2005. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/estimating-maximum-safe-starting-dose-initial-clinical-trials-therapeutics-adult-healthy-volunteers. Accessed Aug 2018.
Van Gerven J, Bonelli MJBJoCP. Commentary on the EMA guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products. Br J Clin Pharmacol. 84(7):1401–09
Ramirez MA, Borja NL. Epalrestat: an aldose reductase inhibitor for the treatment of diabetic neuropathy. Pharmacotherapy. 2008;28(5):646–55. https://doi.org/10.1592/phco.28.5.646.
Ono Pharmaceutical Co, Ltd. Kinedak (epalrestat) package insert. Chuo-ku, Osaka, Japan; 2005.
Steele JW, Faulds D, Goa KL. Epalrestat. A review of its pharmacology, and therapeutic potential in late-onset complications of diabetes mellitus. Drugs Aging. 1993;3(6):532–55. https://doi.org/10.2165/00002512-199303060-00007.
Acknowledgements
We thank the participants of the study.
Funding
This research was funded by the New Medicine Clinical Research Fund (4246Z512) and CSPC ZhongQi Pharmaceutical Technology Co., Ltd. CSPC ZhongQi Pharmaceutical Technology Co., Ltd. had involved in the study design. The New Medicine Clinical Research Fund (4246Z512) is funding the Rapid Service Fee.
Author Contributions
2Beibei Liang contributed to the conception and design of study, laboratory data acquisition, data analysis and drafting of article. Jin Wang contributed to the design of study and critical revision. Guanxuanzi Zhang and Rui Wang contributed to the laboratory data acquisition and data analysis. Yun Cai contributed to the conception and design of study, analysis of data and drafting of article and critical revision.
Prior Publication
For the purpose of communicating with peers on the results as soon as possible, this manuscript was preprinted on Research Square on 15 September 2022 [https://doi.org/10.21203/rs.3.rs-2059704/v1].
Disclosures
All authors declare no other competing interests.
Compliance with Ethics Guidelines
This clinical trial was conducted in accordance with the Helsinki Declaration of 1964 and its later amendments and with Good Clinical Practice (GCP) requirements [8]. The pilot scheme/protocol was approved by the Ethics Committee of the Chinese PLA General Hospital before implementation (No. C2019-009–01), with the researchers guaranteeing that the clinical trial would be conducted in accordance with the laws, regulations, scientific and ethical standards of the People’s Republic of China concerning medical trials. Subjects were required to sign a written informed consent form (ICF) before entering the trial (screening or any other trial-related activities). The trial is registered on ClinicalTrials.gov (https://clinicaltrials.gov/; Identifier: NCT03988413).
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Liang, B., Wang, J., Zhang, G. et al. Safety, Tolerability and Pharmacokinetics of Single-Dose Oral SYHA1402 in Healthy Chinese Subjects. Neurol Ther 12, 947–959 (2023). https://doi.org/10.1007/s40120-023-00480-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-023-00480-x