
Abstract
Antibiotic residues can cause serious health risks by contaminating products for human consumption, such as meat and milk. The present study developed a reliable and sensitive method based on spectrofluorometric detections to determine Florfenicol (FF) residues in milk samples. N–S co-doped carbon dots (CDs) were synthesized through a fast and straightforward microwave-assisted method with tri-ammonium citrate as precursor and thiourea as the sulfur source. The synthesis was performed under microwave irradiation at 400 W for 2.5 min. The CDs were incorporated into a molecularly imprinted polymer (MIP) prepared with (3-aminopropyl)triethoxysilane and tetraethoxysilane as the monomer and cross-linker, respectively. The CDs@MIP composite demonstrated excellent fluorescence stability with specific binding sites, which selectively accumulated FF as the target analyte and was used as a selective sensor for the fluorometric determination of FF. The photoluminescence intensity of the CDs@MIP composite was quenched in the presence of FF but not with other drugs or substances. This fact indicates the selective behavior of the new material toward FF as the template. Under optimal conditions, the relative fluorescence intensity of MIP-coated CDs decreased linearly with increasing the concentration of FF from 3.00 µmol L−1 to 1.50 × 102 µmol L−1, and its detection limit was 1.11 µmol L−1. The new sensor was evaluated for FF detection in spiked milk samples and showed recoveries of 90.86–97.71%. The method proposed the advantages of simplicity, sensitivity, selectivity, and low cost.
Similar content being viewed by others

Introduction
The widespread use of antibiotics in food-producing animals as growth promotions or treatment of infections has raised public concerns about their residues. It can generate allergic reactions, change the intestinal microflora, and increase antibiotic-resistance microorganisms posing a severe threat to human health [1].
Amphenicols are low-cost and readily available antibiotics with widespread antimicrobial activity. Florfenicol (FF) was specially developed for veterinary medicine. The lipophilicity of FF promotes its tissue penetration and improves activity against bovine respiratory pathogens. Chloramphenicol (CAP), with analogous action to FF, was extensively used in human medicine but entirely replaced with FF. Due to the reported risk of human aplastic anemia for CAP, some countries approved FF and thiamphenicol (TAP) as alternatives in food-producing animals [2, 3].
Contamination of food products with amphenicols was reported in different considerations. In a case study in Iran, FF residue in the range of 0.4–3.6 µg g−1 was detected in rainbow trout muscles. In 12.5% of the examined samples, concentrations higher than 100 µg kg−1 (recommended maximum residual level, MRL, by the European Medicines Agency) were identified [4]. In another study, 67.0% of broiler meat and liver samples were contaminated with FF residues higher than regulated MRLs (100 and 2500 µg kg−1, respectively) [5]. Similarly, residues of FF higher than MRLs (100 µg kg−1) were detected in the liver, kidney, and fat (170, 790, and 120 µg kg−1, respectively) of the treated pigs [6]. The results also confirmed the insignificant degradations of antibiotics in temperatures achieved during the usual cooking. However, microwave heating of amphenicols in water (at least 5.0 min) showed a decline compatible with boiling in a water bath for 30.0–60.0 min [3].
Amphenicols can diffuse into human cells and interfere with the synthesis of proteins. So, rapid and reliable methods are necessary to quantify their residues in foodstuffs. Various analytical methods are developed for the detection of this family of antibiotics like enzyme-linked immunosorbent assay (ELISA) [7], electrochemical sensors [1], capillary electrophoresis (CE) [3], and gas-chromatography [8]. High-performance liquid chromatography (HPLC) and LC tandem mass spectrometry (LC–MS) showed high sensitivity and identification ability [2, 4]. Still, they required complicated operation procedures, expert technical skills, and large volumes of toxic organic solvents.
Fluorescent detections are widely accepted in analytical applications owing to operational convenience, low cost, real-time detection, and high sensitivity [9, 10]. Carbon quantum dots (CDs) are an attractive class of nano-carbons with photoluminescence characteristics relevant to the particle size and excitation wavelength. They have carboxylic acid groups at the surface that are suitable for subsequent functionalization and induce excellent water solubility [11, 12]. The unique characteristics of CDs make them ideal candidates for novel fluorescent sensors compared to organic dyes and metal-based quantum dots (QDs). Different synthesis strategies of carbon dots are classified into two main categories. In top-down methods, CDs are broken from a starting carbon structure that usually involves complex processes or needs expensive materials and harsh conditions. In the bottom-up techniques, CDs are produced from a molecular precursor. Pyrolysis of a carbon source through microwave irradiation is a simple, rapid, and flexible bottom-up synthesis method [13].
Poor selectivity is usually associated with CDs-based sensors, and the development of more selective probes is desirable. Molecular imprinting is a particular technology to prepare compound-selective (or class-selective) materials. After the analyte (template molecule) and functional monomer reaction in the pre-polymerization mixture, polymerization occurs in the presence of a cross-linker [14, 15]. In molecularly imprinted polymers (MIPs), selectivity is related to the recognition cavities generated in the polymeric structure, complementary to the analyte in shape, size, and chemical functionality. Modifying CDs with MIPs can improve selectivity toward a specific molecule and its structurally related substances [16, 17]. Its advantage is integrating the selective adsorption property of MIPs with the sensitivity of fluorescence detection of CDs.
The present study aimed to prepare a novel MIP-CDs composite for selective recognition and fluorescent detection of FF. CDs were synthesized by the microwave-assisted method, and the imprinted layer was fabricated through simple sol–gel hydrolysis and condensation reaction. The prepared composite exhibited good selectivity and more accessible locations for the adsorption of FF than the non-imprinted polymer-coated CDs. The fluorescence probes based on MIP-modified CDs allowed rapid and un-expensive screening.
Materials and methods
Instrumentation
Fluorescence determinations were performed with Cary Eclipse Fluorescence Spectrophotometer (Agilent, USA) equipped with a Xenon lamp and 10.0 mm quartz cell. Ultraviolet–visible spectra (200–800 nm) were recorded on a 2501 PC UV–Vis spectrophotometer (Shimadzu, Japan). Fourier transform infrared (FT-IR) spectroscopic measurements (4000–400 cm−1) were performed on a Shimadzu 8400-IR spectrometer. The morphology and microstructure of composites were analyzed by a scanning electron microscope (SEM model Stereoscan 360, Cambridge Scientific Instrument Company, Britain) equipped with energy-dispersive X-ray chemical analysis (EDX).
Chemicals and reagents
Florfenicol was donated by the TEMAD pharmaceutical company in Mashhad, Iran. Chloramphenicol, tri-ammonium citrate (TAC), thiourea, dipotassium hydrogen phosphate (K2HPO4), (3-aminopropyl)triethoxysilane (APTES), and tetraethoxysilane (TEOS) were purchased from Sigma (Missouri, USA). Ethanol (EtOH), methanol (MeOH), acetic acid (HOAc), ammonium hydroxide (NH4OH), trichloroacetic acid, and other chemicals were obtained from Merck (Darmstadt, Germany). Analytical grade reagents and solvents were used as received without any further purification. Ultrapure water (resistivity of 18.2 MΩ cm−1) was obtained from an Aqua Max ultrapure water system (Young Lin, Korea).
Synthesis of N–S-doped carbon dots
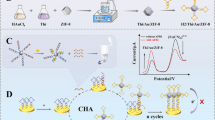
The CDs were synthesized according to earlier work [18] with a slight modification. Briefly, tri-ammonium citrate (1.00 g), thiourea (0.10 g), and dipotassium hydrogen phosphate (0.20 g) were dissolved in 2.5 mL ultrapure water. The solution was sonicated for two minutes and then heated in a microwave oven (Panasonic, NN-GD371) at 400 W and 2.5 min. The crude product was cooled to room temperature (23.0–25.0 °C) and then washed with anhydrous ethanol (5 times). Finally, the as-synthesized CDs were separated and redispersed in 30.0 mL of EtOH.
Synthesis of MIP-coated carbon dots
The CDs were coated with a MIP layer by a typical sol–gel polymerization [19]. At first, FF (40.0 mg) was dissolved in ethanol (5.0 mL) and mixed with APTES (0.1 mL). The mixture was stirred at room temperature for two hours. Then, CDs solution (5.0 mL), TEOS (0.5 mL), and ammonium hydroxide solution (2.5 mL, 5.0%v/v) were added. The resulting mixture was stirred for the next 12 h at 60.0 °C. After cooling to room temperature, the product (CDs@MIP) was collected and subjected to ultrasound-assisted solvent extraction with acidified methanol (MeOH-HOAc, 10.0%v/v). The particles were rinsed with ultrapure water and dried at 60.0 °C overnight, and the final product was kept at 4.0 °C until further use. A non-imprinted composite (CDs@NIP) was also synthesized in the same procedure without the template addition.
Fluorescence measurements
To a series of calibrated tubes, 0.5 mL of CDs@MIP or CDs@NIP suspension (4.0 g L−1) was mixed with different amounts of FF standard solution. The mixtures were diluted to 5.0 mL and ultrasonicated for 5 min before photoluminescence (PL) measurement. PL measurements were performed using an excitation wavelength of 340 nm (slit width of 10 nm) and recording an emission range of 300–800 nm (maximum fluorescence emission at 440 nm). All the measurements were carried out at room temperature (23.0–25.0 °C) and with three replicates. The fluorescence intensity was reduced gradually by raising the concentration of FF, and the variations were plotted based on the Stern–Volmer analysis (Eq. 1).
where KSV is the Stern–Volmer constant, [CFF] is the concentration of FF as the quencher, and F0 and F are the fluorescence intensity of CDs@MIP (or CDs@NIP) in the absence and presence of FF, respectively [20].
Pretreatment of milk samples
Milk samples were purchased from local markets in Mashhad and kept under refrigeration until use. The proteins were precipitated by the addition of trichloroacetic acid (1.0%v/v). The resulting mixture was sonicated for 15.0 min and then centrifuged at 9100 × g for 10 min. The clear supernatant was filtered (0.22 μm Millipore membrane filter), and the filtrate was stored at 4.0 °C [21].
HPLC analysis
The chromatographic analysis of FF in milk samples was carried out with a Waters HPLC system (Waters, USA) equipped with a binary HPLC pump (Waters 1525) and a manual sample injector with a 20 μL loop. The chromatographic separations were performed on a C18 (150 × 4.6 mm, 5 μm particle size) column for 15 min at 25 °C (column temperature) with a linear isocratic elution using a solvent system of water: acetonitrile (75:25 v/v) and a flow rate of 1 mL min−1. Detection was done with the UV–Vis detector (Waters 2489) set at 223 nm [22]. The experimental data from the CDs@MIP sensor and HPLC were evaluated by one-way analysis of variance (ANOVA), and averages of each method were compared using Duncan's test. Statistical analysis was done at 95% confidence level (p < 0.05), and all tests were repeated three times.
Results and discussion
Microwave-assisted synthesis of carbon dots
Different strategies have been practiced to improve the PL characteristics of CDs, involving multi-step synthesis, separation, and treatments with strong acids. Therefore, an efficient and easy procedure to prepare unmodified CDs without further modifications is challenging [23]. In this study, fluorescent N–S-doped CDs were synthesized through fast and straightforward microwave pyrolysis. Microwave irradiation provides intensive, homogeneous, and efficient energy transfer and shortly reaches elevated temperatures. Microwave heating initiates thermal carbonization and the formation of carbon nuclei which subsequently grow into larger particles [24]. Increasing the irradiation time would result in excessive carbonization and the formation of non-fluorescent structures [18, 25]. A transparent solution containing TAC, thiourea, and K2HPO4 was heated in a microwave oven for 2.5 min. The solvent was evaporated quickly, and a yellow and friable substance formed.
Organic molecules containing hydroxyl groups, such as citric acid [23], dextrin [26], and glycerol [25], are commonly used in the microwave synthesis of CDs because they undergo easier carbonization. Phosphate salt increases the ionic conductivity of the solution, generates higher temperatures, and accelerates the reaction [18]. Also, it was observed that the phosphate salt catalyzes the carbon dots formation and affects the formation rate and quantum yield without a significant effect on the photoluminescence characteristic of CDs [25].
Starting materials with amino and carboxyl groups are superior to synthesis CDs with excellent PL characteristics and water solubility. The fluorescence of CDs originated from the quantum confinement of conjugated π-electrons in the SP2 carbon network, and heteroatom doping provides the possibility to modify the PL properties of CDs in different aspects. Doping with nitrogen is the most studied way to modify the PL characteristics of CDs. The nitrogen atom has a comparable atomic side with five valence electrons for bonding with carbon atoms. But, sulfur atoms are larger than carbon and with a slight difference in electronegativity. So, the significant charge transfer in carbon and sulfur seems scarcely possible, and the chemical doping of sulfur into CDs would appear to be rather challenging [27]. In this study, N–S-doped CDs were synthesized from TAC as the precursor and thiourea as the sulfur source. The sol–gel reaction was employed as the most common pathway to integrate nanoparticles into an imprinted layer. It is a suitable strategy to protect CDs with a transparent silica matrix and create recognition sites that support further interactions with the analyte [28].
Characterization of carbon dots and CDs@MIP
Optical properties
The CDs were synthesized at different radiation powers (and times), and the resulting particles were compared for PL characteristics. The prepared CDs were water-soluble and showed absorption peaks at 240 and 335 nm (Fig. 1a), not observed in TAC and thiourea as the starting materials. The peak at about 335 nm is attributed to the n-π* transition from the surface state region [29].

UV–Vis (a) and PL spectra (b) of TAC-CDs synthesized at different radiation powers (The insets display CDs images under daylight (a) and (b) 365 nm UV light)
The corresponding N–S-doped CDs showed a sharp emission peak centered at 450 nm, with a nearly constant PL intensity when excitation was carried out in the range of 350–370 nm (Fig. 1b). Since tri-ammonium citrate, thiourea, and phosphate are not luminescent, the results confirm the formation of carbon dots. As presented in Fig. 1b, higher PL intensity was observed for N–S-doped CDs synthesized at 400 W, and the prepared CDs showed a bright blue fluorescence when exposed to 360 nm UV light that was seen with the naked eye. The higher irradiation power (or increasing the irradiation time) leads the product to change to a dark brown and black powder with weak PL characteristics (Supporting information Fig. S1). When the wavelength of the excitation light gradually increased from 300 to 400 nm, the emission peak position of CDs also showed a red shift accompanied by variations in the PL intensity (Supporting information Fig. S2). It indicates the presence of emissive particles of different sizes and the distribution of emissive trap sites on each CD [30].
The prepared CDs@MIP and CDs@NIP composites showed fluorescence emission at 435–445 nm when excited at 340 nm. The typical UV–Vis absorption spectrum of CDs@MIP showed a characteristic absorption peak around 250–300 nm (Supporting information Fig. S3), corresponding to the aromatic π system of CDs [31]. The results suggested silica coating is optically transparent, without unfavorable effect on the PL properties of CDs.
FT-IR spectra, SEM images, and elemental analysis
In the FT-IR spectra of N–S-doped CDs, the broad absorption band between 3500 and 3100 cm−1 belongs to –OH and –NH stretching vibrations, and the absorption band at 1710 cm−1 belongs to C=O vibration (Fig. 2d). The absorption bands at 3080 and 2982 cm−1 are assigned to –CH stretching vibrations, and peaks at 2750 and 1584 cm−1 are dedicated to –SH stretching and –NH2 bendings, respectively. The C–O stretching vibration was observed at 1200 cm−1 [18, 23]. The C-S bending is also observed at 630 cm−1. Different features of FT-IR spectra for TAC and TAC-CDs indicate complete carbonization during microwave heating. These FT-IR assignments were further verified by EDX analysis. The results showed an elemental composition of 59.57, 13.99, 24.30, and 2.13%wt of C, N, O, and S.

FT-IR spectra of a CDs@NIP, b CDs@MIP, c synthesized silica without the addition of CDs, and d N–S-doped CDs
The CDs@MIP and CDs@NIP composites showed similar characteristics in IR spectra that indicate their analogous structure and successful removal of FF from the imprinted cavities of MIP (Fig. 2a and b). The C–H stretching vibrations are observed in 2980 cm−1 [19]. The bands at 1070 and 1115 cm−1 are attributed to the Si–O–C and Si–O–Si asymmetric stretching vibrations, respectively. The absorption bands at 800 and 460 cm−1 were also assigned to the Si–O vibrations. The N–H stretching vibrations are seen at 3400 and 1565 cm−1 and suggest the aminopropyl groups successfully grafted onto the surface of CDs. The composites also showed absorption bands at 1710 and 1410 cm−1 that do not appear in the pure silica (Fig. 2c) and can be attributed to the presence of CDs. This confirms that the imprinted silicate layer has been successfully grafted on the surface of N–S-doped CDs. The surface morphology of CDs@MIP and CDs@NIP was characterized by SEM images (Supporting information Fig. S4). The composites showed smooth and spherical-shaped particles with a relatively narrow distribution between 60 and 90 nm.
The prepared CDs@MIP and CDs@NIP composites showed a transparent blue fluorescence under a 360 nm UV light that could be observed by the naked eye (Supporting information Fig. S5). Further investigations showed excellent photostability of N–S-doped CDs in consecutive determinations in a day with RSD values of 0.17–0.52%. The initial PL intensity showed a reduction of 9.5% in 8 weeks of storage. For an ethanol solution of CDs@MIP and CDs@NIP (0.40 g L−1), no significant changes in fluorescent intensity and peak characteristics were observed within 20.0 min. The product is stable over two weeks when stored at 4.0 °C as a solid powder.
Chemical sensing of florfenicol with CDs@MIP
The effect of pH and concentration of modified-CDs
The influence of pH on the fluorescence intensity of CDs@MIP and CDs@NIP composites was studied in solutions adjusted in the pH range of 2.0–10.0. The PL intensity was relatively stable in the 6.0–8.0 intervals but considerably decreased in highly acidic and basic media [32]. In high pH values, the hydroxyl groups could nucleophilically attack the surface of CDs@MIP (or CDs@NIP) and create surface defects that lead decrease in the PL intensity [19].
The amount of CDs@MIP can influence the fluorescence intensity and the assay sensitivity. Sensitivity was significantly reduced at concentrations of more than 0.6 g L−1 (Fig. 3a). It is mainly due to the aggregation of particles and the self-quenching of fluorescence intensity. At lower concentrations, the insufficient active sites on the nanoparticles prevent the complete adsorption of analytes, a narrow linear range, and low intensity. Similar considerations were also conducted in other solvents (EtOH and MeOH), and the results confirmed maximum PL intensities achieved in the concentrations of 0.3–0.6 g L−1 of CDs@MIP and CDs@NIP composites. Accordingly, 0.4 g L−1 was selected as the optimum value for future investigations.

a Variation of fluorescence intensity of CDs@MIP and CDs@NIP at different concentrations and b effect of rebinding solvent on the fluorescence quenching of CDs@MIP (0.4 g L−1)
Rebinding solvent and mixing time
Usually, solvent properties influence photoluminescence intensity and the binding of the analyte to the imprinted cavities. Therefore, CDs@MIP or CDs@NIP composite was dispersed in three different solvents, and variations in the PL intensity were determined in the presence of FF. The analyte caused a fluorescence quenching, and the maximum differences occurred in EtOH (Fig. 3b). So, ethanol was selected for further studies. The effect of mixing time on the fluorescence quenching of CDs@MIP and CDs@NIP was also studied. The results showed a gradual decrease in PL intensity (or increase in ΔF) with the extension of the mixing time until 8.0 min, which remained nearly constant within 15.0 min (Fig. 4a). It is believed that the fluorescence properties of CDs@MIP (and CDs@NIP) arise from the radiative recombination of electron–hole pairs at the surface of the composite material. In the presence of FF, the functional groups of the template can bind to APTES amino groups, mainly by hydrogen bonding. The new interactions generated at the surface can lead to quenching the intrinsic fluorescence [20].

a Fluorescence quenching of CDs@MIP and CDs@NIP (0.4 g L−1) at the presence of FF (10.0 µmol L−1) and different mixing time and b fluorescence emission of CDs@MIP in different concentrations of FF (3–200 µmol L−1)
Analytical performance and method validation
Under optimized conditions, the as-synthesized CDs@MIP (and CDs@NIP) were directly employed as a fluorescence probe to detect FF. The addition of analyte caused fluorescence quenching of the CDs@MIP (Fig. 4b), and complete quenching was observed with 2.00 × 102 µmol L−1 of FF.
As previously mentioned, the fluorescence quenching can be described by the Stern–Volmer equation. A good linear relationship of (F0/F) against the concentration of FF was observed in the range of 3.00–1.50 × 102 μmol L−1 (Fig. 5), with a correlation coefficient of 0.998 and Stern–Volmer constant of 0.074 for CDs@MIP. For CDs@NIP, the Stern–Volmer plot showed a linear portion in the 1.50 × 101–1.50 × 102 μmol L−1 with a correlation coefficient of 0.995 and Stern–Volmer constant of 0.031. These differences revealed that imprinted cavities at CDs@MIP gave more chance to bind FF. The LOD and LOQ values of 1.11 and 3.69 μmol L−1 were obtained for FF based on 3σ/k (and 10σ/k) criteria which σ is the standard deviation of blank measurements (%RSD = 2.78, n = 7) and k is the slope of the calibration curve. The %RSD values of 4.8 and 2.5 were obtained for three replicate detections of 2.00 × 101 and 4.00 × 101 μmol L−1 FF concentrations, respectively.

Linear calibration curves in the determination of FF by the CDs@MIP and CDs@NIP composites
The analytical performances of the suggested method were compared with some other studies described in the literature. The new sensor showed excellent linearity and low detection limit with satisfying precision and accuracy comparable to or better than other studies (Table 1). On the other hand, the new PL sensor demonstrates the advantages of simplicity and low-cost than complicated chromatographic methods. Moreover, our probe does not contain heavy metal ions and exhibits less toxicity and is eco-friendly with a time-saving and straightforward synthesis process.
Selectivity
To evaluate the ability of the proposed sensor to analyze FF in real samples, the effect of some potentially interfering species was investigated. In this regard, the variation of PL intensity of CDs@MIP was determined in a solution containing FF and an interfering compound. The tolerance limit was defined as the concentration of the added species making a relative error of < 10%. The results (Table 2) showed the most studied species have a slight or negligible quenching effect. This result suggests that the prepared composite has high selectivity toward FF over other chemicals. The selectivity tests were also carried out for the CDs@NIP composite, but the results showed low selectivity of the CDs@NIP versus CDs@MIP.
Analytical applications
The prepared sensor was applied to detect FF in tap water and milk samples by the standard addition method. The recovery studies were carried out by spiking samples with FF in the three different concentrations and showed recoveries in the range of 90.86% to 97.71%. The FF concentration in spiked samples was analyzed by HPLC. As presented in Table 3, the results showed a good correlation between the two methods which confirms the ability of the proposed method to accurately determine FF in real samples.
Conclusion
In this study, fluorescent N–S-doped CDs were prepared through a rapid and straightforward method. The starting mixture was exposed to microwave radiation (2.5 min at 400 W), and the resulting CDs showed blue luminescence without needing any surface passivation. The synthesized N–S-doped CDs were incorporated into an imprinted silica layer. The PL intensity of CDs@MIP showed significant quenching in the presence of the template molecule. The composite was used as a sensor to analyze FF in water and milk samples, and satisfactory results were obtained. In this system, the CDs and FF molecules acted as the electron acceptor and electron donor, respectively. The synthesis method and detection process are straightforward, green, and economical. The sensor integrated the advantages of both CDs and MIPs. It has simple synthesis, bright fluorescence, excellent stability, and low cytotoxicity of CDs, along with the specific recognition ability of MIPs.
Data availability
All data generated or analyzed during this study are included in this manuscript and its supplementary information file.
Code availability
A statement regarding code availability is not applicable.
Abbreviations
- APTES:
-
(3-Aminopropyl)triethoxysilane
- CAP:
-
Chloramphenicol
- CDs:
-
Carbon dots
- CE:
-
Capillary electrophoresis
- EDX:
-
Energy-dispersive X-ray
- ELISA:
-
Enzyme-linked immunosorbent assay
- EtOH:
-
Ethanol
- FF:
-
Florfenicol
- HOAc:
-
Acetic acid
- HPLC:
-
High performance liquid chromatography
- LC–MS:
-
Liquid chromatography tandem–mass spectrometry
- MeOH:
-
Methanol
- MIP:
-
Molecularly imprinted polymer
- MRL:
-
Maximum residual level
- PL:
-
Photoluminescence
- QDs:
-
Quantum dots
- TAC:
-
Tri-ammonium citrate
- TEOS:
-
Tetraethoxysilane
- TAP:
-
Thiamphenicol
References
S. Pilehvar, K. Gielkens, S.A. Trashin, F. Dardenne, R. Blust, K. De Wael, Crit. Rev. Food Sci. Nutr. 56, 2416 (2016)
C. Power, R. Sayers, M. Danaher, M. Moloney, B. O’Brien, A. Furey, K. Jordan, Int. Dairy J. 39, 270 (2014)
C.A. Franje, S.-K. Chang, C.-L. Shyu, J.L. Davis, Y.-W. Lee, R.-J. Lee, C.-C. Chang, C.-C. Chou, J. Pharm. Biomed. Anal 53, 869 (2010)
M. Ansari, M. Raissy, E. Rahimi, Comp. Clin. Pathol. 23, 61 (2014)
A. Nasim, B. Aslam, I. Javed, A. Ali, F. Muhammad, A. Raza, Z.-U.-D. Sindhu, J. Sci. Food Agric. 96, 1284 (2016)
D. Damte, H.-J. Jeong, S.-J. Lee, B.-H. Cho, J.-C. Kim, S.-C. Park, Food Chem. Toxicol. 50, 773 (2012)
S. Shi-Yuan, Y.-K. Wang, Y.-T. Tai, Y.-C. Lei, T.-H. Chang, C.-H. Yao, T.-F. Kuo, J. Immunoass. Immunochem. 34, 438 (2013)
Y. Yikilmaz, A. Filazi, Food Anal. Methods 8, 1027 (2015)
J. Jana, H.J. Lee, J.S. Chung, M.H. Kim, S.H. Hur, Anal. Chim. Acta 1079, 212 (2019)
X. Wang, X. Li, X. Li, Y. Wang, Q. Han, J. Li, Anal. Chim. Acta 1098, 170 (2020)
Y. Yan, J.H. Liu, R.S. Li, Y.F. Li, C.Z. Huang, S.J. Zhen, Anal. Chim. Acta 1063, 144 (2019)
E. Darvishi, Z. Shekarbeygi, S. Yousefinezhad, Z. Izadi, A.A. Saboury, H. Derakhshankhah, B.S. Varnamkhasti, J. Iran. Chem. Soc. 18, 2863 (2021)
N. Amin, A. Afkhami, L. Hosseinzadeh, T. Madrakian, Anal. Chim. Acta 1030, 183 (2018)
M. Roushani, M. Sarabaegi, A. Rostamzad, J. Iran. Chem. Soc. 17, 2407 (2020)
R. Ahmadi, E. Noroozian, A.R. Jassbi, J. Iran. Chem. Soc. 17, 1153 (2020)
E. Iranmanesh, M. Jahani, A. Nezhadali, M. Mojarrab, J. Sep. Sci. 43, 1164 (2020)
N. Kazemifard, A.A. Ensafi, Z.S. Dehkordi, New J. Chem. 45, 10170 (2021)
J. Hou, J. Yan, Q. Zhao, Y. Li, H. Ding, L. Ding, Nanoscale 5, 9558 (2013)
S. Sadeghi, M. Jahani, F. Belador, Spectrochim. Acta, Part A 159, 83 (2016)
R. Jalili, M. Amjadi, RSC Adv. 5, 74084 (2015)
X. Yang, S. Zhu, Y. Dou, Y. Zhuo, Y. Luo, Y. Feng, Talanta 122, 36 (2014)
S. Sadeghi, M. Jahani, Food Anal. Methods 7, 2084 (2014)
Q. Liu, N. Zhang, H. Shi, W. Ji, X. Guo, W. Yuan, Q. Hu, New J. Chem. 42, 3097 (2018)
M.S. Hosseini, F. Belador, Analyst 139, 5007 (2014)
X. Wang, K. Qu, B. Xu, J. Ren, X. Qu, J. Mater. Chem. 21, 2445 (2011)
N. Puvvada, B.N.P. Kumar, S. Konar, H. Kalita, M. Mandal, A. Pathak, Sci. Technol. Adv. Mater. 13, 045008 (2012)
J. Zhou, H. Zhou, J. Tang, S. Deng, F. Yan, W. Li, M. Qu, Microchim. Acta 184, 343 (2017)
M. Zakery, A.A. Ensafi, B. Rezaei, J. Iran. Chem. Soc. 17, 601 (2020)
Y. Xu, H. Li, B. Wang, H. Liu, L. Zhao, T. Zhou, M. Liu, N. Huang, Y. Li, L. Ding, Y. Chen, Microchim. Acta 185, 252 (2018)
H. Zhu, X. Wang, Y. Li, Z. Wang, F. Yang, X. Yang, Chem. Commun. 5118 (2009)
K. Xie, L. Jia, Y. Yao, D. Xu, S. Chen, X. Xie, Y. Pei, W. Bao, G. Dai, J. Wang, Z. Liu, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 879, 2351 (2011)
J. Hou, H. Li, L. Wang, P. Zhang, T. Zhou, H. Ding, L. Ding, Talanta 146, 34 (2016)
E.A. Orlando, A.G. Costa Roque, M.E. Losekann, A.V. Colnaghi Simionato, J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 1035, 8 (2016)
İ. Taşkın, Ö. Güngör, S. Titretir Duran, Polym. Bull. 78, 4721 (2021)
A. Fan, G. Yang, H. Yang, F. Zhao, Mater. Today Commun. 25, 101448 (2020)
X. Li, H. Zhou, L. Wang, H. Wang, A. Adili, J. Li, J. Zhang, J. Food Compos. Anal. 115, 104911 (2023)
Acknowledgements
The authors gratefully acknowledge the Iran National Science Foundation (INSF) for support of this work under project No. 94023433. The authors also thank all the research laboratories in the research institute of food science and technology (RIFST) for the help of chemical analysis.
Funding
The study was funded by Iran National Science Foundation (INSF).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Consent to participate
Not applicable.
Consent for publication
The authors hereby consent to the publication of the work.
Ethical approval
This article does not contain any studies with human or animal subjects.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jahani, M., Feizy, J. Development of a fluorescence sensing assay based on N–S-doped carbon dots and molecularly imprinted polymer for selective and sensitive detection of florfenicol in milk. J IRAN CHEM SOC 20, 485–494 (2023). https://doi.org/10.1007/s13738-022-02684-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-022-02684-8