
Abstract
Purpose of Review
Neutrophilic dermatoses are defined by the presence of a sterile neutrophilic infiltrate on histopathology. This review focuses on the pathogenesis, epidemiology, clinicopathological features, diagnosis, and management of four disorders: Sweet syndrome, pyoderma gangrenosum, Behçet syndrome, and neutrophilic eccrine hidradenitis.
Recent Findings
Recent studies have provided insight into the complex pathogenesis of neutrophilic dermatoses. Evidence supports an intricate interplay of abnormal neutrophil function and inflammasome activation, malignant transformation into dermal infiltrating neutrophils, and genetic predisposition.
Summary
Neutrophilic dermatoses have diverse cutaneous and extracutaneous manifestations and may be associated with significant morbidity and mortality. Common underlying associations include infectious, inflammatory, and neoplastic disorders, as well as drug reactions. Emerging diagnostic and therapeutic frameworks identify an expanding role for biologic and targeted anti-inflammatory therapies.
Similar content being viewed by others

Introduction
Neutrophilic dermatoses (ND) are a group of heterogenous inflammatory disorders united by a sterile neutrophilic infiltrate on histopathology. The cutaneous manifestations of neutrophilic dermatoses are diverse even with several clinical presentations found in the same patient. The location of the neutrophilic infiltrate (epidermal, dermal, and/or subcutaneous), clinical appearance, and disease chronicity help to distinguish each neutrophilic dermatosis. However, modern classification systems recognize that neutrophilic dermatoses can occur on a spectrum. This review showcases four disorders: Sweet syndrome (SS), pyoderma gangrenosum (PG), Behçet syndrome (BS), and neutrophilic eccrine hidradenitis (NEH). Other neutrophilic dermatoses are beyond the scope of this review.
Pathogenesis of Neutrophilic Dermatoses
Neutrophils are the most abundant leukocyte cell type and a critical component of the innate immune system. Originating in the bone marrow, neutrophils are released in the blood stream as terminally differentiated polymorphonuclear cells, morphologically with multi-lobulated nuclei and granular cytoplasm containing lysosomes with acid hydrolases and antimicrobial peptides [1]. Neutrophils quickly mobilize to sites of inflammation and infection. Production and differentiation are primarily regulated by granulocyte colony-stimulating factor (G-CSF). When activated, neutrophils are critical in targeting and destroying microbes through a variety of mechanisms including phagocytosis, production of neutrophil extracellular traps (NETs), and release of antimicrobial substances. Furthermore, neutrophils release cytokines, promoting recruitment of additional innate and adaptive immune cells, thereby providing a positive feedback loop that perpetuates the inflammatory immune response. In normal conditions, phagocytosis of neutrophils and subsequent downregulation of interleukin (IL)-23 and IL-17 serve as a regulatory loop leading to decreased G-CSF production, which suppresses the neutrophilic response [2].
A full understanding of the pathogenesis of neutrophilic dermatoses remains incomplete; however, recent evidence suggests that abnormal neutrophil function and inflammasome activation, malignant transformation into dermal infiltrating neutrophils, and genetic predisposition are important contributors to disease [3, 4]. A variety of inflammatory cell markers and cytokines have been shown to be overexpressed in neutrophilic dermatoses, including CD3, CD163, interferon-γ, IL-1 (α and β), IL-2, IL-6, IL-8, IL-17, myeloperoxidase, and TNF-α [5,6,7]. Autoinflammation is increasingly thought to play a role in the pathogenesis of neutrophilic dermatoses [8]. Important animal models have characterized pathologic variants in tyrosine-protein phosphatase non-receptor type 6 (PTPN6) and have led to the identification of the role of inflammatory mediators: CARD9, IL-1α, MAP3K5, and MAP3K7 [9, 10].
Other investigations have shown evidence of neutrophilic clonality in patients with underlying myeloid malignancies in which skin biopsy specimens demonstrate evidence of neutrophils differentiating from the underlying malignant clone [11, 12, 13•]. Differentiation from the malignant clone can also be induced by drugs including all-trans retinoic acid (ATRA) and fms-like tyrosine kinase 3 (FLT-3) inhibitors [14,15,16,17].
Lastly, widespread availability of genomic data has led to discoveries on the role of genetics in the pathogenesis of NDs and other inflammatory diseases. Human leukocyte antigen (HLA)-B51 and HLA-B54 have been linked to BS and SS respectively [3, 18]. Pathologic variants in a classic autoinflammatory gene, proline-serine-threonine phosphatase-interacting protein 1 (PSTPIP1), leading to hyperactivation of the inflammasome, has been identified in patients with syndromic forms of PG [10]. More recently, peripheral blood exome sequencing identified a mutation in UBA1, a gene encoding the ubiquitin-activating enzyme 1, in a severe adult-onset inflammatory disorder coined VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome. This disorder is characterized by recurrent fevers, pulmonary symptoms, and dermatologic manifestations, including neutrophilic dermatoses [19•, 20].
Sweet Syndrome
Epidemiology
First described as “acute febrile neutrophilic dermatosis” by Dr. Robert Douglas Sweet in 1964, SS is a prototypical neutrophilic dermatosis [21]. In his original report, Dr. Sweet described acute onset of fever, leukocytosis, and painful edematous plaques in eight middle-aged women; histopathology of the lesions showed a dermal neutrophilic infiltrate. The eponym “Sweet syndrome” was termed by Whittle and colleagues in 1968 [22]. SS can occur at any age, but the average age of onset is between 30 and 60 years, with a female predominance [23]. Given the association with a broad range of comorbid conditions, many authors group SS into three main subtypes: classic (idiopathic), malignancy-associated, and drug-induced.
Classic SS encompasses idiopathic SS and SS associated with infections, vaccinations, inflammatory disorders, and pregnancy. Upper respiratory infections are most commonly cited as an infectious trigger; however, a range of bacterial, viral, and fungal infections have been implicated [24,25,26,27,28,29,30,31]. Since 2019, SS has been reported following coronavirus disease 2019 (COVID-19) and rarely following vaccination with the Oxford-AstraZeneca, Moderna, and Pfizer-BioNTech vaccines [32•, 33,34,35,36]. Inflammatory conditions like inflammatory bowel disease (IBD), systemic lupus erythematous (SLE), and other rheumatologic diseases are associated with the development of classic SS [37]. Other states of immune dysregulation have been linked to SS, including human immunodeficiency virus infection and common variable immunodeficiency [28, 38, 39]. A recent retrospective multicenter study of SS and PG in pediatric patients found that, when compared to adults, pediatric patients more frequently have extracutaneous involvement, a broader range of associated general conditions, and genetic autoinflammatory syndromes [40••]. Malignancy-associated SS is more commonly reported with hematologic malignancies and myeloproliferative or myelodysplastic disorders compared to solid-organ malignancies. The most common associations are AML followed by MDS. SS may precede or follow a diagnosis of malignancy. SS may also signal cancer recurrence [37]. While most commonly associated with colony-stimulating factors (e.g., G-CSF), drug-induced SS has been reported to a variety of medications. Notable cancer therapies include ATRA, FLT-3 inhibitors, immune checkpoint inhibitors, and tyrosine kinase inhibitors [3, 14, 16, 17, 41,42,43,44,45].
Clinicopathologic Features
SS (Fig. 1) is characterized by rapidly developing, tender erythematous edematous papules or plaques in a febrile patient; however, the clinical phenotype can vary widely. Variants of SS include pustular SS, bullous SS, cellulitis-like SS, necrotizing SS, subcutaneous SS, and neutrophilic dermatosis of the dorsal hands. SS can present as a single lesion or multiple lesions, often in an asymmetric distribution, most frequently involving the upper extremities, neck, and face [46]. Involvement of the oral cavity and mucosa is rare, occurring in 2% of classic SS cases and 12% of malignancy-associated SS cases, with a higher frequency in hematologic as compared to solid malignancies [37]. Pathergy can be seen in up to 25% of cases and can be a helpful clinical clue [3]. Fever is the most common associated symptom, sometimes preceding the cutaneous findings [37]. Patients can be ill-appearing with other associated constitutional symptoms including arthralgia, fatigue, headache, and myalgia [46]. Neutrophilic infiltration can rarely involve ocular, pulmonary, cardiac, gastrointestinal, and neurologic systems and can lead to significant morbidity and mortality [3]. Clinical features that more commonly occur in patients with malignancy-associated SS include advanced age, vesiculobullous variant, absence of arthralgia, leukopenia, anemia, thrombocytopenia, and elevated erythrocyte sedimentation rate (ESR) [3, 47,48,49,50,51].

Sweet syndrome. A Edematous pink papules and plaques on the thigh. B Papillary dermal edema and an underlying dense infiltrate of mature neutrophils (40×, hematoxylin and eosin staining). C Higher magnification of mature neutrophils (400×, hematoxylin and eosin staining)
Unifying the diverse clinical presentations of SS are the typical histological findings of a dense dermal infiltrate predominately composed of mature neutrophils and papillary dermal edema. Secondary vascular injury and leukemic cells are variable [52]. Infectious stains and cultures are negative. Pathologic variants include cryptococcoid SS, histiocytoid SS, lymphocytic SS, subcutaneous SS, and xanthomatized SS [53,54,55,56,57]. Histiocytoid Sweet syndrome (HSS) demonstrates histiocytoid mononuclear immature myeloid cells. A recent systematic review found that HSS had a higher risk of underlying hematologic malignancy, especially in patients with older age at disease onset and systemic symptoms [58]. This has been corroborated in some studies [47, 59,60,61] but refuted by others [50, 55, 62].
Diagnosis
The diagnosis of SS is based on clinical and laboratory findings after excluding alternative infectious, inflammatory, and neoplastic disorders. Careful attention should be paid to any history of infectious symptoms, vaccinations, inflammatory disorders, pregnancy, malignancy, or drug exposures. Additionally, the patient should be examined for pathergy, especially at sites of skin manipulation or venipuncture sites, and counseled on the risk of pathergy development in the future [47]. Complete blood count (CBC) with differential, C-reactive protein (CRP), and ESR are commonly evaluated. As SS is a diagnosis of exclusion, patients with suspected SS should receive a skin biopsy and sterile tissue culture for bacterial, fungal, and mycobacterial organisms to exclude infection.
Table 1 outlines the diagnostic criteria for SS, originally proposed by Su and Liu in 1986 and subsequently updated by Moschella and Davis [23, 63]. Walker and Cohen developed distinct diagnostic criteria for drug-induced SS, which include the temporal relationship between drug exposure and development of SS and subsequent resolution with drug withdrawal [64].
When SS is diagnosed, the presence of underlying associated diseases should be considered. Universally accepted guidelines for additional work-up have not been developed; however, all patients should be offered age-appropriate cancer screening. Additional testing for pregnancy, IBD, and autoimmune conditions is reasonable depending on the clinical context. Given the association with hematologic disorders, a low threshold for malignancy evaluation including peripheral blood smear, assessment for paraproteinemia, and bone marrow biopsy may be warranted [3].
Management
Treatment of any underlying disorder or malignancy is crucial; however, SS-directed therapy is often needed. Systemic corticosteroids are first line. Usually, prednisone 0.5 to 1 mg/kg/day is started with rapid improvement of symptoms, followed by a slow taper over 4–6 weeks. For limited disease, topical or intralesional corticosteroids can be used; however, a theoretical risk of pathergy at the injection site should be discussed prior to the injection. For severe or refractory disease, pulse dosed methylprednisolone daily for up to 5 days may be indicated initially. Potassium iodide (900 mg/day), colchicine (1.5 mg/day), and dapsone (100–200 mg/day) are common steroid-sparing agents [3, 65, 66•]. Tumor necrosis factor alpha (TNF-α) inhibitors are effective and increasingly utilized in IBD-associated SS or rheumatologic-associated SS [67, 68]. A recent multicenter retrospective study of ustekinumab for Crohn disease (CD) associated neutrophilic dermatoses demonstrated remission in 6 out of 7 patients treated and is promising given its dual efficacy [69••]. Treatment with anakinra, an IL-1 receptor antagonist, has also been reported for refractory SS [70]. Drug-induced and pregnancy-induced SS can resolve after the corresponding trigger is removed [71]. Unfortunately, Sweet syndrome and other neutrophilic dermatoses are lacking in high-quality evidence and treatment should be tailored to the individual patient.
Pyoderma Gangrenosum
Epidemiology
PG was first described by Brocq in 1916 and named by Brunsting, Goeckerman, and O’Leary in 1930 [72, 73]. A recent United States (US) cross-sectional analysis reported a prevalence of 5.8 cases per 100,000 adults, with a female predominance and an adjusted prevalence in women almost twice that of men [74••]. Fifty to sixty-seven percent of cases are associated with systemic diseases, most commonly IBD, arthritis, and hematologic cancers [75,76,77]. Drug-induced PG has been reported to a variety of medications including colony-stimulating factors, levamisole tainted cocaine, and a multitude of immunomodulating agents [78]. In addition to increased morbidity from the painful PG wounds, a population-based study in the UK demonstrated a threefold increase in mortality compared to age- and sex-matched controls [79]. Recent US data on the inpatient burden of PG identified female sex, black race/ethnicity, and multiple chronic conditions as predictors of PG-associated hospitalizations in both adults fand children [80••, 81]. Moreover, PG was the leading etiology of ulcers seen by US inpatient consultative dermatology services [82].
Clinicopathologic Features
The five clinical subtypes of PG (Fig. 2) are ulcerative, bullous, vegetative, pustular, and peristomal. Ulcerative PG is the most common and classically presents with a tender nodule or pustule that rapidly expands into a well-defined, violaceous ulcer with an undermined, inflammatory erythematous border and an exudative, fibrinous base. Lesions can develop anywhere on the body and exhibit pathergy. PG can present as single or multiple lesions and can be exquisitely painful. Lesions may heal with cribriform scarring. Bullous PG is a controversial entity (variably classified as a form of SS) characterized by rapidly developing blue-gray bullae that degrade into superficial ulcerations. Compared to ulcerative PG, bullous PG has a predilection for the face and upper extremities and is more frequently associated with malignancy. Vegetative (superficial granulomatous) PG is characterized by a single superficial ulcer with verrucous features and lacks the classic violaceous, undermined border of ulcerative PG. On histopathology, pseudoepitheliomatous hyperplasia with layered neutrophils, histiocytes, and plasma cells is seen [83]. Pustular PG tends to present with multiple painful pustules with a surrounding erythematous halos [76]. Finally, peristomal PG occurs near a stoma site as a painful papule that rapidly evolves into a classic PG ulcer. Peristomal PG is thought to be a pathergic response to trauma from surgery or ostomy care [3]. Rarely, PG can affect extracutaneous sites and has been reported in the eyes, lungs, spleen, and musculoskeletal system [84•]. Recent studies have shed light on the association between PG and depression, pain, and significant negative impact on quality of life [85,86,87].

Pyoderma gangrenosum. A Irregular slate-gray ulcer with central bullae with an expanding violaceous to red border on the lower leg. The border may not be obviously violaceous in darker skin types. B Neutrophils predominate in the dermis below the ulceration (100×, hematoxylin and eosin staining). C The border of the ulcer shows a neutrophilic infiltrate in the dermis (400×, hematoxylin and eosin staining)
Diagnosis
Definitive diagnosis of PG can be challenging, with controversy over its status as a diagnosis of exclusion. There have been three proposed diagnostic frameworks to help aid the diagnosis: Su criteria, Delphi consensus criteria for ulcerative PG, and the PARACELSUS score (Table 2) [88,89,90]. Notable variations in these frameworks include the approach to ruling out alternative diagnoses and the importance of requiring a neutrophilic infiltrate on histopathology. A recent comparative study found that the PARACELSUS score identified the highest proportion of patients in a PG cohort; however, further research would be valuable [91••]. The diagnostic work-up may include skin biopsy and sterile tissue culture from the ulcer edge. A proposed age-focused initial evaluation for PG is summarized in Table 3 [75]. As venous insufficiency and peripheral artery disease are common culprits for lower-extremity ulcers, duplex ultrasonography and ankle-brachial index testing can be helpful to distinguish vascular etiologies from PG and to address any contribution of underlying vascular disease to delayed wound healing.
Management
Treatment of PG should first focus on halting the neutrophilic inflammation then followed by wound healing. This section will focus on stopping neutrophilic inflammation using a combination of topical and systemic therapies. For localized or mild disease, high-potency topical corticosteroids, intralesional corticosteroids, topical calcineurin inhibitors, and topical dapsone can be used [75, 92]. Given the theoretical risk of pathergy, intralesional steroids should be injected into the ulcer, toward the active edge, rather than into intact skin. Multiple systemic therapies have reported efficacy for PG. A 2018 systematic review found that systemic corticosteroids, cyclosporine, infliximab, and canakinumab had the strongest level of evidence for treating PG [93]. Systemic corticosteroid, typically prednisone 0.5 to 2.0 mg/kg/day, characteristically leads to rapid stabilization of lesions. In aggressive and severe cases, higher doses of systemic steroids or pulse dosing of methylprednisolone may be used. In the 2015 STOP GAP trial, systemic cyclosporine 4 mg/kg/day was shown to have a similar healing time and rate of recurrence compared to oral prednisolone 0.75 mg/kg/day, with fewer serious adverse reactions [94]. TNF-α inhibitors, supported by a randomized controlled trial of infliximab, have been increasingly used [95]. A 2021 study of 64 patients from Denmark found that patients treated with systemic corticosteroids had a higher mortality rate and TNF-α inhibitors had shortest time to PG remission [96]. TNF-α inhibitors can also treat co-existing underlying inflammatory conditions (e.g., IBD, arthritis).
Response to therapy is signaled by improved pain, edema, and erythema at the ulcer edges, correlating with resolution of the underlying neutrophilic inflammation. A residual wound may take additional weeks to months to heal, and treatment should be tapered cautiously prior to complete wound healing. Local wound care is imperative in PG treatment; however, it is beyond the scope of this review. For patients with active PG undergoing surgical procedures, prophylactic immunosuppression can be considered; however, there is a lack of consensus on the type and duration of therapy [97, 98].
Behçet Syndrome
Epidemiology
Traditionally classified as a neutrophilic disorder, BS is more common along the ancient Silk Road, from eastern Asia to the Mediterranean, with the highest prevalence in Turkey (80 to 370 cases per 100,000) [99]. It is less commonly found in the US and Europe, with an estimated prevalence ranging from 0.12 to 14.61 per 100,000 [100, 101]. BS typically presents in the 3rd-4th decade of life [3]. Endemic areas have an equal gender distribution; however, in the US and Europe, there is a female predominance. Earlier age of onset is associated with more severe disease. Additionally, men, patients from Middle or Far Eastern Asia, and patients with multi-organ involvement tend to have a more complicated and severe disease course. Similar to other inflammatory diseases, BS has been recently associated with an increased risk of cardiovascular disease, thromboembolic disease, and mortality [101]. Recent studies have also implicated an overall increased risk of malignancy [102–104].
Clinicopathologic Features
BS (Fig. 3) is a chronic, relapsing multisystem inflammatory syndrome characterized by recurrent mucocutaneous ulcers with a variety of heterogeneous systemic manifestations involving ocular, neurological, gastrointestinal, rheumatological, and vascular systems. The most common clinical feature of BS is recurrent painful mucocutaneous ulcerations. Patients tend to present first with painful, recurrent oral aphthous ulcerations, ranging in size from a few millimeters to centimeters, most commonly on the lips, buccal mucosa, tongue, and soft palate. Lesions start as erythematous papules or pinpoint pustules and progress to round-to-oval ulcers with rolled borders and a white-yellow necrotic base with surrounding erythema. Individual ulcers tend to heal spontaneously without scarring within 1 to 3 weeks; however, patients may have continued outbreaks that severely impact eating, drinking, and speaking [3]. Oral aphthosis may precede other manifestations of BS and are indistinguishable from those seen in complex aphthosis or IBD. Genital ulcerations most commonly arise on the vulva in women or scrotum and penis in men and are the most specific finding for BS [105]. Compared to oral lesions, anogenital aphthae tend to be larger, with irregular margins, and heal with scarring [23]. Other cutaneous manifestations include non-follicular papulopustular eruptions, erythema nodosum-like lesions, superficial thrombophlebitis, PG-like lesions, and cutaneous pathergy. In patients with coexistent relapsing polychondritis, mouth and genital ulcers with inflamed cartilage (MAGIC) syndrome can be considered [106].

Behçet syndrome. A Oral aphthous ulcer. B Histopathologic findings can show an acute neutrophilic infiltrate, as with any ulcer (40×, hematoxylin and eosin staining)
Most extracutaneous findings of BS are thought to arise from a systemic vasculitis, with involvement of both the arterial and venous circulation and vessels of all sizes (small, medium, and large). The multisystem manifestations of BS vary widely and can affect every organ system. Specifically, ocular disease is commonly seen, usually presenting with uveitis and retinal vasculitis. Significant morbidity and mortality arise from vascular involvement, especially of the gastrointestinal tract, potentially leading to bowel perforation [3]. Aneurysm of the large proximal pulmonary artery branches is relatively specific to BS and carries a mortality of 25% [107].
Oral and genital ulcers show nonspecific histopathologic findings. Neutrophils may be prominent, as can be true for any acutely inflamed ulcer. Some authors have proposed that more specific findings in about 50% of BS patients include venulitis, either leukocytoclastic or lymphocytic [108].
Diagnosis
Diagnosis of BS is based on the constellation of clinical findings, as histologic and laboratory tests are nonspecific. More than a dozen diagnostic criteria have been proposed. The two most widely accepted diagnostic criteria are compared in Table 4 [109, 110]. It is important to rule out infectious mimickers, specifically viral etiologies like herpetic ulcerations or reactive infectious mucocutaneous eruption.
Management
Given its chronic, waxing and waning, heterogeneous disease course, the treatment of BS should be tailored to the most significant disease manifestation, as well as individual patient characteristics, most importantly patient age and sex. The treatment goal is to ameliorate any negative effects on a patient’s quality of life and to suppress inflammation that could lead to irreversible damage. In 2018, the European League Against Rheumatism (EULAR) updated recommendations for the management of BS [111••]. This review will focus on treatment of the mucocutaneous manifestations; however, a multidisciplinary approach is often necessary for management of multisystem involvement.
For oral and genital ulcerations, topical steroids used in conjunction with supportive measures such as topical sucralfate and topical anesthetics are first line [112]. Intralesional corticosteroids can also be considered for localized disease, although the theoretical risk of pathergy should be discussed. Topical pimecrolimus has been shown to improve healing time in genital ulcerations [113]. Colchicine is first-line therapy for recurrent oral and genital ulcerations and other cutaneous lesions, including erythema nodosum [111••]. Topical and systemic therapies for acne vulgaris can be trialed for the acne-like eruptions of BS [111••]. Leg ulcerations require special attention, often necessitating involvement of both dermatology and vascular surgery. Treatment should be directed toward the underlying causes of ulceration (e.g., venous stasis, vasculitis, PG-like lesions). BS severity may improve with time, and patients can potentially taper off medications.
For severe and refractory mucocutaneous lesions, systemic immunomodulatory agents such as corticosteroids, azathioprine, thalidomide, and TNF-α inhibitors can be used [111••, 114, 115]. A 2019 phase 3, randomized control trial of apremilast, an oral phosphodiesterase-4 inhibitor, showed statistically significant reduction in the number of oral ulcers compared to placebo [116••]. A recent systematic review showed control of mucocutaneous and ocular BS using the IL-1 blocking agents anakinra and canakinumab [117]. TNF-alpha, IL-17, and IL 12 and 23 inhibitors are being used with increasing frequency for severe and refractory cases [118–121]. IL-6 blockade with tocilizumab is controversial and has been reported to worsen mucocutaneous lesions [122, 123].
Neutrophilic Eccrine Hidradenitis
Epidemiology
NEH is a self-limited disorder of the eccrine sweat glands. It was originally described in 1982 by Harrist et al. [124]. in a patient with AML treated with cytarabine and doxorubicin who developed tender edematous plaques shortly after initiation of therapy. Since its original report, it has been described in a variety of contexts, including drug-induced (both antineoplastic and non-antineoplastic), in association with malignancy or altered immunity, and in healthy young children without a known trigger. In the absence of chemotherapies or other medications, new-onset NEH could indicate underlying malignancy or its relapse [125].
The pathogenesis of NEH is uncertain; however, it is thought to be due to direct cytotoxicity of the eccrine sweat glands and generation of toxic byproducts which stimulate neutrophilic inflammation [126]. The most frequently implicated antineoplastic agents include antimitotic agents, followed by anthracyclines, BRAF inhibitors, the epidermal growth factor receptor (EGFR) inhibitor cetuximab, and the tyrosine kinase inhibitor imatinib [3]. Other associated medications include antipyretics (acetaminophen), TNF inhibitors (adalimumab), antiretrovirals, immunosuppressants (azathioprine), and anticonvulsants (carbamazepine) [127]. Recently, NEH has been reported with infliximab, ticagrelor, and pegfilgrastim [128–130]. In healthy children, NEH is an idiopathic benign, self-limited eruption that tends to occur in summer months called idiopathic plantar hidradenitis (IPH) [131]. In these cases, it is hypothesized that the combination of trauma and high temperatures provoke sweat gland rupture leading to an inflammatory cascade and neutrophilic recruitment [132].
The name NEH reflects its historical classification as a neutrophilic dermatosis. Some authors argue that NEH may be better classified under the rubric of toxic erythema of chemotherapy.
Clinicopathologic Features
Classically, NEH (Fig. 4) presents with erythematous edematous papules or plaques most commonly on the trunk, but it can also involve the extremities or face. NEH clinical presentations are polymorphic, including linear or annular edematous plaques, erythema multiforme-like lesions, or even purpuric or pustular eruptions. The average latency time to presentation following chemotherapy exposure is 10 days, often associated with fever and neutropenia in the setting of chemotherapy [127]. IPH presents as an eruption of urticaria-like erythematous papules, plaques, and occasionally nodules. It is usually provoked by physical activity, wet footwear, and friction, and is self-limited, resolving within days to weeks [133].

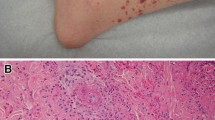
Neutrophilic eccrine hidradenitis. A Erythematous macules and papules on the sole. B In this case associated with chemotherapy, there is vacuolar change and peri-eccrine inflammation (40×, hematoxylin and eosin staining). C Higher magnification shows a mixed infiltrate with scattered neutrophils and squamous metaplasia of the eccrine coils (400×, hematoxylin and eosin staining)
Classic histologic findings include peri-eccrine neutrophilic inflammation as well as alteration of eccrine gland epithelium, including vacuolar change, pyknosis, and/or dyskeratosis. Intraluminal neutrophilic abscesses may also be identified. Less commonly, apocrine glands have also been reported to be involved. Over time, the diagnosis of NEH has evolved, and a prominent peri-eccrine neutrophilic infiltrate is not required for the diagnosis [134]. IPH can be identical to NEH on histopathology; however, some authors consider it a distinct entity given its characteristic clinical presentation and localization of neutrophilic infiltrate to dermal eccrine coils.
Diagnosis
Given the spectrum of clinical presentations, the diagnosis of NEH is dependent on histopathology. It is essential to rule out infection; thus, infectious stains and sterile tissue culture should be negative [127].
Management
NEH is self-limited, with non-scarring spontaneous resolution within days to weeks. Descriptive studies have shown improved fever and pain and possible reduced duration of lesions with nonsteroidal anti-inflammatory drugs and systemic corticosteroids [3]. Chemotherapy-induced NEH can recur with subsequent cycles, and dapsone has been used prophylactically to prevent recurrence [135].
Conclusions
In summary, neutrophilic dermatoses are a heterogenous group of inflammatory disorders defined by a sterile neutrophilic infiltrate on histopathology. Neutrophilic dermatoses have diverse cutaneous and extracutaneous manifestations and may be associated with significant morbidity and mortality. Common associations include infectious, inflammatory, and neoplastic disorders as well as drugs. Scientific research has continued to unravel the complex pathogenesis of neutrophilic dermatoses involving abnormal neutrophil function and inflammasome activation, malignant transformation into dermal infiltrating neutrophils, and genetic predisposition. As new evidence emerges, targeted novel therapies for neutrophilic dermatoses are on the horizon.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Bainton DF, Ullyot JL, Farquhar MG. The development of neutrophilic polymorphonuclear leukocytes in human bone marrow. J Exp Med. 1971;134(4):907–34.
Marzano AV, Ortega-Loayza AG, Heath M, Morse D, Genovese G, Cugno M. Mechanisms of inflammation in neutrophil-mediated skin diseases. Front Immunol. 2019;10:1059.
Nelson CA, Stephen S, Ashchyan HJ, James WD, Micheletti RG, Rosenbach M. Neutrophilic dermatoses. J Am Acad Dermatol. 2018;79(6):987–1006.
Heath MS, Ortega-Loayza AG. Insights into the pathogenesis of Sweet’s syndrome. Front Immunol. 2019;10:414.
Marzano AV, Cugno M, Trevisan V, Fanoni D, Venegoni L, Berti E, et al. Role of inflammatory cells, cytokines and matrix metalloproteinases in neutrophil-mediated skin diseases. Clin Exp Immunol. 2010;162(1):100–7.
Giasuddin AS, El-Orfi AH, Ziu MM, El-Barnawi NY. Sweet’s syndrome: is the pathogenesis mediated by helper T cell type 1 cytokines? J Am Acad Dermatol. 1998;39(6):940–3.
Reuss-Borst MA, Pawelec G, Saal JG, Horny HP, Müller CA, Waller HD. Sweet’s syndrome associated with myelodysplasia: possible role of cytokines in the pathogenesis of the disease. Br J Haematol. 1993;84(2):356–8.
Satoh TK, Mellett M, Contassot E, French LE. Are neutrophilic dermatoses autoinflammatory disorders? Br J Dermatol. 2018;178(3):603–13.
Tartey S, Gurung P, Samir P, Burton A, Kanneganti T-D. Cutting edge: dysregulated CARD9 signaling in neutrophils drives inflammation in a mouse model of neutrophilic dermatoses. J Immunol. 2018;201(6):1639–44.
Maverakis E, Marzano AV, Le ST, Callen JP, Brüggen M-C, Guenova E, et al. Pyoderma gangrenosum Nat Rev Dis Primers. 2020;6:81.
Magro CM, Kiani B, Li J, Crowson AN. Clonality in the setting of Sweet’s syndrome and pyoderma gangrenosum is not limited to underlying myeloproliferative disease. J Cutan Pathol. 2007;34(7):526–34.
Sujobert P, Cuccuini W, Vignon-Pennamen D, Martin-Garcia N, Albertini AF, Uzunov M, et al. Evidence of differentiation in myeloid malignancies associated neutrophilic dermatosis: a fluorescent in situ hybridization study of 14 patients. J Investig Dermatol. 2013;133(4):1111–4.
• Passet M, Lepelletier C, Vignon-Pennamen M-D, Chasset F, Hirsch P, Battistella M, et al. Next-generation sequencing in myeloid neoplasm-associated Sweet’s syndrome demonstrates clonal relation between malignant cells and skin-infiltrating neutrophils. Journal of Investigative Dermatology. 2020;140(9):1873–6. J invest Derm article using next-generation sequencing to demonstrate a common clonal progenitor relationship between malignant myeloid clone and dermal infiltrating PMNs.
Piette WW, Trapp JF, O’Donnell MJ, Argenyi Z, Talbot EA, Burns CP. Acute neutrophilic dermatosis with myeloblastic infiltrate in a leukemia patient receiving all-trans-retnoic acid therapy. J Am Acad Dermatol. 1994;30(2):293–7.
Mo W, Wang X, Wang Y, Li Y, Zhang R. Clonal neutrophil infiltrates in concurrent Sweet’s syndrome and acute myeloid leukemia: a case report and literature review. Cancer Genet. 2018;226–227:11–6.
Fathi AT, Le L, Hasserjian RP, Sadrzadeh H, Levis M, Chen Y-B. FLT3 inhibitor-induced neutrophilic dermatosis. Blood. 2013;122(2):239–42.
Varadarajan N, Boni A, Elder DE, Bagg A, Micheletti R, Perl AE, et al. FLT3 inhibitor–associated neutrophilic dermatoses. JAMA Dermatol. 2016;152(4):480.
Mizoguchi M. Human leukocyte antigen in Sweet’s syndrome and its relationship to Behçet’s disease. Arch Dermatol. 1988;124(7):1069.
• Beck DB, Ferrada MA, Sikora KA, Ombrello AK, Collins JC, Pei W, et al. Somatic mutations in UBA1 and severe adult-onset autoinflammatory disease. N Engl J Med. 2020;383(27):2628–38. Using a genotype-driven approach, the authors identified a disorder that connects seemingly unrelated adult-onset inflammatory syndromes, named VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome.
Zakine E, Schell B, Battistella M, Vignon-Pennamen M-D, Chasset F, Mahévas T, et al. UBA1 variations in neutrophilic dermatosis skin lesions of patients with VEXAS Syndrome. JAMA Dermatol. 2021;157(11):1349–54.
Sweet RB. An acute febrile neutrophtlic dermatosts. Br J Dermatol. 1964;76(8–9):349–56.
Whittle CH, Beck GA, Champion RH. Recurrent neutrophilic dermatosis of the face—a variant of Sweet’s syndrome. Br J Dermatol. 1968;80(12):806–10.
Moschella SL. Neutrophilic dermatoses. In: Bolognia JL, Jorizzo JL, Schaffer JV, editors. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012. p. 423–38.
Amouri M, Masmoudi A, Ammar M, Boudaya S, Khabir A, Boudawara T, et al. Sweet’s syndrome: a retrospective study of 90 cases from a tertiary care center. Int J Dermatol. 2016;55(9):1033–9.
Sulk M, Ehrchen J. Sweet syndrome in association with enterobiasis. J Dermatol. 2019;46(3):e106–7.
Vithoosan S, Thanushah B, Piranavan P, Gamlaksha D, Karunatilake H, Jayanaga A. A rare case of Sweet syndrome secondary to melioidosis. BMC Dermatol. 2019;19:16.
Yan G, Tan KB, Chandran NS, Chai L, Chew KL, Somani J, et al. Histoplasmosis presenting with Sweet’s syndrome. Clin Microbiol Infect. 2020;26(6):795–6.
Mudroch SM, Rohan C, Conger NG, Lindholm DA. Sweet syndrome in an elderly man with well-controlled human immunodeficiency virus. Cureus. 2020;12(9):e10330.
De Lima ÍMF, Ferraz CE, Gonçalves De Lima-Neto R, Takano DM. Case report: Sweet syndrome in patients with sporotrichosis: a 10-case series. Am J Trop Med Hyg. 2020;103(6):2533–8.
Rochael MC, Pantaleão L, Vilar EAG, Zacaron LH, Spada EQ, Xavier MHSB, et al. Síndrome de Sweet: estudo de 73 casos, com ênfase nos achados histopatológicos. An Bras Dermatol. 2011;86(4):702–7.
Rochet NM, Chavan RN, Cappel MA, Wada DA, Gibson LE. Sweet syndrome: clinical presentation, associations, and response to treatment in 77 patients. J Am Acad Dermatol. 2013;69(4):557–64.
• Taşkın B, Vural S, Altuğ E, Demirkesen C, Kocatürk E, Çelebi İ, et al. Coronavirus 19 presenting with atypical Sweet’s syndrome. J Eur Acad Dermatol Venereol. 2020;34(10). First reported case of SS following COVID-19 infection.
Majid I, Mearaj S. Sweet syndrome after Oxford-AstraZeneca COVID-19 vaccine (AZD1222) in an elderly female. Dermatol Ther. 2021;34(6):e15146.
Capassoni M, Ketabchi S, Cassisa A, Caramelli R, Molinu AA, Galluccio F, et al. AstraZeneca (AZD1222) COVID-19 vaccine-associated adverse drug event: a case report. J Med Virol. 2021;93(10):5718–20.
Torrealba-Acosta G, Martin JC, Huttenbach Y, Garcia CR, Sohail MR, Agarwal SK, et al. Acute encephalitis, myoclonus and Sweet syndrome after mRNA-1273 vaccine. BMJ Case Rep. 2021;14(7):e243173.
Darrigade AS, Théophile H, Sanchez-Pena P, Milpied B, Colbert M, Pedeboscq S, et al. Sweet syndrome induced by SARS-CoV-2 Pfizer-BioNTech mRNA vaccine. Allergy. 2021;76(10):3194–6.
Cohen PR. Sweet’s syndrome – a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2(1):34.
Cook QS, Zdanski CJ, Burkhart CN, Googe PB, Thompson P, Wu EY. Idiopathic, refractory Sweet’s syndrome associated with common variable immunodeficiency: a case report and literature review. Curr Allergy Asthma Rep. 2019;19:32.
Berger TG, Dhar A, McCalmont TH. Neutrophilic dermatoses in HIV infection. J Am Acad Dermatol. 1994;31(6):1045–7.
•• Bucchia M, Barbarot S, Reumaux H, Piram M, Mahe E, Mallet S, et al. Age‐specific characteristics of neutrophilic dermatoses and neutrophilic diseases in children. J Eur Acad Dermatol Venereol. 2019;33(11):2179–87. A retrospective multi-center study of SS and pyoderma gangrenosum in pediatric patients found that when compared to adults, ND in pediatric patients may be more associated with extracutaneous neutrophilic infiltration of other organ systems and a broad range of associated general conditions, and genetic autoinflammatory syndromes.
Park JW, Mehrotra B, Barnett BO, Baron AD, Venook AP. The Sweet syndrome during therapy with granulocyte colony-stimulating factor. Ann Intern Med. 1992;116(12 Pt 1):996–8.
Ravi V, Maloney NJ, Worswick S. Neutrophilic dermatoses as adverse effects of checkpoint inhibitors: a review. Dermatol Ther. 2019;32(5):e13074.
Apalla Z, Papageorgiou C, Lallas A, Delli F, Fotiadou C, Kemanetzi C, et al. Cutaneous adverse events of immune checkpoint inhibitors: a literature review. Dermatol Pract Concept. 2021;11:e2021155.
Yang JJ, Maloney NJ, Nguyen KA, Worswick S, Smogorzewski J, Bach DQ. Sweet syndrome as an adverse reaction to tyrosine kinase inhibitors: a review. Dermatol Ther. 2021;34(1):e14461.
Cox NH, O’Brien HAW. Sweet’s syndrome associated with trans-retinoic acid treatment in acute promyelocytic leukemia. Clin Exp Dermatol. 1994;19(1):51–2.
Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42(10):761–78.
Nelson CA, Noe MH, McMahon CM, Gowda A, Wu B, Ashchyan HJ, et al. Sweet syndrome in patients with and without malignancy: a retrospective analysis of 83 patients from a tertiary academic referral center. J Am Acad Dermatol. 2018;78(2):303–9.
Marcoval J, Martín-Callizo C, Valentí-Medina F, Bonfill-Ortí M, Martínez-Molina L. Sweet syndrome: long-term follow-up of 138 patients. Clin Exp Dermatol. 2016;41(7):741–6.
Bourke JF, Keohane S, Long CC, Kemmett D, Davies M, Zaki I, et al. Sweet’s syndrome and malignancy in the U.K. Br J Dermatol. 1997;137(4):609–13.
Zheng S, Li S, Tang S, Pan Y, Ding Y, Qiao J, et al. Insights into the characteristics of Sweet syndrome in patients with and without hematologic malignancy. Front Med. 2020;7:20.
El-Khalawany M, Aboeldahab S, Mosbeh A-S, Thabet A. Clinicopathologic, immunophenotyping and cytogenetic analysis of Sweet syndrome in Egyptian patients with acute myeloid leukemia. Pathol Res Pract. 2017;213(2):143–53.
Ratzinger G, Burgdorf W, Zelger BG, Zelger B. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29(2):125–33.
Kamimura A, Yanagisawa H, Tsunemi Y, Kusano T, Arai E, Tsuchida T, et al. Normolipemic xanthomatized Sweet’s syndrome: a variant of Sweet’s syndrome with myelodysplastic syndrome. J Dermatol. 2021;48(5):695–8.
Ferris GJ, Fabbro S, Gru A, Kaffenberger J. Xanthomatized neutrophilic dermatosis in a patient with myelodysplastic syndrome. Am J Dermatopathol. 2017;39(5):384–7.
Alegría-Landa V, Rodríguez-Pinilla SM, Santos-Briz A, Rodríguez-Peralto JL, Alegre V, Cerroni L, et al. Clinicopathologic, immunohistochemical, and molecular features of histiocytoid Sweet syndrome. JAMA Dermatol. 2017;153(7):651.
Chan MP, Duncan LM, Nazarian RM. Subcutaneous Sweet syndrome in the setting of myeloid disorders: a case series and review of the literature. J Am Acad Dermatol. 2013;68(6):1006–15.
Mazzei ME, Guerra A, Dufrechou L, Vola M. Cryptococcoid Sweet syndrome: a clinical and histologic imitator of cryptococcosis. Actas Dermosifiliogr. 2021;112(1):79–80.
Haber R, Feghali J, El Gemayel M. Risk of malignancy in histiocytoid Sweet syndrome: a systematic review and reappraisal. J Am Acad Dermatol. 2020;83(2):661–3.
Merlant M, Lepelletier C, Battistella M, Vignon-Pennamen M-D, Duriez P, Moguelet P, et al. Acute myeloid leukemia and myelodysplastic syndrome–associated Sweet syndrome: a comparative multicenter retrospective study of 39 patients. J Am Acad Dermatol. 2021;84(3):838–40.
Magro CM, Momtahen S, Nguyen GH, Wang X. Histiocytoid Sweet’s syndrome: a localized cutaneous proliferation of macrophages frequently associated with chronic myeloproliferative disease. Eur J Dermatol. 2015;25(4):335–41.
Ghoufi L, Ortonne N, Ingen-Housz-Oro S, Barhoumi W, Begon E, Haioun C, et al. Histiocytoid Sweet syndrome is more frequently associated with myelodysplastic syndromes than the classical neutrophilic variant: a comparative series of 62 patients. Medicine (Baltimore). 2016;95(15):e3033.
Bush JW, Wick MR. Cutaneous histiocytoid Sweet syndrome and its relationship to hematological diseases. J Cutan Pathol. 2016;43(4):394–9.
Su WP, Liu HN. Diagnostic criteria for Sweet’s syndrome. Cutis. 1986;37(3):167–74.
Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34(5):918–23.
Cohen PR. Neutrophilic dermatoses. Am J Clin Dermatol. 2009;10(5):301–12.
• Hrin ML, Feldman SR, Huang WW. Dapsone as corticosteroid-sparing therapy for Sweet syndrome. J Am Acad Dermatol. 2021. Retrospective single institution case series of 11 SS patients treated with dapsone monotherapy.
Calabrese L, Caldarola G, Peris K, De Simone C. Recalcitrant Sweet syndrome successfully treated with adalimumab. J Dtsch Dermatol Ges. 2021;19(1):122–4.
Watson IT, Haugh I, Gardner AR, Menter MA. Histiocytoid Sweet syndrome successfully treated with etanercept. Bayl Univ Med Cent Proc. 2018;31(3):347–9.
•• De Risi-Pugliese T, Seksik P, Bouaziz J-D, Chasset F, Moguelet P, Gornet J-M, et al. Ustekinumab treatment for neutrophilic dermatoses associated with Crohn’s disease: a multicenter retrospective study. J Am Acad Dermatol. 2019;80(3):781–4. A multicenter retrospective study of Ustekinumab and Crohn’s disease (CD) associated neutrophilic dermatoses (ND) demonstrated remission of ND in 6 out of 7 patients treated and is a promising given its efficacy in both CD and ND.
Shahid Z, Kalayanamitra R, Patel R, Groff A, Jain R. Refractory Sweet syndrome treated with anakinra. Cureus. 2019;11(4):e4536.
Corbeddu M, Pilloni L, Pau M, Pinna AL, Rongioletti F, Atzori L. Treatment of Sweet’s syndrome in pregnancy. Dermatol Ther. 2018;31(4):e12619.
Brunsting LA, Goeckerman WH, O’Leary PA. Pyoderma (echthyma) gangrenosum: clinical and experimental observations in five cases occurring in adults. Arch Dermatol. 1982;118(10):743–68.
Brocq A. new contribution to the study of geometric phagedenism. Ann Dermatol Syphiligr. 1916;9:1–39.
•• Xu A, Balgobind A, Strunk A, Garg A, Alloo A. Prevalence estimates for pyoderma gangrenosum in the United States: an age- and sex-adjusted population analysis. Journal of the American Academy of Dermatology. 2020;83(2):425–9. A recent US cross-sectional analysis reported a prevalence of 5.8 cases per 100,000 adults, most commonly affecting women and those aged ≥ 50 years.
Ashchyan HJ, Nelson CA, Stephen S, James WD, Micheletti RG, Rosenbach M. Neutrophilic dermatoses. J Am Acad Dermatol. 2018;79(6):1009–22.
Ahronowitz I, Harp J, Shinkai K. Etiology and management of pyoderma gangrenosum. Am J Clin Dermatol. 2012;13(3):191–211.
Gillard M, Anuset D, Maillard H, Senet P, Cuny JF, Mahe E, et al. Comorbidities of pyoderma gangrenosum: a retrospective multicentric analysis of 126 patients. Br J Dermatol. 2018;179(1):218–9.
Wu BC, Patel ED, Ortega-Loayza AG. Drug-induced pyoderma gangrenosum: a model to understand the pathogenesis of pyoderma gangrenosum. Br J Dermatol. 2017;177(1):72–83.
Langan SM, Groves RW, Card TR, Gulliford MC. Incidence, mortality, and disease associations of pyoderma gangrenosum in the United Kingdom: a retrospective cohort study. J Investig Dermatol. 2012;132(9):2166–70.
•• Narla S, Silverberg JI. The inpatient burden and comorbidities of pyoderma gangrenosum in adults in the United States. Arch Dermatol Res. 2021;313(4):245–53. Authors analyzed prevalence, length of stay (LOS), cost of care, and any risk factors for admission and associated comorbidities in adults hospitalized for PG in the U.S.
Narla S, Silverberg JI. The inpatient burden of pyoderma gangrenosum and associated comorbidities in children in the United States. Arch Dermatol Res. 2021.
Haynes D, Hammer P, Malachowski SJ, Kaffenberger B, Yi JS, Vera N, et al. Characterisation and diagnosis of ulcers in inpatient dermatology consultation services: a multi-centre study. Int Wound J. 2019;16(6):1440–4.
Wilson-Jones E, Winkelmann RK. Superficial granulomatous pyoderma: a localized vegetative form of pyoderma gangrenosum. J Am Acad Dermatol. 1988;18(3):511–21.
• Borda LJ, Wong LL, Marzano AV, Ortega-Loayza AG. Extracutaneous involvement of pyoderma gangrenosum. Arch Dermatol Res. 2019;311(6):425–34. 2019 review of the extra-cutaneous manifestations of PG.
Hobbs MM, Byler R, Latour E, Bonomo L, Hennessy K, Cruz-Diaz CN, et al. Treatment of pyoderma gangrenosum: a multicenter survey-based study assessing satisfaction and quality of life. Dermatol Ther. 2021;34(2):e14736.
Nusbaum KB, Ortega-Loayza AG, Kaffenberger BH. Health-related domains of quality of life in pyoderma gangrenosum: a qualitative analysis. J Am Acad Dermatol. 2021.
Ighani A, Al-Mutairi D, Rahmani A, Weizman AV, Piguet V, Alavi A. Pyoderma gangrenosum and its impact on quality of life: a multicentre, prospective study. Br J Dermatol. 2019;180(3):672–3.
Su WPD, Davis MDP, Weenig RH, Powell FC, Perry HO. Pyoderma gangrenosum: clinicopathologic correlation and proposed diagnostic criteria. Int J Dermatol. 2004;43(11):790–800.
Maverakis E, Ma C, Shinkai K, Fiorentino D, Callen JP, Wollina U, et al. Diagnostic criteria of ulcerative pyoderma gangrenosum: a Delphi Consensus of International Experts. JAMA Dermatol. 2018;154(4):461–6.
Jockenhöfer F, Wollina U, Salva KA, Benson S, Dissemond J. The PARACELSUS score: a novel diagnostic tool for pyoderma gangrenosum. Br J Dermatol. 2019;180(3):615–20.
•• Haag C, Hansen T, Hajar T, Latour E, Keller J, Shinkai K, et al. Comparison of three diagnostic frameworks for pyoderma gangrenosum. J Investig Dermatol. 2021;141(1):59–63. Comparison of the 3 diagnostic frameworks for PG. PARACELSUS identified highest proprotion of PG cases compared to Su and Delphi criteria.
Handler MZ, Hamilton H, Aires D. Treatment of peristomal pyoderma gangrenosum with topical crushed dapsone. J Drugs Dermatol. 2011;10(9):1059–61.
Partridge ACR, Bai JW, Rosen CF, Walsh SR, Gulliver WP, Fleming P. Effectiveness of systemic treatments for pyoderma gangrenosum: a systematic review of observational studies and clinical trials. Br J Dermatol. 2018;179(2):290–5.
Ormerod AD, Thomas KS, Craig FE, Mitchell E, Greenlaw N, Norrie J, et al. Comparison of the two most commonly used treatments for pyoderma gangrenosum: results of the STOP GAP randomised controlled trial. BMJ. 2015;350(3):h2958.
Brooklyn TN, Dunnill MG, Shetty A, Bowden JJ, Williams JD, Griffiths CE, et al. Infliximab for the treatment of pyoderma gangrenosum: a randomised, double blind, placebo controlled trial. Gut. 2006;55(4):505–9.
Schøsler L, Fogh K, Bech R. Pyoderma gangrenosum: a retrospective study of clinical characteristics, comorbidities, response to treatment and mortality related to prednisone dose. Acta Derm Venereol. 2021;101(4):adv00431.
Haag CK, Bacik L, Latour E, Morse DC, Fett NM, Ortega-Loayza AG. Perioperative management of pyoderma gangrenosum. J Am Acad Dermatol. 2020;83(2):369–74.
Xia FD, Liu K, Lockwood S, Butler D, Tsiaras WG, Joyce C, et al. Risk of developing pyoderma gangrenosum after procedures in patients with a known history of pyoderma gangrenosum-A retrospective analysis. J Am Acad Dermatol. 2018;78(2):310–4.
Yurdakul S, et al. Behçet syndrome. Curr Opin Rheumatol. 2004;16(1):38–42.
Calamia KT, Wilson FC, Icen M, Crowson CS, Gabriel SE, Kremers HM. Epidemiology and clinical characteristics of Behçet’s disease in the US: a population-based study. Arthritis Rheum. 2009;61(5):600–4.
Thomas T, Chandan JS, Subramanian A, Gokhale K, Gkoutos G, Harper L, et al. Epidemiology, morbidity and mortality in Behçet’s disease: a cohort study using The Health Improvement Network (THIN). Rheumatology. 2020;59(10):2785–95.
Guven DC, Bolek EC, Altintop SE, Celikten B, Aktas BY, Kiraz S, et al. Cancer incidence in Behçet’s disease. Irish J Med Sci. 2020;189(4):1209–14.
Wang X, Peng Y, Gao J, Han S, Li Y. Risk of malignancy in Behcet disease: a meta-analysis with systematic review. Medicine (Baltimore). 2019;98(44):e17735.
Na SJ, Kang MJ, Yu DS, Han KD, Lee JH, Park YG, et al. Cancer risk in patients with Behçet disease: a nationwide population-based dynamic cohort study from Korea. J Am Acad Dermatol. 2018;78(3):464–70.
International Study Group for Behçet’s Disease. Criteria for diagnosis of Behcet’s disease. Lancet. 1990;335(8697):1078–80.
Firestein GS, Gruber HE, Weisman MH, Zvaifler NJ, Barber J, O’Duffy JD. Mouth and genital ulcers with inflamed cartilage: MAGIC syndrome. Five patients with features of relapsing polychondritis and Behçet’s disease. Am J Med. 1985;79(1):65–72.
Hamuryudan V, Er T, Seyahi E, Akman C, Tüzün H, Fresko I, et al. Pulmonary artery aneurysms in Behçet syndrome. Am J Med. 2004;117(11):867–70.
Chen KR, Kawahara Y, Miyakawa S, Nishikawa T. Cutaneous vasculitis in Behçet’s disease: a clinical and histopathologic study of 20 patients. J Am Acad Dermatol. 1997;36(5 Pt 1):689–96.
Criteria for diagnosis of Behçet’s disease. International Study Group for Behçet’s Disease. Lancet. 1990;335(8697):1078–80.
International Team for the Revision of the International Criteria for Behçet’s Disease (ITR‐ICBD). The International Criteria for Behçet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol. 2014;28(3):338–47.
•• Hatemi G, Christensen R, Bang D, Bodaghi B, Celik AF, Fortune F, et al. 2018 update of the EULAR recommendations for the management of Behçet’s syndrome. Ann Rheum Dis. 2018;1;77(6):808–18. Update of the EULAR recommendations for the management of Behçet’s syndrome.
Nakamura K, Iwata Y, Asai J, Kawakami T, Tsunemi Y, Takeuchi M, et al. Guidelines for the treatment of skin and mucosal lesions in Behçet’s disease: a secondary publication. J Dermatol. 2020;47(3):223–35.
Chams-Davatchi C, Barikbin B, Shahram F, Nadji A, Moghaddassi M, Yousefi M, et al. Pimecrolimus versus placebo in genital aphthous ulcers of Behcet’s disease: a randomized double-blind controlled trial. Int J Rheum Dis. 2010;13(3):253–8.
Braun-Moscovici Y, Tavor Y, Markovits D, Toledano K, Rozin A, Nahir MA, et al. The effects of adalimumab in Behçet disease patients on clinical manifestations and on pro-inflammatory cytokines milieu: long-term follow-up. Isr Med Assoc J. 2020;22(5):289–93.
Alpsoy E. Behçet’s disease: a comprehensive review with a focus on epidemiology, etiology and clinical features, and management of mucocutaneous lesions. J Dermatol. 2016;43(6):620–32.
•• Hatemi G, Mahr A, Ishigatsubo Y, Song Y-W, Takeno M, Kim D, et al. Trial of apremilast for oral ulcers in Behçet’s syndrome. New England Journal of Medicine. 2019;381(20):1918–28. A Phase 3, randomized control trial studying Apremilast, an oral phosphodiesterase-4 inhibitor, showed statistically significant reduction in number of oral ulcers compared to placebo.
Bettiol A, Silvestri E, Di Scala G, Amedei A, Becatti M, Fiorillo C, et al. The right place of interleukin-1 inhibitors in the treatment of Behçet’s syndrome: a systematic review. Rheumatol Int. 2019;39(6):971–90.
Mirouse A, Barete S, Desbois AC, Comarmond C, Sène D, Domont F, et al. Long-term outcome of ustekinumab therapy for Behçet’s disease. Arthritis Rheumatol. 2019;71(10):1727–32.
Desbois AC, Biard L, Addimanda O, Lambert M, Hachulla E, Launay D, et al. Efficacy of anti-TNF alpha in severe and refractory major vessel involvement of Behcet’s disease: a multicenter observational study of 18 patients. Clin Immunol. 2018;197:54–9.
Lopalco G, Fabiani C, Venerito V, Lapadula G, Iannone F, Cantarini L. Ustekinumab efficacy and safety in mucocutaneous multi-refractory Behçet’s disease. Clin Exp Rheumatol. 2017;35(6):130–1.
Vitale A, Emmi G, Lopalco G, Gentileschi S, Silvestri E, Fabiani C, et al. Adalimumab effectiveness in Behçet’s disease: short and long-term data from a multicenter retrospective observational study. Clin Rheumatol. 2017;36(2):451–5.
Cantarini L, Lopalco G, Vitale A, Coladonato L, Rigante D, Lucherini OM, et al. Paradoxical mucocutaneous flare in a case of Behçet’s disease treated with tocilizumab. Clin Rheumatol. 2015;34(6):1141–3.
Diamantopoulos AP, Hatemi G. Lack of efficacy of tocilizumab in mucocutaneous Behçet’s syndrome: report of two cases. Rheumatology. 2013;52(10):1923–4.
Harrist TJ. Neutrophilic eccrine hidradenitis. Arch Dermatol. 1982;118(4):263.
Pierson JC, Helm TN, Taylor JS, Elston DM, Tuthill RJ. Neutrophilic eccrine hidradenitis heralding the onset of acute myelogenous leukemia. Arch Dermatol. 1993;129(6):791–2.
Crane JS, Krishnamurthy K. Neutrophilic eccrine hidradenitis. Treasure Island (FL): StatPearls Publishing LLC; 2021. Copyright© 2021.
Bachmeyer C, Aractingi S. Neutrophilic eccrine hidradenitis. Clin Dermatol. 2000;18(3):319–30.
Feraru G, Dodiuk-Gad R, Krausz J, Ziv M. Infliximab-induced neutrophilic eccrine hidradenitis in a patient with hidradenitis suppurativa. Dermatol Ther. 2020;33(6):e13900.
Bishnoi A, Daroach M, Aggarwal D, Radotra BD, Panda P, Parsad D. Ticagrelor induced neutrophilic eccrine hidradenitis: a unique adverse effect of a new antiplatelet drug. Postgrad Med J. 2019;95(1123):279–80.
Puar N, Scheele A, Perez Marques F, Panicker J. Neutrophilic eccrine hidradenitis secondary to pegfilgrastim in a patient with synovial sarcoma. Clin Case Rep. 2019;7(3):533–6.
Arteaga JEC, Gonçalves DLM, Giannotti MA, Samorano LP. Neutrophilic eccrine hidradenitis in a healthy Brazilian child. J Dtsch Dermatol Ges. 2019;17(8):834–6.
Lee WJ, Kim CH, Chang SE, Lee MW, Choi JH, Moon KC, et al. Generalized idiopathic neutrophilic eccrine hidradenitis in childhood. Int J Dermatol. 2010;49(1):75–8.
Piqué E, Aguilar A, Olivares M, Palacios S, Roman V, Gallego M, et al. Idiopathic palmoplantar hidradenitis. Dermatology. 1997;195(4):379–81.
Bolognia JL, Cooper DL, Glusac EJ. Toxic erythema of chemotherapy: a useful clinical term. J Am Acad Dermatol. 2008;59(3):524–9.
Shear NH, Knowles SR, Shapiro L, Poldre P. Dapsone in prevention of recurrent neutrophilic eccrine hidradenitis. J Am Acad Dermatol. 1996;35(5):819–22.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors have no relevant financial or non-financial interests to disclose. Dr. Mostaghimi reports grants and personal fees from Pfizer, personal fees and other from Concert, personal fees and other from Lilly, personal fees from AbbVie, personal fees from ACON Labs, personal fees from Bioniz, personal fees from hims and hers, and personal fees from Digital Diagnostics, outside the submitted work. Dr. Rosenbach reports personal fees from Merck, personal fees from Janssen, personal fees from Novartis, personal fees from Eli Lilly, and grants and personal fees from Processa, outside the submitted work.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Hospital-Based Dermatology
Rights and permissions
About this article

Cite this article
Weiss, E.H., Ko, C.J., Leung, T.H. et al. Neutrophilic Dermatoses: a Clinical Update. Curr Derm Rep 11, 89–102 (2022). https://doi.org/10.1007/s13671-022-00355-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13671-022-00355-8