
Abstract
Bacteria may adhere to and develop biofilm structures onto dairy surfaces trying to protect themselves from adverse conditions such as pasteurization and CIP processes. Thus, biofilms are considered common sources of food contamination with undesirable bacteria. The purpose of this study was to evaluate the diversity of the microbiota attached to stainless steel surfaces in pre- and post-pasteurization pipe lines of a milk-processing plant. Seventy Gram-positive isolates were identified as Enterococcus faecalis (33), Bacillus cereus (26), Staphylococcus hominis (8), Staphylococcus saprophyticus (2), and Staphylococcus epidermidis-Staphylococcus aureus (1) species. Fifty-five Gram-negative isolates were identified to the species Escherichia coli (18), Klebsiella pneumoniae (13), Acinetobacter calcoaceticus (6), Serratia marcescens (6), Enterobacter spp. (5), Pseudomonas aeruginosa (4), Escherichia vulneris (2), and Proteus mirabilis (1). Fifty-five different strains were detected by the RAPD technique. These were subjected to an in vitro assay to evaluate their biofilm-forming capability. E. faecalis (7), A. calcoaceticus (4), K. pneumoniae (3), S. hominis (3), and P. aeruginosa (2) were the species in which more biofilm producer strains were encountered. The adhered microbiota was also assessed by the PCR-DGGE culture-independent technique. This analysis revealed a greater bacterial diversity than that revealed by culturing methods. In this way, in addition to the bacteria detected by culturing, DNA bands belonging to the genera Chrysobacterium and Streptomyces were also identified. This study emphasizes that knowledge of attached microorganisms to dairy surfaces may help develop strategies to improve optimal operational parameters for pasteurization and CIP processes in dairy plants.
Similar content being viewed by others

1 Introduction
Milk is a complex substrate that can support the growth of a wide variety of both Gram-positive and Gram-negative bacteria, as well as that of yeast and moulds (Lafarge et al. 2004; Martins et al. 2006). Milk pasteurization is a common practice for safeguarding consumers from food-borne pathogenic bacteria (Ranieri et al. 2009). This practice is used worldwide in order to increase the shelf-life of this highly perishable food product (Ivy et al. 2012). After pasteurization, cleaning in place (CIP) processes are designed in order to maintain a clean and hygienic environment, including piping and fitting systems (Bayoumi et al. 2012). Bacteria surviving pasteurization and CIP processes could compromise quality and safety of the pasteurized milk and dairy products manufactured with pasteurized milk. In addition, the surviving bacteria can potentially attach to piping and fitting surfaces, where they could promote the development of biofilm structures that enable protection against high temperatures and chemical compounds applied during pasteurization and sanitization procedures. Bacteria within biofilms may attach to tools and equipment at other positions of the plant, thus persisting a longer time in the dairy environment (Brooks and Flint 2008).
Biofilms are bacterial communities embedded in an extracellular matrix composed by proteins, exopolysaccharides, DNA, and/or lipopeptides (Donlan 2002). The presence of biofilms is a widespread phenomenon in many ecosystems, including dairy plants (Latorre et al. 2010). Biofilm development takes place when planktonic cells adhere to a surface in a reversible and nonspecific manner due to electrostatic interactions and begin to secrete a complex extracellular matrix. This process is affected by many factors, such as the type of surface, temperature, pH, physical interaction between the constituents, physicochemical characteristics of bacterial and spore surfaces, etc. (Brooks and Flint 2008; Rickard et al. 2003). The presence of biofilms in equipment and tools of milk-processing plants, such as bends in pipes, gaskets, floors, milk handling devices, etc., has been well documented (Brooks and Flint 2008). Biofilms are considered as a source of microbial contamination leading to food spoilage, shelf life reduction, and are also considered as a potential way of pathogen transmission (Brooks and Flint 2008; Latorre et al. 2010). Bacteria embedded in a biofilm have been considered as more resistant to cleaning and sanitizing chemicals than the corresponding planktonic cells (Anand and Singh 2013; Peng et al. 2002). Biofilms can further allow attachment of nonbiofilm-producing microbial types, thus increasing microbial contamination (Brooks and Flint 2008). Additionally, the development of biofilms creates serious problems in dairy plants due to enhanced corrosion rates of metallic surfaces, reduced heat transfer efficacy, decreased flow of the pipelines and increased fluid frictional resistance; aspects that reduce the microbiological quality of the final products and also lead to economic losses (Mittelman 1998). For all these reasons, the removal of biofilm-embedded microorganisms continues to be a major challenge in dairy industry. A large number of food spoilage and/or pathogenic bacteria, including Enterococcus faecalis, Pseudomonas spp., Klebsiella spp., Staphylococcus aureus, Listeria monocytogenes, Bacillus cereus, and others, have already been associated with biofilms from dairy niches (Brooks and Flint 2008; Latorre et al. 2010; Sharma and Anand 2002).
This work aimed to investigate the microbial diversity attached to milk-processing surfaces in a dairy plant before and after pasteurization using culturing and culture-independent techniques for a better description of the adherent microbiota. The isolated microorganisms were identified by molecular methods, which included digestion of ribosomal amplicons with restriction endonucleases (ARDRA) and 16S rDNA sequencing and sequence comparison. Additionally, the biofilm-forming capability of the identified strains was assessed by analyzing their ability to attach onto polystyrene surfaces.
2 Material and methods
2.1 Sampling procedures and isolation of microorganisms
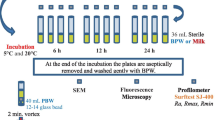
Four to six samples from each sampling point were collected over summer seasons of 2010, 2011, and 2012. Two different positions were sampled along the production pipeline of the milk-processing plant. The sampling point A was located before the pasteurizer and receives raw milk. Thus, bacteria recovered from this point represent those able to attach to stainless steel surfaces. The sampling point B was located immediately after the pasteurizer. In addition to be able to attach to the surface of pipes, microorganisms isolated from this sampling point must survive pasteurization. Samples were collected by swabbing a surface of 5 cm2, following the procedure described by Mattila et al. (1990). Swabs were refrigerated and transferred immediately to the laboratory for analysis.
Swabs from each sampling point were used to inoculate in 5 mL of nutrient broth (NB; Fluka), which were subsequently incubated overnight at 37 °C. Overnight cultures were plated onto nutrient agar plates (NA; Fluka). After an incubation period of 24 h at 37 °C, colonies were picked at random from the plates, purified by subculturing and stored at 4 °C on the same media. For long-term storage, isolates were cultured in Brain Heart Infusion (BHI, Merck) broth, a 25% glycerol was added (Merck), and kept frozen at −80 °C.
2.2 Phenotypic identification isolates
Isolates were examined for cell morphology and Gram reaction. According to these, isolates were separated in three groups, Gram-positive rods, Gram-positive cocci, and Gram-negative rods.
2.3 Molecular identification by ARDRA and 16S rDNA sequencing
DNA extracts of the purified isolates were obtained by suspending a colony in 100 μL of molecular-biology-grade water (Sigma-Aldrich), heating at 98 °C for 15 min, and then treating with the same volume of chloroform. After centrifugation at 16,100 g for 3 min, DNA extracts were harvest from the supernatants and were used directly in PCR reactions. For some isolates in which this procedure did not result in amplification, total genomic DNA was purified using the ATP Genomic Mini Kit (ATP Biotech) following the manufacturer’s recommendations. DNA extracts or purified genomic DNA was used as a template to amplify a 1.5 kb DNA fragment of the 16S rRNA gene (16S rDNA) using the universal bacterial primer S-D-Bact0008-a-S-20 27 F (5′-AGAGTTTGATYMTGGCTCAG-3′) and the universal prokaryotic primer S-*-Univ1492R-b-A-21 1492R (5′-GGTTACCTTGTTACGACTT-3′) (Lane 1991). The PCR conditions were as follows: one cycle at 95 °C for 5 min, 35 cycles at 94 °C for 30 s, 55 °C for 45 s, and 72 °C for 2 min, and a final extension cycle at 72 °C for 10 min. For amplified ribosomal DNA restriction analysis (ARDRA), amplicons were purified to remove unincorporated primers and nucleotides using ATP™ Gel/PCR Extraction Kit (ATP Biotech), and subjected to restriction with the restriction enzymes HinfI, HhaI, Sau3AI, and HaeIII (Takara), all of them with a short-length (4–5 bp) recognition sequence. Digestion fragments were separated in 1% agarose, stained with ethidium bromide (0.5 mg mL−1) and photographed under UV light.
Representative 16S rDNA amplicons of all different ARDRA profiles were sequenced by cycle extension in an ABI 373 DNA sequencer (Applied Biosystems). Approximately 800 bp of sequence was obtained per amplicon; these were compared with those deposited in the GenBank database, using the online BLAST program (http://www.ncbi.nlm.nih.gov/BLAST/) and with those in the Ribosomal Database Project (http://rdp.cme.msu.edu/index.jsp). Sequences sharing a percentage of identity of 97% or higher to those in databases were considered to belong to the same species (Stackebrandt et al. 2002).
2.4 PCR fingerprinting
The intraspecies genetic diversity of the isolates was assessed by independent PCR fingerprinting with primers BOXA2R (5′-ACGTGGTTTGAAGAGATTTTCG-3′), as reported by Koeuth et al. (1995), and M13 (5′-GAGGGTGGCGGTTCT-3′), as described by Rossetti and Giraffa (2005). PCR reaction mixtures contained 5 μL of each DNA extract or purified genomic DNA, 25 μL of Taq master Mix (Ampliqon), 5 μL of primer (10 μmol L−1) and 15 μL of molecular-biology-grade water in a total volume of 50 μL. The PCR conditions for the RAPD analysis were the same as those described above with an annealing temperature of 40 °C for primer BOXA2R and 42 °C for primer M13. Reproducibility studies of the PCR fingerprinting technique for the mentioned primers (independent amplification with the same DNA) showed a percentage of similarity of over 90%. PCR profiles were visualized after 90 min of electrophoresis (75 V) in agarose gels (1.2%) after staining with ethidium bromide as above. Profiles were clustered using the unweighted pair group method using arithmetic averages (UPGMA) and their similarity expressed by the Sørensen–Dice’s coefficient.
2.5 Detection and quantification of biofilm production
Biofilm production on polystyrene surface was determined using 96-well microtiter plates (Nunc), following the quantitative method described by Stepanovic et al. (2000) with minor modifications. In short, 10 μL of an overnight culture at 37 °C in Tryptic Soy Broth (TSB) supplemented with 0.25% glucose (for optimal growth of all species) was used to inoculate at 5% (cell concentration of ≈106 CFU mL−1) independent microtiter plate wells with 200 μL of the same medium. Plates were incubated aerobically for 24 h at 37 °C. Then, two rounds of vigorous washings with phosphate-buffer saline (PBS) were realized to remove nonadhered cells. Microtiter plates were subsequently dried at room temperature for 15 min prior to staining with a 0.1% crystal violet solution for 15 min. Excess of stain was rinsed off by dipping the microtiter plates in tap water. After further drying, adherence of the cells was measured as the absorbance released at 595 nm by using an automatic microtiter plate reader (Bio-Rad) after solubilization of the dye bound to the plates with a 33% acetic acid solution. Based upon the absorbance, strains were classified into the four following categories: no biofilm producer (OD ≤ ODc), and weak (ODc < OD ≤ 2X ODc), moderate (2X ODc < OD ≤ 4X ODc), or strong (OD > 4X ODc) biofilm producer (Stepanovic et al. 2000), where ODc is the optical density measured for the negative control. Each strain was tested in quadruplicate and average results are presented. Negative (uninoculated broth) and positive (Staphylococcus epidermidis B, DG2Ñ and YLIC17, strong biofilm-forming strains) (Delgado et al. 2009) controls were assayed in the same conditions.
2.6 Analysis of adherent bacteria by PCR-DGGE
The composition and dynamics of the dominant bacterial populations in the biofilms was analyzed by the culture-independent PCR-DGGE technique. To do this, samples were collected in three consecutive days from identical positions as those analyzed by culturing. Total DNA of the samples was extracted using QIAamp DNA Stool Mini kit (Qiagen) following the manufacturer’s instructions. Amplification of the V3 region of the bacterial 16S rRNA gene was performed using two universal primers 357F (5′-CCTACGGGAGGCAGCAG-3′) and 518R (5′-GTATTACCGCGGCTGCTGG-3′). A GC clamp was attached to the 5′ end of the forward primer (CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGG), as reported by Muyzer et al. (1993). The PCR conditions were as follows: 95 °C for 5 min, 35 cycles at 95 °C for 30 s, 56 °C for 30 s, and 72 °C for 1 min, and a final extension cycle at 72 °C for 10 min. The DGGE analysis was performed at 60 °C in 8% polyacrylamide gels with a formamide-urea denaturing range of 40–60% using a DCode apparatus (Bio-Rad). Electrophoresis was conducted at 75 V for 17 h, and the DGGE patterns were visualized after staining with ethidium bromide as above. The most intense bands were excised from the acrylamide gels and identified after reamplification with the original pair of primers without the GC clamp, DNA purification, and sequencing as above.
3 Results
3.1 Culture analysis of pipe-adhered microorganisms
A total of 125 isolates representative of all colony morphologies was selected from the two different sampling points analyzed in this study. Isolates were firstly grouped as Gram-negative rods (55 isolates; 44%), Gram-positive cocci (44 isolates; 35%), and Gram-positive rods (26 isolates; 21%) (Table 1). Most microbial types were found in both A (after pasteurization) and B (before pasteurization) sampling points. As can be clearly seen, the number of Gram-negative bacteria recovered decreased after pasteurization (from 63 to 31%). In contrast, numbers of Gram positive isolates increased after this process (from 37 to 69%).
The ARDRA profiles obtained after digestion of the amplicons with the restriction enzyme HinfI gave 8 different profiles (Fig. 1). Digestion of the amplicons with the restriction enzymes HhaI, Sau3AI, and HaeIII further separated some of the profiles to a final total number of 13 different restriction patterns. Representative amplicons of all different profiles were sequenced and their sequences compared with those deposited on databases. Extending the sequencing results to all isolates, 41 out of the 44 Gram-positive cocci isolates were allocated to the species Enterococcus faecalis (33) and Staphylococcus hominis (8) (Table 1). The other three Gram-positive isolates could not be identified with confidence to the species level. Two of these were identified as members of the Staphylococcus saprophyticus species group and one as belonging to the Staphylococcus epidermidis-Staphylococcus aureus species group. A single ARDRA profile was obtained from all Gram-positive rods, which was identified by sequencing as belonging to the species Bacillus cereus (Table 1). Fifty out of the 55 Gram-negative isolates were classified as Escherichia coli (18), Klebsiella pneumoniae (13), Acinetobacter calcoaceticus (6), Serratia marcescens (6), Pseudomonas aeruginosa (4), Escherichia vulneris (2), and Proteus mirabilis (1) (Table 1). As before, five of the isolates could only be identified to the genus level; they all were assigned to Enterobacter spp.

Differentiation between species using ARDRA profiles obtained after amplification of 16S rDNA and digestion of the amplicons with the restriction enzymes: a HinfI, b HhaI, c Sau3AI, and d HaeIII. Lines: M, GRS Universal Ladder (Grip); 1, K. pneumoniae; 2, A. calcoaceticus; 3, Enterobacter spp; 4, E. coli; 5, E. vulneris; 6, P. aeruginosa; 7, S. marcescens; 8, P. mirabilis; 9, B. cereus; 10, E. faecalis; 11, S. hominis; 12, S. saprophyticus species group; 13, S. epidermidis-S. aureus species group
Using a threshold of 90% identity obtained in the reproducibility study, 55 different RAPD profiles were independently obtained with both M13 and BoxA2R primer (data not shown). As an example, Fig. 2 depicts all profiles obtained for the Gram-positive isolates. Therefore, all these 55 profiles were considered to belong to different strains. In this way, 44 Gram-positive cocci isolates resulted in 15 different strains, of which 7 belonged to the species E. faecalis, 5 to S. hominis, and 2 and 1 to S. saprophyticus and S. epidermidis-S. aureus species group, respectively. Five profiles were considered among the 26 B. cereus isolates. Finally, 35 RAPD strains were found among the 55 Gram-negative isolates, as follows: 9 of E. coli, 8 of K. pneumoniae, 5 of Enterobacter spp., 4 of A. calcoaceticus, 3 of P. aeruginosa, 3 of S. marcescens, 2 of E. vulneris, and 1 of P. mirabilis.

Representative fingerprinting PCR profiles obtained with primer M13, as follows: a Gram-positive rods isolates, b Gram-positive cocci isolates. M molecular weight marker; lines 1–5, B. cereus strains; 6–12, E. faecalis strains; 13–17, S. hominis strains; 18–19, S. saprophyticus group strains; 20, S. epidermidis-S. aureus group strain
3.2 Biofilm-forming ability of the strains
One strain of each of the 55 RAPD profiles was tested for its biofilm-forming ability on polystyrene surfaces; this was considered an indirect proof of biofilm production. The results of this assay are summarized in Table 2. Under the study conditions, a majority of the strains (38) were considered as no biofilm producers (27 strains) or weak biofilm producers (11 strains). In contrast, 17 strains showed moderate (15) or strong (2) capacity for biofilm formation on polystyrene surfaces. None of the strains of the species E. coli (9), E. vulneris (2), and S. marcescens (3) were able to form biofilm. In contrast, strains of all other species showed at least a certain ability to produce biofilm on the polystyrene surface of the plates. The strongest biofilm producers belonged one strain each to the species E. faecalis and S. hominis.
3.3 Culture-independent analysis of pipe-adhered microorganisms
As the pre-enrichment (resuscitation) step could introduce some bias on the microorganisms originally present in the biofilms at the two sampling points analyzed, a PCR-DGGE approach was performed to assess by a culture-independent method the composition of the bacterial populations. Three samples in consecutive days were taken from a single site before pasteurization (A1) and from two points after pasteurization (B1 and B2) and analyzed by PCR-DGGE (Fig. 3). A high bacterial diversity was discovered not only among samples at the different sampling points but also between samples from the same sampling point at different dates. Bands at the same level (belonging to the same species) and with similar intensity were only observed occasionally. A total of 19 bands were identified, after DNA elution, reamplification, sequencing, and sequence comparison against databases. In addition to sequences of the genera identified by the conventional culture approach (Enterobacteriaceae, Bacillus, Pseudomonas), sequences belonging to previously undetected genera (Chryseobacterium, Streptomyces) were also identified.

DGGE profiles of PCR amplicons of the V3 region of the bacterial 16S rDNA representing the biodiversity of the bacterial communities attached to milk pipes in a dairy. Samples were taken in three consecutive days at one position before (A) and at two position after (B1 and B2) pasteurization. Bands with a number were identified at the genus level after DNA isolation, reamplification, sequencing, and sequence comparison. Identity of the bands: 1, Enterobacteriaceae; 2, Bacillus spp.; 3, Lactobacillus spp.; 4, Pseudomonas spp.; 5, Shewanella spp.; 6, Streptomyces spp.; 7, Serratia spp.; 8, Chryseobacterium spp.
4 Discussion
In this study, pipe-adhered microorganisms from a milk-processing dairy were recovered in culture and identified from two different positions, before (sampling point A) and after pasteurization (sampling point B). A varied microbiota composed of both Gram-positive and Gram-negative bacteria belonging to 13 species was isolated from the two positions. This strongly suggests that the pasteurization process does not select for specific bacteria among those present in raw milk. High genetic diversity was found among isolates recovered before and after pasteurization from all bacterial species, which further supports the view that pasteurization does not make a selection for certain genetic profiles. All microbial types identified in this work have already been reported to attach to stainless steel pipe surfaces (Anand and Singh 2013; Mattila et al. 1990; Sharma and Anand 2002). The presence of microorganisms on dairy surfaces in post-pasteurization lines is a cause of concern, as it may cause spoilage of processed dairy products and/or be involved in food safety issues.
The bacterial diversity, both at the species and strain levels, was maintained from the raw milk section (A) to the pasteurization section (B), as 11 different species were obtained from both positions. High microbial loads, post-pasteurization recontamination, and heat resistance of the strains may all contribute to the presence of high microbial diversity on post-pasteurization pipe line surfaces. However, as concerns the recovery of isolates, those of E. faecalis and B. cereus increased their numbers after pasteurization. The heat resistance of enterococci from milk and dairy products has been reported before by several authors (Martinez et al. 2003; McAuley et al. 2012). Resistance of B. cereus to pasteurization conditions is neither surprising, given the ability of this species to form heat-resistant endospores (Huck et al. 2007). The increasing recovery of these species is maintained even after the CIP process (data not shown), suggesting these bacteria are resistant to different stressful conditions.
Resistance to heat (pasteurization) and cleaning (CIP) processes might be linked to the ability of the microorganisms to form biofilms, as has been reported by many authors (Anand and Singh 2013; Flint et al. 2002; Peng et al. 2002). In this context, all E. faecalis strains analyzed in this work were able to form biofilms on polystyrene surfaces. Similar results have been reported before for enterococci strains from raw milk and fermented meat Jahan and Holley (2014). Similarly, many of the tested Staphylococcus strains exhibited a moderate to strong capacity to form biofilms. Staphylococci species have been frequently shown to harbour ica genes (Gutierrez et al. 2012; Szweda et al. 2012), which are involved in biofilm formation. However, surprisingly, and in contrast to previous reported results (Faille et al. 2001), most B. cereus strains were found to be nonbiofilm producers, even though isolates of these species were a majority population in all samples. The abundance of potential nonbiofilm producers strains in all samples argues for attachment of these bacteria to the real biofilm producers forming mixed-species biofilms (Habimana et al. 2010; Lourenco et al. 2011; Simoes et al. 2007) or for these bacteria being attached to inert milk constituents (fat, protein) precipitating on the stainless steel surfaces. This is not surprising since the methodology followed in this study selected for attachment not for biofilm formation. Nevertheless, half of the isolates were shown to produce biofilms in polystyrene plates. In this context, previous studies on biofilm formation on polystyrene surfaces have been found to be positively (Moretro et al. 2003) or negatively (Rivas et al. 2007) correlated with biofilm formation on stainless steel. The ability of the strains of this study to form biofilms onto stainless surfaces has yet to be demonstrated.
Different bacterial profiles in samples before (A) and after (B) pasteurization, in different pipe sections (B1 and B2) and at different dates (1, 2, and 3) were revealed by the PCR-DGGE technique. This indicates major changes in the types and numbers of populations in the different sampling points and from consecutive samples of the same point. This strongly suggests that a resident biofilm-forming microbiota was not established in this dairy. It was surprising not to find DNA bands corresponding to E. faecalis, as it was a dominant species among the cultures. Disagreement between culturing and culture-dependent approaches could be due to the pre-enrichment step, which may select for species in good physiological conditions (more thermophilic) and/or those growing faster in the culture conditions of this study. Due to the small 16S rDNA segment amplified for the DGGE analysis, sequences could only be assigned to a genus level; therefore, bands with the same number may belong to different species. The DGGE technique is considered semi-quantitative, as the intensity of individual bands is thought to be an indirect measure of the abundance of their DNA in the population (Muyzer et al. 1993). Though the technique does not distinguish between DNA coming from dead or alive bacteria, the DGGE is considered a valuable tool for the molecular fingerprinting of the microbiota associated to pipes in a dairy plant.
In conclusion, culturing and culture-independent methods were applied to study the pipe-associated microbiota at different positions in a dairy. A high inter- and intra-species microbial diversity was found among the bacteria recovered from sampled positions. The results suggest that a biofilm-producing microbiota was not established in the analyzed dairy plant. Instead, the recovered bacteria can be a reflection of the day-to-day microbial variation of both bacterial types and numbers. In spite of this, the presence of high numbers and types of Gram-positive and Gram-negative bacteria should be taken into account to implement stronger hygiene routines. The establishment of optimal operational parameters (pasteurization temperature, type, and concentration of sanitizers) to improve the overall quality, shelf life, and safety of the milk requires further investigation.
References
Anand S, Singh D (2013) Resistance of the constitutive microflora of biofilms formed on whey reverse-osmosis membranes to individual cleaning steps of a typical clean-in-place protocol. J Dairy Sci 96(10):6213–6222
Bayoumi MA, Kamal RM, Abd El Aal SF, Awad EI (2012) Assessment of a regulatory sanitization process in Egyptian dairy plants in regard to the adherence of some food-borne pathogens and their biofilms. Int J Food Microbiol 158(3):225–231
Brooks JD, Flint SH (2008) Biofilms in the food industry: problems and potential solutions. Int J Food Sci Technol 43(12):2163–2176
Delgado S, Arroyo R, Jimenez E, Marin ML, del Campo R, Fernandez L, Rodriguez JM (2009) Staphylococcus epidermidis strains isolated from breast milk of women suffering infectious mastitis: potential virulence traits and resistance to antibiotics. BMC Microbiol 9:82
Donlan RM (2002) Biofilms: Microbial life on surfaces. Emerg Infect Dis 8(9):881–890
Faille C, Fontaine F, Benezech T (2001) Potential occurrence of adhering living Bacillus spores in milk product processing lines. J Appl Microbiol 90(6):892–900
Flint S, Brooks J, Bremer P, Walker K, Hausman E (2002) The resistance to heat of thermo-resistant streptococci attached to stainless steel in the presence of milk. J Ind Microbiol Biotechnol 28(3):134–6
Gutierrez D, Delgado S, Vazquez-Sanchez D, Martinez B, Cabo ML, Rodriguez A, Herrera JJ, Garcia P (2012) Incidence of Staphylococcus aureus and analysis of associated bacterial communities on food industry surfaces. Appl Environ Microbiol 78(24):8547–8554
Habimana O, Heir E, Langsrud S, Asli AW, Moretro T (2010) Enhanced surface colonization by Escherichia coli O157:H7 in biofilms formed by an Acinetobacter calcoaceticus isolate from meat-processing environments. Appl Environ Microbiol 76(13):4557–4559
Huck JR, Woodcock NH, Ralyea RD, Boor KJ (2007) Molecular subtyping and characterization of psychrotolerant endospore-forming bacteria in two New York state fluid milk processing systems. J Food Prot 70(10):2354–2364
Ivy RA, Ranieri ML, Martin NH, den Bakker HC, Xavier BM, Wiedmann M, Boor KJ (2012) Identification and characterization of psychrotolerant sporeformers associated with fluid milk production and processing. Appl Environ Microbiol 78(6):1853–1864
Jahan M, Holley RA (2014) Incidence of virulence factors in enterococci from raw and fermented meat and biofilm forming capacity at 25 °C and 37 °C. Int J Food Microbiol 170:65–69
Koeuth T, Versalovic J, Lupski JR (1995) Differential subsequence conservation of interspersed repetitive Streptococcus pneumoniae BOX elements in diverse bacteria. Genome Res 5(4):408–418
Lafarge V, Ogier JC, Girard V, Maladen V, Leveau JY, Gruss A, Delacroix-Buchet A (2004) Raw cow milk bacterial population shifts attributable to refrigeration. Appl Environ Microbiol 70(9):5644–5650
Lane DJ (1991) 16S/23S rRNA Sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic Acid Techniques in Bacterial Systematics. Wiley, Chichester, pp 115–175
Latorre AA, Van Kessel JS, Karns JS, Zurakowski MJ, Pradhan AK, Boor KJ, Jayarao BM, Houser BA, Daugherty CS, Schukken YH (2010) Biofilm in milking equipment on a dairy farm as a potential source of bulk tank milk contamination with Listeria monocytogenes. J Dairy Sci 93(6):2792–2802
Lourenco A, Machado H, Brito L (2011) Biofilms of Listeria monocytogenes produced at 12 °C either in pure culture or in co-culture with Pseudomonas aeruginosa showed reduced susceptibility to sanitizers. J Food Sci 76(2):143–148
Martinez S, Lopez M, Bernardo A (2003) Thermal inactivation of Enterococcus faecium: effect of growth temperature and physiological state of microbial cells. Lett Appl Microbiol 37(6):475–481
Martins ML, Pinto CL, Rocha RB, de Araujo EF, Vanetti MC (2006) Genetic diversity of Gram-negative, proteolytic, psychrotrophic bacteria isolated from refrigerated raw milk. Int J Food Microbiol 111(2):144–148
Mattila T, Manninen M, Kylasiurola AL (1990) Effect of cleaning-in-place disinfectants on wild bacterial strains isolated from a milking line. J Dairy Res 57(1):33–39
McAuley CM, Gobius KS, Britz ML, Craven HM (2012) Heat resistance of thermoduric enterococci isolated from milk. Int J Food Microbiol 154(3):162–168
Mittelman MW (1998) Structure and functional characteristics of bacterial biofilms in fluid processing operations. J Dairy Sci 81(10):2760–2764
Moretro T, Hermansen L, Holck AL, Sidhu MS, Rudi K, Langsrud S (2003) Biofilm formation and the presence of the intercellular adhesion locus ica, among staphylococci from food and food processing environments. Appl Environ Microbiol 69(9):5648–5655
Muyzer G, de Waal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59(3):695–700
Peng JS, Tsai WC, Chou CC (2002) Inactivation and removal of Bacillus cereus by sanitizer and detergent. Int J Food Microbiol 77(1–2):11–18
Ranieri ML, Huck JR, Sonnen M, Barbano DM, Boor KJ (2009) High temperature, short time pasteurization temperatures inversely affect bacterial numbers during refrigerated storage of pasteurized fluid milk. J Dairy Sci 92(10):4823–4832
Rickard AH, Gilbert P, High NJ, Kolenbrander PE, Handley PS (2003) Bacterial coaggregation: an integral process in the development of multi-species biofilms. Trends Microbiol 11(2):94–100
Rivas L, Dykes GA, Fegan N (2007) A comparative study of biofilm formation by Shiga toxigenic Escherichia coli using epifluorescence microscopy on stainless steel and a microtitre plate method. J Microbiol Methods 69(1):44–51
Rossetti L, Giraffa G (2005) Rapid identification of dairy lactic acid bacteria by M13-generated, RAPD-PCR fingerprint databases. J Microbiol Methods 63(2):135–144
Sharma M, Anand SK (2002) Characterization of constitutive microflora of biofilms in dairy processing lines. Food Microbiol 19(6):627–636
Simoes LC, Simoes M, Vieira MJ (2007) Biofilm interactions between distinct bacterial genera isolated from drinking water. Appl Environ Microbiol 73(19):6192–6200
Stackebrandt E, Frederiksen W, Garrity GM, Grimont PA, Kampfer P, Maiden MC, Nesme X, Rossello-Mora R, Swings J, Truper HG, Vauterin L, Ward AC, Whitman WB (2002) Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int J Syst Evol Microbiol 52(3):1043–1047
Stepanovic S, Vukovic D, Dakic I, Savic B, Svabic-Vlahovic M (2000) A modified microtiter-plate test for quantification of staphylococcal biofilm formation. J Microbiol Methods 40(2):175–179
Szweda P, Schielmann M, Milewski S, Frankowska A, Jakubczak A (2012) Biofilm production and presence of ica and bap genes in Staphylococcus aureus strains isolated from cows with mastitis in the eastern Poland. Pol J Microbiol 61(1):65–69
Acknowledgments
Financial support for this work was provided by projects from CICYT (Ref. AGL2011-24300-ALI) and INIA (Ref. RM2011-00005-00-00). A.B Flórez was supported by a research contract under Juan de la Cierva Program (Ref. JCI-2010-07457).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Cherif-Antar, A., Moussa–Boudjemâa, B., Didouh, N. et al. Diversity and biofilm-forming capability of bacteria recovered from stainless steel pipes of a milk-processing dairy plant. Dairy Sci. & Technol. 96, 27–38 (2016). https://doi.org/10.1007/s13594-015-0235-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13594-015-0235-4