
Abstract
In order to protect biodiversity, conservation genetics are of great importance. Until now, a few population genetic studies of Neotropical bumblebees are available but studies of temporal stability in genetic diversity are lacking. Here, genetic variability of three South Brazilian species Bombus bellicosus, B. pauloensis, and B. morio was investigated over time. Hereto, museum collection specimens of 1946 until 2012, from eight locations in Rio Grande do Sul, were genotyped at 16 microsatellite loci. During an additional sampling in the foraging season of 2015, no bumblebees could be collected possibly due to the super El Niño of 2015–2016. Our results on the collection specimens demonstrated a significantly higher genetic diversity in B. morio than in B. pauloensis. Genetic variability in B. pauloensis gradually and significantly decreased over time from 1946 until 2012; while in B. morio, genetic variability remained stable until the last time period (2010–2012). For B. bellicosus, not enough data was available. Although the studied populations became more vulnerable over time, for the conservation of Neotropical bumblebees, still more information is needed and could include more frequent monitoring of bumblebees. Adding B. bellicosus to the Brazilian list of threatened species is suggested.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Many studies in Europe and North America investigated the genetic variability in populations of declining and contemporary stable bumblebee (Bombus) species, and observed a lower genetic diversity in the populations of the declining species (Darvill et al. 2006; Ellis et al. 2006; Goulson et al. 2008; Charman et al. 2010; Cameron et al. 2011). The difference is supposed to be directly or indirectly caused by the combined impact of the different drivers of bee decline, such as loss of food resources, pesticide use, climate change, pathogen spill-over, and landscape modifications (e.g., Potts et al. 2010, 2015; Carvalheiro et al. 2013; Goulson et al. 2015). In the Neotropical region, the main threats are the loss of habitat due to deforestation with the continuous process of urbanization and agricultural development, and the introduction of exotic species (Freitas et al. 2009; Martins et al. 2013; Schmid-Hempel et al. 2014). In Chile and Argentina, populations of B. dahlbomii (Guérin-Meneville 1835) showed a fast decline in consequence of the introduction of the exotic species, B. terrestris (Linnaeus 1758) and B. ruderatus (Fabricius 1775), and of the impact of their hitchhiking parasites (Arbetman et al. 2013; Schmid-Hempel et al. 2014).
Bumblebee populations which are affected by these drivers, may become small and isolated (Reed and Frankham 2003; Frankham 2005; Zayed 2009). Small populations will possess lower genetic diversity levels due to the impact of genetic drift and reduced gene flow, which in turn will reduce their potential to adapt to current and future changes in the environment (Frankham 2005; Zayed 2009; Habel et al. 2014). Furthermore, inbreeding and inbreeding depression will increase in isolated populations due to the reduced gene flow and increased change of brother-sister matings (Frankham 2005; Zayed 2009; Habel et al. 2014).
Recently, this general hypothesis came under debate as multiple studies revealed similar differences of genetic diversity between declining and stable bumblebee species determined only in historical populations (for North America, Lozier et al. 2011; in Europe, Maebe et al. 2015), and by comparing both historical and recent populations (in North America, Lozier and Cameron 2009; and in Europe, Maebe et al. 2016). Furthermore, the latter two studies showed, in contrast with the general hypothesis, no reduction of genetic diversity over time (Lozier and Cameron 2009; Maebe et al. 2016). Even more, the difference in genetic diversity between species was already present before the alleged recent drivers of bumblebee decline could have acted (for Europe, this is generally accepted to have started around the 1950s; Maebe et al. 2016). The lower genetic variability in populations of declining species may be due to past population dynamics, such as the occurrences of bottlenecks during glacial oscillations (Lecocq et al. 2013), while the temporal stability of the reduced genetic diversity could then be maintained by countering drift effects by dispersal (as discussed in Maebe et al. 2016). Furthermore, as genetically pauperized bumblebees are more vulnerable, for instance more susceptible to diseases (Cameron et al. 2011; Whitehorn et al. 2011), it is essential to determine the genetic variation patterns in bumblebees. This knowledge will allow us to make and improve conservation strategies, and further secure the pollination services of wild bumblebees (Goulson et al. 2008; Zayed 2009; Maebe et al. 2016).
Here, we performed a genetic study on the populations of the three native bumblebee species of the Rio Grande do Sul State, South Brazil. The genetic diversity of the declining species, B. bellicosus (Smith 1879), and that of the two more abundant species, B. pauloensis (Friese 1913) and B. morio (Swederus 1787), was determined through time from 1946 to 2012 using museum collection material. An additional sampling attempt during the foraging season of 2015 was conducted to be able to compare historical with recent genetic diversity levels. Our sampling design allowed us to search if temporal changes in genetic diversity also occur in South America, and in turn gain knowledge on which species may need special attention and future conservation measures.
2 Material and methods
2.1 Study species
In Southern Brazil, where climate is predominantly moist subtropical, three species are mainly found: B. bellicosus, B. morio, and B. pauloensis (Cameron et al. 2007; Martins and Melo 2010; De Paula and Melo 2015). A fourth species native to South Brazil, B. brasiliensis (Lepeletier 1836) is not included in this study because it is a very rare species with only a few records in this area (Abramovich et al. 2004). B. bellicosus has the most limited distribution in South America, being present only in Uruguay, Southern Brazil, and most of Argentina (Abrahamovich et al. 2001). The two other species, B. pauloensis, formerly named B. atratus (Franklin 1913) (Moure and Melo 2012), and B. morio, are widespread in South America, with their distribution ranging from north-western South America to south-eastern Brazil, Uruguay, until central Argentina (Abrahamovich et al. 2004). In Brazil, B. bellicosus occurs in sympatry with B. pauloensis and B. morio in most regions of its distribution (Moure and Sakagami 1962; Françoso and Arias 2012). This species reaches its northern limit in the Paraná state (Brazil), where it was relatively abundant until the early 1980s, but it is now assumed to be locally extinct there (Martins and Melo 2010; Martins et al. 2015). Hence, 50 years ago, B. bellicosus outnumbered B. pauloensis and B. morio at some places, but nowadays B. pauloensis is generally the most abundant and widely distributed species, having a higher climatic and altitudinal tolerance and a broader distribution (Abrahamovich et al. 2004; Martins and Melo 2010).
2.2 Sampling
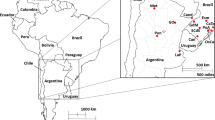
Specimens of the three species were obtained from multiple Brazilian collections. Most historical material was gathered from the entomological (Hymenoptera) collection of the Museum of Science and Technology at Pontifícia Universidade Católica do Rio Grande do Sul (PUCRS). Additional specimens were sampled from the entomological collection at Museu de Ciências Naturais da Fundação Zoobotânica do Rio Grande do Sul (curator—Aline Barcellos), from the entomological collection at UNISC in Santa Cruz do Sul (curator—Andreas Köhler) and the private collection of Mardiore Pinheiro (collaborator of Prof. Blochtein). Four time periods with enough specimens and/or multiple locations were selected: 1946–1959, 1991–1994, 1999–2004, and 2010–2012. Sampled locations were Esmeralda (Esm), Candiota (Can), Guarani das Missões (GdM), and Santa Cruz do Sul (SCdS). As bumblebee annual dispersal distance can range up to 50 km (Schmid-Hempel et al. 2007), samples from locations in the close proximity and within the same time frame were grouped together. This resulted in the grouped locations of Cambará do Sul (Cam) and Torres (Tor) as one location—CATO. Hence, locations Osório (Oso) and Capão da Canoa (CdC) as OSCA, and Porto Alegre (PA) with Viamão (Via), and Guaíba (Gua) as POA (Figure 1 and Table I). In order to collect recent bumblebee specimens, several field trips were performed across the state from the end of October until the end of December 2015 (Supplementary Table 1). To allow comparison of bumblebee-populations over time, sampling locations for current specimens were deduced from the most sampled collection locations.

Overview of the bumblebee species sampled in Rio Grande do Sul State (RS), South Brazil. The blue location is where only B. pauloensis is sampled, the red location with both B. pauloensis and B. bellicosus specimens, and in green B. pauloensis and B. morio specimens. Sampled locations were Esm, Esmeralda; Can, Candiota; GdM, Guarani das Missões; SCdS, Santa Cruz do Sul; CaTo, Cambará do Sul (Cam) and Torres (Tor); OsCa, Osório (Osó) and Capão da Canoa (CdC); and PoA, Porto Alegre (PA), Viamão (Via), and Guaíba (Gua).
2.3 DNA extraction
From collected specimens, one middle leg was used for DNA extraction. Individual DNA extraction was performed with a Chelex DNA extraction protocol as described in Maebe et al. (2015). The DNA extractions were stored in microcentrifuge tubes and frozen at – 20 °C for their later use.
2.4 Microsatellite protocol
Individual specimens were genotyped with 16 microsatellite (MS) loci which were originally developed for European bumblebee species. These 16 MS loci gave reliable signals in previous research using bumblebee museum samples (Maebe et al. 2015, 2016): BL13, BT02, BT23, BT24, BL02, BT04, BT05, BT08, and BT10 (Reber-Funk et al. 2006); B100, B11, B126, and B132 (Estoup et al. 1993); and 0294, 0304, and 0810 (Stolle et al. 2011; Supplementary Table 2). MS were amplified by multiplex PCR in 10 μl using the Type-it QIAGEN PCR kit. Each reaction contained 1.33 μl template DNA, Type-it Multiplex PCR Master Mix (2×, Qiagen), and the forward and reverse primer of four MS loci for each of four multiplex mixes (Supplementary Table 2) which were already successfully established by Maebe et al. (2016). The PCR protocol, and capillary electrophoreses on an ABI-3730xl sequencer (Applied Biosystems), was performed with the method as described in Maebe et al. (2013). The fragments were examined and scored manually using Peak Scanner Software v 2.0 (Applied Biosystems).
2.5 Data analysis
Due to some extra validation steps, described in Maebe et al. (2015), several genotyped individuals were excluded prior to data analyses. These validation steps included removal of specimens which could not be scored in a reliable manner for at least 10 loci, and exclusion of identified sisters with the programs Colony 2.0 (Wang 2004) employing corrections for genotyping errors (5% per locus), and by the 2-allele algorithm and consensus method implemented in Kinanalyzer (Ashley et al. 2009). Hence, genotypic linkage disequilibrium was tested using the program Fstat 2.9.3 (Goudet 2001), deviations from Hardy-Weinberg equilibrium (HW) with GenAlEx 6.5 (Peakall and Smouse 2006), and evidence of null alleles with Microchecker (Van Oosterhout et al. 2004).
Genetic diversity of each population was determined based on Nei’s unbiased expected heterozygosity (HE) and observed heterozygosity (HO) (Nei 1978) performed with the program GenAlEx 6.5 (Peakall and Smouse 2006), and the sample size-corrected private allelic richness (AR) estimated with the program Hp-Rare 1.1 (Kalinowski 2002). The latter parameter was calculated and normalized to a population of 10 diploid specimens. In order to detect a change in genetic diversity through time, we conducted linear mixed models (LMMs) in RStudio (R Development Core Team 2008) with R package lme4 version 1.1-10 (Bates et al. 2015). LMMs were performed for each species and both genetic diversity parameters (AR and HE) separately. Models started with location and time period as fixed factors and microsatellite loci as random factor. The latter was chosen to account for inter-locus variability, as is performed in Soro et al. (2017). With the dredge command within the MUMIn package, we selected the model that best fitted the pattern in genetic diversity based on Akaike’s Information Criterion (AIC) (Barton 2015; Maebe et al. 2016). Then, the selected LMMs were run in RStudio with R package lme4 version 1.1-10 (Bates et al. 2015) following the guidelines of a free available tutorial (http://popgen.nescent.org/LMM-Genetic-Diversity.html) at popgen.nescent.org, an online community resource which gives guidance and tutorials for population genetic analyses in R (Kamvar et al. 2017). By performing likelihood ratio tests (LTR), in which we compared the selected model including all factors to a null model without these factors, we analyzed the main effect of the factor of interest (see tutorial; Soro et al. 2017). Furthermore, we computed the variance of location and/or time period alone vs the variance for loci variability, as the marginal and conditional coefficient of determination, respectively (see tutorial; Nakagawa and Schielzeth 2013; Soro et al. 2017). Finally, by Tukey HSD post hoc comparisons using the R package multcomp (Hothorn et al. 2008), we searched for differences in genetic diversity between locations and/or time periods (see tutorial; Soro et al. 2017).
Population structure within each Bombus species was inferred, based on two different Bayesian clustering methods. The R package, Geneland 4.0.6 (Guillot et al. 2005) was used to estimate the number of groups by including the coordinates of each sample in the model. A spatial model with correlated allele frequency and null allele correction was run with K = 1 to K = 7 (maximum seven locations for a species) with 1,000,000 iterations, 100 thinning, and 1000 burn-in. The uncertainty of coordinates was set to 0.1 because the exact location of some specimens was not certain, while all other parameters were set as default.
The software Structure v. 2.3.3 (Pritchard et al. 2000) was used to perform a Bayesian approach to determine the number of populations in each species’ dataset separately. In this analysis, the number of populations (K) was estimated from 1 to 10 for both B. morio and B. pauloensis, and from 1 to 5 for B. bellicosus. Each K-value was calculated with a burn-in of 250,000 iterations and 750,000 MCMC data collecting steps, and was repeated nine times. The open-source program Structure Harvester v. 0.6.93 (Earl and vonHoldt 2012) was used to determine the best value of K (Evanno et al. 2005), and the program Distruct v.1.1 (Rosenberg 2004) was used for graphical visualization of the population structure.
3 Results
3.1 Sampling of recent Bombus specimens
Despite our survey efforts, no bumblebees could be collected in Rio Grande do Sul State, Brazil, during the 2015 foraging season (Supplementary Table 1). Thus, all genetic analysis consists only out of 265 specimens originated from the Brazilian museum collections. Based on the Colony 2.0 and Kinalyzer analyses, 66 specimens were identified as full sibs and removed from our dataset (Table I). Of the 16 microsatellites, only two loci (BT08 and 0294) were difficult to amplify and score reliably in B. morio. These loci were excluded from all further analyses for all three species to keep comparison of genetic parameters based on the same MS loci. Furthermore, 13 specimens which showed missing data at more than 5 out of 14 loci were also excluded (Table I). No significant linkage disequilibrium between loci were detected. Testing for genotype frequencies against Hardy-Weinberg equilibrium (HWE) expectations displayed several loci with heterozygote deficits (Supplementary Table 3) that may be due to the subpopulation structuring (=Wahlund effect; Wahlund 1928) or the presence of null alleles. However, only very low frequencies of null alleles (< 5%) were detected in these MS loci.
3.2 Population genetic diversity
For B. pauloensis, the allelic richness ranged from 2.017 to 2.842, with a mean AR of 2.313 (Table II). Overall populations observed heterozygosity values ranged from 0.214 to 0.498, while mean HO was 0.371. Mean expected heterozygosity was 0.458, with values ranging from 0.353 to 0.623 within the individual populations (Table II). In B. morio, the estimated mean AR, HO, and HE values were higher than in B. pauloensis (2.830, 0.475, and 0.609 vs 2.313, 0.371, and 0.458, respectively; Table II). The single population of B. bellicosus showed genetic diversity values which could fall within the higher range of the observed values for B. pauloensis (AR = 2.339, HO = 0.351, and HE = 0.474; Table II).
3.3 Changes in genetic diversity over time
By comparing the AIC scores of the different LMM models, best models in both species and for both genetic diversity parameters included time period as the only fixed factor (delta > 2; and Figure 2). The importance of time period on AR and HE can also be seen in B. pauloensis when comparing the models with and without time period as a fixed factor (LRT, AR, and HE: χ2 = 26.297, d.f. = 3, p < 0.001; and χ2 = 31.360, d.f. = 3, p < 0.001, respectively) (Table III). The marginal R2 accounted for time period was 4.7% for AR, and 6.7% for HE, while the conditional R2 was much higher with 77.6 and 73.8%, respectively (Nakagawa and Schielzeth 2013). The latter clearly showed the importance of taking inter-locus variability into account when performing LMM (as is discussed in Soro et al. 2017). In B. pauloensis, AR decreased for 26.65% over time (Table II and Figure 2) with the significant difference situated around 1991–1994 and 1999–2004 (Tukey HSD post hoc test comparisons: 1946–1959 vs 1999–2004, z = − 4.027, p < 0.001; 1946–1959 vs 2010–2012, z = − 4.821, p < 0.001; and 1991–1994 vs 2010–2012, z = − 3.458, p = 0.003; Table IV). A similar result was found for HE: a decreased of 40.14% over time (Table II and Figure 2) with the significant effect between 1991–1994 and 1999–2004 (Tukey HSD post hoc test comparisons: 1946–1959 vs 1999–2004, z = − 3.989, p < 0.001; 1946–1959 vs 2010–2012, z = − 4.980, p < 0.001; 1991–1994 vs 1999–2001, z = − 2.965, p = 0.015; and 1991–1994 vs 2010–2012, z = − 4.366, p < 0.001; Table IV).

Temporal comparison of genetic diversity in B. pauloensis and B. morio. For each location, the expected heterozygosity (HE) and the allelic richness (AR) over all microsatellite loci in each time period are given. In blue, B. pauloensis and in red, B. morio.
For B. morio, no similar effect over time was detected for AR (LRT: χ2 = 5.404, d.f. = 3, p = 0.145). As can be seen from Figure 2, AR remained fairly stable through time (Tukey HSD post hoc tests, p > 0.05; Table IV). However, also significant changes of HE over time were detected in B. morio (LRT: χ2 = 12.191, d.f. = 3, p = 0.007). Tukey HSD post hoc test comparisons showed a decrease of 19.2% (Table II and Figure 2) between 1991–1994 and 2010–2012 (z = − 3.174, p = 0.008; Table IV), and of 17.3% (Table II and Figure 2) between 1999–2004 and 2010–2012 (z = − 2.793, p = 0.026; Table IV).
3.4 Genetic structure
The Geneland analysis grouped the B. pauloensis populations into three clusters (K = 3), while B. morio populations were structured into four clusters (K = 4) (Figure 3). For B. pauloensis, the populations of CaTo and OsCa both of 1991–1994 were grouped together with Can of 1999–2004 in cluster 1. The 2nd cluster was formed by two populations of 1999–2004 (SCdS and PoA) and all recent populations (2010–2012) (Esm, GdM, and PoA). The two older populations of PoA (of 1946–1959 and 1991–1994) were grouped in cluster 3. For B. morio, the recent populations of GdM and PoA of 2010–2012 were grouped in cluster 1. Older populations (CaTo, OsCa both of 1991–1994, and PoA of 1946–1959) were grouped together in cluster 2, while populations of SCdS (1999–2004 and 2010–2012) and the populations of PoA (1999–2004 and 2010–2012) were grouped in clusters 3 and 4, respectively.

Population clustering estimated by Geneland analysis. For B. pauloensis, K = 4 (a), while K = 3 in B. morio (b). Each map represents a detected cluster with population membership indicated in color: a high membership, light yellow; a low membership, dark red.
For B. pauloensis, the Evanno method identified ΔK = 2, which is the best value of K (or number of populations) that fitted our data (Supplementary Table 4; Supplementary Fig. 1). Although for both B. morio and B. bellicosus, the Bayesian analysis via Structure Harvester also identified ΔK = 2 (Supplementary Table 4; Supplementary Fig. 1), we adapted the best K-value for B. bellicosus to K = 1. This change in final K-value is based on two reasons: (i) the Evanno method does not calculate, ΔK = 1, and (ii) in Structure, when ΔK = 2, each specimen of B. bellicosus belongs for ± 50% to both groups (Figure 4). In general, these structuring results showed temporal clustering of populations in B. pauloensis and in a lower degree also in B. morio.

Bayesian clustering of B. pauloensis, B. morio, and B. bellicosus populations by Structure. Each vertical line represents a specimen, while allele frequencies are colored. With indication of the “original” populations (or sampling locations).
4 Discussion
The objective of this study was to investigate the temporal stability of genetic diversity in three South Brazilian bumblebee species: in B. bellicosus, of which local extinction has been reported in literature (Martins and Melo 2010; Martins et al. 2015), and compared in to two more stable bumblebee species occurring in the same region, B. pauloensis and B. morio. For the latter two species, our results based on collection material clearly showed temporal instability in genetic diversity. Indeed, for B. pauloensis, our results showed a decrease of both AR and HE over time (26.65 and 40.14%, respectively; Table II and Figure 2). Although AR remained fairly stable through time for B. morio, also, a decrease of HE over time was detected between 1991–1994 and 2010–2012 (19.2%), and between 1999–2004 and 2010–2012 (17.3%; Table II and Figure 2). The structuring results showed also a temporal clustering of populations in B. pauloensis around 1991–1994 and 1999–2004, and also in a lower degree in the populations of B. morio (Figures 3 and 4).
These results are in contrast with the results on temporal stability of genetic diversity observed in multiple bumblebee species of Europe and North America (Maebe et al. 2016; Lozier and Cameron 2009). Although still very speculative, we hypothesize here that temporal differences in land-use practices between these continents, such as the long history of deforestation in Europe vs the more recent strong deforestation in South America (Hansen et al. 2013), could have caused the difference in temporal stability of genetic diversity in these bumblebee populations. However, further research will be needed to test this hypothesis.
In B. morio, the genetic variability was higher than in B. pauloensis (for AR, 2.830 vs 2.313 and for HE, 0.609 vs and 0.458, respectively; Table II). Although comparison of results from different studies should be treated with caution due to the use of different loci, the observed values of genetic diversity for B. morio obtained here were similar to those described by Francisco et al. (2015) (HE 0.558–0.745 vs 0.530–0.708; AR 3.73–4.85 vs 2.580–3.180; Francisco et al. (2015) vs this study, respectively). In addition to a high genetic diversity, Francisco et al. (2015) also found a low genetic divergence between distant B. morio populations. Their results suggested no effect of long-term isolation on population viability possibly due to a high dispersal ability and capacity to survive in urban environments and highly fragmented landscapes. However, our results showed recent losses of genetic diversity and population structuring, indicating some impact on population viability.
For B. pauloensis, our results demonstrated a gradual decrease of genetic diversity since the 1950s. Although B. pauloensis is supposed to be a stable species (Martins and Melo 2010), these results seem to indicate that B. pauloensis populations are becoming more vulnerable. Although further research is needed to confirm this result, it should be taken into account in future conservation strategies. If genetic diversity would decrease further, and populations become more isolated, this species might become sensitive to local extinction due to an increased vulnerability to changes and stressors in the environment, and susceptibility to diseases and pathogens (Whitehorn et al. 2011; Zayed 2009; Cameron et al. 2011).
For the declining species B. bellicosus, not all genetic parameters could be calculated due its scarcity numbers in the collections, compared to the other two species. The underrepresentation of this species in the collections could be due to its rarity in nature or may be an extra indication for its disappearance in South Brazil as described in recent literature (e.g., Martins and Melo 2010; Martins et al. 2015) which in a way represents a major loss of genetic diversity.
Unfortunately, during the 2015 foraging season, no bumblebees could be collected in the Southern Brazilian state Rio Grande do Sul. Although their absence could be part of the worldwide bee decline (e.g., Potts et al. 2010, 2015; Carvalheiro et al. 2013), there is here also an alternative explanation, namely, the “super” El Niño of 2015–2016. The region encountered frequent storm systems with rainfall averaged 100–250% above normal (NOAA/NWS/NCEP Climate Prediction Center 2015; Weather underground 2016; Supplementary Fig. 2 and Supplementary Fig. 3). Although the effect of the impact of El Niño on bees in literature is mixed (Wcislo and Tierney 2009), a study in Northwestern Costa Rica showed that the previous “super” El Niño in 1997–1999 caused a decline of large bees (Frankie et al. 2005). It also had great negative effects on vegetation, and caused a reduction or flowering delay in some of these bees’ key resources for building and provisioning of their nests which probably caused their decline (Frankie et al. 2005).
Here, without the extra recent specimens, we performed the best possible sampling and setup considering the difficulties when using and genotyping collection material. But still, the sampling size is rather small. Although this could limit the strength of some of our results, which is especially the case for B. morio, we were still able to make some clear conclusions regarding the temporal instability of genetic diversity within the populations of these bumblebee species in Rio Grande do Sul. For the accurate conservation of bumblebee species in the tropical and subtropical regions, it will be important to assemble more information on changes in genetic diversity and to assess the possible link with environmental influences such as deforestation. Furthermore, monitoring bumblebee species should be performed in a frequent basis (e.g., at least every 2 years) and species like B. bellicosus should be added to future editions of the Brazilian list of threatened animal species.
References
Abrahamovich, A.H., Díaz, N.B., Morrone, J.J. (2004) Distributional patterns of the neotropical and andean species of the genus Bombus (Hymenoptera: Apidae). Acta Zool. Mex. 20, 99–117.
Abrahamovich, A.H., Tellería, M.C., Díaz, N.B. (2001) Bombus species and their associated flora in Argentina. Bee World 82, 76–87.
Arbetman, M.P., Meeus, I., Morales, C.L., Aizen, M.A., Smagghe, G. (2013) Alien parasite hitchhikes to Patagonia on invasive bumblebee. Biol. Invasions, 15, 489–494.
Ashley, M.V., Caballero, I.C., Chaovalitwongse, W., Das Gupta, B., Govindan, P., et al. (2009) Kinalyzer, a computer program for reconstructing sibling groups. Mol. Ecol. Resour. 9, 1127–1131.
Barton, K. (2015) MuMIn: multi - model inference. R package version 1.15.1. (http://CRAN.R - project.org/package=MuMIn).
Bates, D., Maechler, M., Bolker, B., Walker, S. (2015) Fitting linear mixed - effects models using lme4. J. Stat. Softw. 67(1), 1–48.
Cameron, S.A., Hines, H.M., Williams, P.H.A. (2007) A comprehensive phylogeny of the bumble bees (Bombus). Biol. J. Linn. Soc. 91, 161–188.
Cameron, S.A., Lozier, J.D., Strange, J.P., Koch, J.B., Cordes, N., et al. (2011) Patterns of widespread decline in North American bumble bees. Proc. Natl. Acad. Sci. U. S. A. 108, 662–667.
Carvalheiro, L.G., Kunin, W.E., Keil P., Aguirre-Gutiérrez J., Ellis W.N., et al. (2013) Species richness declines and biotic homogenization have slowed down for NW - European pollinators and plants. Ecol. Lett. 16, 870–878.
Charman, T.G., Sears, J., Green, R.E., Bourke, A.F.G. (2010) Conservation genetics, foraging distance and nest density of the scarce Great Yellow Bumblebee (Bombus distinguendus). Mol. Ecol. 19, 2661–2674.
Darvill, B., Ellis, J.S., Goulson, D. (2006) Population structure and inbreeding in a rare and declining bumblebee, Bombus muscorum (Hymenoptera: Apidae). Mol. Ecol. 15, 601–611.
de Paula, G.A.R., Melo, G.A.R. (2015) Inferring sex and caste seasonality patterns in three species of bumblebees from Southern Brazil using biological collections. Neotrop. Entomol. 44, 10–20.
Earl, D.A., vonHoldt, B.M. (2012) Structure haverster: a website and program for visualizing structure output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361.
Ellis, J.S., Knight, M.E., Darvill, B., Goulson, D. (2006) Extremely low effective population sizes, genetic structuring and reduced genetic diversity in a threatened bumblebee species, Bombus sylvarum (Hymenoptera: Apidae). Mol. Ecol. 15, 4375–4386.
Estoup, A., Solignac, M., Harry, M., Cornuet, J.-M. (1993) Characterization of (GT)n and (CT)n microsatellites in two insect species Apis mellifera and Bombus terrestris. Nucleic Acids Res. 21, 1427–1431.
Evanno, G., Regnauts, S., Goudet, J. (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620.
Francisco, F.O., Santiago, L.R., Mizusawa, Y.M., Oldroyd, B.P., Arias, M.C. (2015) Genetic structure of island and mainland populations of a Neotropical bumble bee species. J. Insect Conserv. 20, 383.
Françoso, E., Arias, M.C. (2012) Characterization of microsatellite loci for Bombus pauloensis (Hymenoptera, Apidae, Bombini). Mol. Ecol. Resour. Available at http://biomath.trinity.edu/manuscripts/12-4/mer-11-0403.pdf.
Frankham, R. (2005). Genetics and extinction. Biol. Conserv. 126, 131–140.
Frankie, G.W., Rizzardi, M., Vinson, S.B., Griswold, T.L., Ronchi, P. (2005) Changing bee composition and frequency on a flowering legume, Andira inermis (Wright) Kunth ex DC. during El Niño and La Niña years (1997–1999) in northwestern Costa Rica. J. Kansas Entomol. Soc. 78(2), 100–117.
Freitas, B.M., Imperatriz-Fonseca, V.L., Medina, L.M., Kleinert, A.D.M.P., Galetto, L., et al. (2009) Diversity, threats and conservation of native bees in the Neotropics. Apidologie 40, 332–346.
Goudet, J. (2001) Fstat: a program to estimate and test gene diversities and fixation indices (version 2.9.3). Updated from Goudet, J (1995): Fstat (version 1.2): a computer program to calculate F - statistics. J. Hered. 86, 485–486.
Goulson, D., Lye, G.C., Darvill, B. (2008) Decline and conservation of bumble bees. Annu. Rev. Entomol. 53, 191–208.
Goulson, D., Nicholls, E., Botías, C., Rotheray, E.L. (2015) Bee declines driven by combined stress from parasites, pesticides, and lack of flowers. Science, 347(6229): 1255957.
Guillot, G., Mortier, F., Estoup, A. (2005) Geneland: A program for landscape genetics. Mol. Ecol. Notes 5, 712–715.
Habel, J.C., Hisemann, M., Finger, A., Danley, P.D., Zachos, F.E. (2014) The relevance of time series in molecular ecology and conservation biology. Biol. Rev. 89, 484–492.
Hansen, M.C., Potapov, P.V., Moore, R., Hancher, M., Turubanova, S.A. et al. (2013) High-resolution global maps of 21st-century forest cover change. Science 342(6160), 850–853. Maps are online available at: https://earthenginepartners.appspot.com/science-2013-global-forest (Accessed 5 November 2017).
Hothorn, T., Bretz, F., Westfall, P. (2008) Simultaneous inference in general parametric models. Biom. J. 50, 346–363.
Kalinowski, S.T. (2002) How many alleles per locus should be used to estimate genetic distances? Heredity 88, 62–65.
Kamvar, Z.N., López-Uribe, M.M., Coughlan, S., Grünwald, N.J., Lapp, H., Manel, S. (2017). Developing educational resources for population genetics in R: An open and collaborative approach. Mol. Ecol. Resour. 17, 120–128.
Lecocq, T., Dellicour, S., Michez, D., Lhomme, P., Vanderplanck, M. et al. (2013) Scent of a break-up: phylogeography and reporductive trait divergences in the red-tailed bumblebee (Bombus lapidarius). BMC Evol. Biol. 13, 263–280.
Lozier, J.D., Cameron, S.A. (2009) Comparative genetic analyses of historical and contemporary collections highlight contrasting demographic histories for the bumblebees Bombus pensylvanicus and B. impatiens in Illinois. Mol. Ecol. 18, 1875–1886.
Lozier, J.D., Strange, J.P., Stewart, I.J., Cameron, S.A. (2011) Patterns of range-wide genetic variation in six North American bumble bee (Apidae: Bombus) species. Mol. Ecol. 20, 4870–4888.
Maebe, K., Meeus, I., Maharramov, J., Grootaert, P., Michez, D., Rasmont, P., Smagghe, G. (2013) Microsatellite analysis in museum samples reveals inbreeding before the regression of Bombus veteranus. Apidologie 44(2), 188–197.
Maebe, K., Meeus, I., Ganne, M., De Meulemeester, T., Biesmeijer, K., et al. (2015) Microsatellite analysis of museum specimens reveals historical differences in genetic diversity between declining and more stable Bombus species. PLoS One 10, e0127870.
Maebe, K., Meeus, I., Vray, S., Claeys, T., Dekoninck, W., et al. (2016) Genetic diversity of restricted wild bumblebees was already low a century ago. Sci. Rep. 6, 38289.
Martins, A.C., Goncalves, R.B., Melo, G.A.R. (2013) Changes in wild bee fauna of a grassland in Brazil reveal negative effects associated with growing urbanization during the last 40 years. Zoologia 30, 157–176.
Martins, A.C., Melo, G.A.R. (2010) Has the bumblebee Bombus bellicosus gone extinct in the northern portion of its distribution range in Brazil? J. Insect Conserv. 14, 207–210.
Martins, A.C., Silva, D.P., De Marco, P., Melo, G.A.R. (2015) Species conservation under future climate change: the case of Bombus bellicosus, a potentially threatened South American bumblebee species. J. Insect Conserv. 19, 33–43.
Moure, J. S., Melo, G. A. R. (2012) Bombini Latreille, 1802. In Moure, J.S., Urban, D., Melo, G.A.R. (Orgs). Catalogue of Bees (Hymenoptera, Apoidea) in the Neotropical Region - online version. Available at http://www.moure.cria.org.br/catalogue. (Accessed 20 May 2016).
Moure, J.S., Sakagami, S.F. (1962) As mamangabas sociais do Brasil (Bombus Latreille) (Hymenoptera, Apoidea). Stud. Entomol. 5, 65–194.
Nakagawa, S., Schielzeth, H. (2013) A general and simple method for obtaining R2 from generalized linear mixed-effects models. Methods Ecol. Evol. 4, 133–142.
Nei, M. (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89, 583–590.
NOAA/NWS/NCEP Climate Prediction Center (2015) Climate diagnostics bulletin of September, October, November and December 2015. Available at: http://www.cpc.ncep.noaa.gov/products/CDB/CDB_Archive_pdf/pdf_CDB_archive.shtml (Accessed 25 June 2016 – April 2017).
Peakall, R., Smouse, F. (2006) GENALEX 6: Genetic Analysis in Excel. Population Genetic Software for Teaching and Research. Australian National University, Canberra, Australia.
Pritchard, J.K., Stephens, M., Donnelly, P. (2000) Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Potts, S.G., Biesmeijer, J.C., Kremen, C., Neumann, P., Schweiger, O., et al. (2010). Global pollinator declines: Trends, impacts and drivers. Trends Ecol. Evol. 25, 345–353.
Potts, S. G., Biesmeijer, J.C., Bommarco, R., Breeze, T., Carvalheiro, L. et al. (2015) Status and trends of European pollinators. Key findings of the STEP project. Pensoft Publishers, Sofia.
R Development Core Team (2008) R: a language and environment for statistical computing. R Foundation for Statistical Computing Vienna http://www.R-project.org.
Reber-Funk, C., Schmidt-Hempel, R., Schmid-Hempel, P. (2006) Microsatellite loci for Bombus spp. Mol. Ecol. Notes 6, 83–86.
Reed, D.H., Frankham, R. (2003) Correlation between fitness and genetic diversity. Conserv. Biol. 17, 230–237.
Rosenberg, N.A. (2004) Distruct: a program for the graphical display of population structure. Mol. Ecol. Notes 4, 137–138.
Schmid-Hempel, P., Schmid-Hempel, R., Brunner, P.C., Seeman, O.D., Allen, G.R. (2007) Invasion success of the bumblebee, Bombus terrestris, despite a drastic genetic bottleneck. Heredity 99, 414–422.
Schmid-Hempel, R., Eckhardt, M., Goulson, D., Heinzmann, D., Lange, C., Plischuk, S., Schmid-Hempel, P. (2014). The invasion of southern South America by imported bumblebees and associated parasites. J. Anim. Ecol. 83, 823–837.
Soro, A., Quezada-Euan, J.J.G., Theodorou, P., Moritz, R.F.A., Paxton, R.J. (2017) The population genetics of two orchid bees suggests high dispersal, low diploid male production and only an effect of island isolation in lowering genetic diversity. Conserv. Genet. 18(3), 607–619.
Stolle, E., Wilfert, L., Schmid-Hempel, R., Schmid-Hempel, P., Kube, M., et al. (2011) A second generation genetic map of the bumblebee Bombus terrestris (Linaeus, 1758) reveals slow genome and chromosome evolution in the Apidae. BMC Genomics 12, 48.
Van Oosterhout, C., Hutchinson, W.F., Wills, D.P.M., Shipley, P. (2004) MICROCHECKER: software for identifying and correcting genotyping errors. Mol. Ecol. Notes 4, 535–538.
Wahlund, S. (1928) Zusammensetzung von Population und Korrelationserscheinung vom Standpunkt der Vererbungslehre aus betrachtet. Hereditas 11, 65–106.
Wandeler, P., Hoeck, P.E.A., Keller, L.F. (2007) Back to the future: museum specimens in population genetics. Trends Ecol. Evol. 22, 634–642.
Wang, J. L. (2004) Sibship reconstruction from genetic data with typing errors. Genetics 166, 1963–1979.
Wcislo, W.T., Tierney, S.M. (2009) Behavioural environments and niche construction: the evolution of dim-light foraging in bees. Biol. Rev. 84, 19–37.
Weather underground (2016) El Niño, https://wunderground.atavist.com/el-nino-forecast (Accessed on 7 Jan 2016).
Whitehorn, P.R., Tinsley, M.C., Brown, M.J.F., Darvill, B., Goulson, D. (2011) Genetic diversity, parasite prevalence and immunity in wild bumblebees. Proc. R. Soc. B 278, 1195–1202.
Zayed, A. (2009) Bee genetics and conservation. Apidologie 40, 237–262.
Acknowledgements
The authors would like to thank all the researchers who helped with field work and with the search for bumblebees during the 2015 foraging season. We also acknowledge the curators of the several Brazilian bumblebee collections: Aline Barcellos (entomological collection at Museu de Ciências Naturais da Fundação Zoobotânica do Rio Grande do Sul), Andreas Köhler (the entomological collection at UNISC in Santa Cruz do Sul), and Mardiore Pinheiro (private collection). Finally, we thank three anonymous reviewers whose constructive remarks helped us to improve this manuscript.
Funding
This study was funded, as part of the BELBEES project, by the Belgian Science Policy (BELSPO; grant BR/132/A1/BELBEES).
Author information
Authors and Affiliations
Contributions
KM and GS conceived and designed the experiments. LG, PNS, and BB collected the specimens. KM and LG performed the experiments and analyzed the data. KM, LG, PNS, BB, and GS wrote the paper.
Corresponding author
Ethics declarations
Data accessibility
Our data set of microsatellite genotypes of each specimen will be archived at DRYAD: https://doi.org/10.5061/dryad.0n1hb08.
Additional information
Manuscript editor: Marina Meixner
Changements temporels de la variabilité génétique chez trois espèces de bourdons de Rio Grande do Sul, Brésil du Sud
Diversité génétique / microsatellites / Bombus / stabilité temporelle / Rio Grande do Sul
Zeitliche Veränderungen der genetischen Variabilität von drei Hummelarten aus Rio Grande do Sul, Südbrasilien
Genetische Diversität / Mikrosatelliten / Bombus / zeitliche Stabilität / Rio Grande do Sul
Electronic supplementary material
ESM 1
(DOCX 5001 kb)
Rights and permissions
About this article

Cite this article
Maebe, K., Golsteyn, L., Nunes-Silva, P. et al. Temporal changes in genetic variability in three bumblebee species from Rio Grande do Sul, South Brazil. Apidologie 49, 415–429 (2018). https://doi.org/10.1007/s13592-018-0567-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13592-018-0567-1