
Abstract
Chronic spontaneous urticaria (CSU) is an unpredictable inflammatory skin condition characterized by the spontaneous onset of itchy wheals, angioedema, or both, which occurs for longer than 6 weeks overall. Despite the relatively straightforward diagnostic algorithm for CSU, relying primarily on a detailed medical history and only limited laboratory tests, patients often wait years to be diagnosed, with many cycling through different healthcare practitioners before a diagnosis is made. Even then, current treatment options for CSU are limited, with approximately half of patients resistant to standard-of-care second-generation antihistamines at standard or higher doses. As such, there is an unmet need for improved, streamlined management for patients with CSU. Here, we review the evidence-based diagnostic algorithm for CSU, consider the required steps of the diagnostic workup, and provide practical, real-world advice on the management of CSU to improve the timely diagnosis and care of patients with this debilitating disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Patients can wait years before receiving a chronic spontaneous urticaria (CSU) diagnosis and treatment options are limited. |
There is an unmet need for improved, streamlined management for patients with CSU. |
Diagnosis is straightforward, based primarily on patient history and self-reporting. Only basic laboratory tests are required. |
We encourage dermatologists to be proactive and adopt straightforward, practical management algorithms to improve the timely diagnosis, personalized care, and lives of patients with CSU. |
Introduction
Chronic spontaneous urticaria (CSU) is an unpredictable inflammatory skin condition characterized by the sudden onset of itchy wheals (also known as hives), angioedema, or both, with a worldwide point prevalence of up to 3% [1]. Although the diagnosis of CSU is relatively straightforward, patients often endure long delays in diagnosis, with wait times ranging from 2 to 15 years [2, 3], and some are misdiagnosed [4]. Furthermore, approximately half of patients with CSU do not respond to guideline-recommended first-line second-generation antihistamines (sgAHs) [2]. In this review, we describe the clinical presentation and clinical burden of CSU, and practical evidence-based recommendations for the diagnosis and treatment of CSU to improve the lives of patients with this debilitating skin disease, focusing on the adult population. This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Characteristics and Clinical Presentation
Urticaria is a systemic condition affecting the skin characterized by the development of wheals (hives) and/or angioedema, and is classified on the basis of its duration (acute or chronic) and its triggers (inducible [CIndU] or spontaneous [CSU]; Fig. 1a) [1, 5]. Wheals are pruritic, often described as itchy and/or burning, are of variable size and shape, and are usually surrounded by erythema [5]. Angioedema is a sudden, pronounced deep swelling in the lower dermis and subcutis or mucous membranes that is often accompanied by tingling, burning, or tightness [5].

Classification of CU (a) and the diagnostic algorithm for CSU (b). Figure adapted under the CC BY 4.0 license from Zuberbier et al. Allergy. 2022;77:734–766 [5]. AAE acquired angioedema due to C1-inhibitor deficiency, AE adverse event, ACEi angiotensin-converting enzyme inhibitor, AID autoinflammatory disease, CIndU chronic inducible urticaria, CSU chronic spontaneous urticaria, CU chronic urticaria, HAE hereditary angioedema
In CIndU, wheals and/or angioedema appear following a known trigger and so cases can be subdivided on the basis of these causes, such as cold urticaria, contact urticaria, and symptomatic dermographism (Fig. 1a) [5]. In CSU, wheals appear spontaneously as a result of known or unknown causes; this pattern lasts for more than 6 weeks, with each isolated case typically resolving within 24 h [5]. Angioedema may also occur, typically resolving within 72 h [5], but this is not a consistent feature of CSU. Approximately 57% of patients present with wheals only, 37% present with wheals and angioedema, and 6% present with angioedema only [1]. Although wheals can appear anywhere on the body, they are usually seen on the arms and legs, whereas angioedema most commonly occurs on the face [1]. Signs and symptoms of CSU can occur daily or almost daily, or have an intermittent–recurrent course, and occur at any time of the day but most commonly during the evening [5, 6].
CSU is the most common subtype of chronic urticaria (CU), accounting for approximately 60–90% of all CU cases, and occurs more commonly in adults than children (point prevalence 0.02–2.7% vs 0.1–0.9%, respectively) [1]. The typical age of CSU onset is 6–9 years in children and 30–50 years in adults, with the majority (approx. 86%) of patients diagnosed with moderate-to-severe disease [1, 7]. Children typically present with lower rates of angioedema (19% vs 60%) and shorter disease duration (5 vs 12 months) compared with adults [8]; a more comprehensive review of CSU management in children is beyond the scope of this paper but is provided elsewhere [9, 10]. Cross-sectional, observational studies have shown that patients suffer a wide-ranging disease duration of 1–10 years [1, 11,12,13,14], with approximately 40% of patients enduring the disease for longer than 10 years [11]. Disease trajectory and characteristics impact CSU duration, with a relapsing/remitting disease course, co-existing angioedema, and systemic complaints associated with prolonged disease duration [15, 16]. Approximately two-thirds of patients with CSU report systemic complaints, including joint pain and fatigue [16]. In an observational study of 155 patients, 52% of patients who experienced systemic complaints had CSU for more than 4 years, compared with 31% of patients without systemic complaints [16].
Many patients with CSU report disease exacerbation in response to triggering factors, both mental, such as stress, and physical stimuli, as in concomitant chronic inducible urticaria. Other reported triggers include diet, pseudoallergens, and parasitic infestations [5]. Patients can also experience more severe disease following treatment with non-steroidal anti-inflammatory drugs (NSAIDs) [5, 17]. This is known as NSAID-exacerbated cutaneous disease and has been reported in up to 30% of patients with CSU [18, 19]. Viral infection can also lead to worsening urticaria symptoms [5, 17, 20]. There is evidence that viral infections such as herpes simplex, norovirus, and HHV-4 and -6 are associated with worsening CSU, though data and understanding of the viral role and relationship with CSU are lacking, despite frequent clinical observations of this association [20].
One virus and its role in the exacerbation of CSU that has been reported frequently in the literature is COVID-19 [20]. Dermatologists have observed that COVID-19 may result in new-onset CU and CSU, with researchers hypothesizing that inflammation triggered by COVID-19 can exacerbate CU symptoms via the direct or indirect activation of mast cells and basophils by SARS-CoV-2 [21, 22]. Furthermore, studies (2022–2023) have shown that COVID-19 vaccination can trigger disease exacerbation and case reports suggest new-onset CSU in some patients [23,24,25,26,27].
Although spontaneous remission of CSU may occur at any time, more than half of patients are not in remission at 5 years [28]. A recent literature review published in 2021 revealed that the cumulative weighted average estimates of spontaneous remission at 1, 5, and 20 years were 17%, 45%, and 73%, respectively [28]. However, limited evidence on rates and definitions of spontaneous remission preclude conclusive data on remission rates in patients with CSU [28].
Underlying Mechanisms and Endotypes
Mast cells are the major driver of CSU, with mast cell activation leading to the release of inflammatory mediators including histamine, type 2 cytokines, and chemokines, as well as synthesis of eicosanoids [1, 29, 30]. This leads to sensory nerve activation, vasodilatation, and an increase in vascular permeability, leading to the formation of wheals and angioedema [1, 5, 31]. The histaminergic pathway transmits acute itch in response to histamine secreted by mast cells, basophils, and keratinocytes, whereas the non-histaminergic pathway transmits chronic itch and is mediated by pruritogens other than histamine such as proteases and cytokines/chemokines that are secreted by mast cells, granulocytes, macrophages, lymphocytes, keratinocytes, and neurons [32, 33]. Peripheral and central sensitization can further amplify the effects of these inflammatory mediators [34].
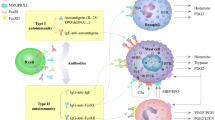
There are two main autoimmune endotypes of CSU: type I and type IIb. In both endotypes, mast cells are the primary drivers and basophils are important secondary contributors [17, 35]. Type I autoimmune CSU, or “autoallergy”, is mediated by immunoglobulin E (IgE) against various autoallergens, such as interleukin-24 and thyroid peroxidase (TPO) [36,37,38], and type IIb autoimmune CSU is mediated predominantly via immunoglobulin G (IgG) directed against the high-affinity IgE receptor FcεRI or FcεRI-bound IgE. Both mechanisms involve cross-linking of FcεRI and the activation of downstream signaling pathways via Bruton’s tyrosine kinase (BTK) [17, 39]. A number of diagnostic tests can be performed that may suggest a patient has type IIb versus type I autoimmune CSU for example, measurement of total IgE [5].
Lastly, these are also non-immunological pathways, such as the Mas-related G-protein coupled receptor X2 (MRGPRX2). This is a multiligand receptor that mediates degranulation of mast cells, basophils, and eosinophils via IgE-independent activation [40].
Burden and Unmet Needs
Despite the straightforward process of diagnosing CSU, relying primarily on a detailed medical history and only limited laboratory tests, patients experience long delays in diagnosis. Contributing factors include a lack of disease awareness and intermittent symptoms causing a delayed approach to patient care [3]. The average time from CU symptom onset to diagnosis is approximately 2–4 years [1], and in one small study (n = 25) patients with CSU waited as long as 15 years [3]. A US study demonstrated that patients with CSU waited approximately 14 weeks from their first physician consultation to diagnosis, despite 45% receiving their first appointment within only 6 weeks of symptom onset [41]. Furthermore, some patients cycle through a number of different healthcare practitioners, including an allergist, dermatologist, endocrinologist, general practitioner, and rheumatologist, before receiving a diagnosis [3]. An Italian study of 190 patients reported that 75% visited three or more physicians before receiving a diagnosis of CSU or having appropriate treatment prescribed [42].
The CSU disease course is unpredictable, with patients often experiencing relapse after months or years of full remission [5]. Recent studies indicate relapse rates of 6–31% [1]. Current treatment options for CSU are sub-optimal, with approximately half of patients resistant to first-line antihistamines at standard or higher doses [2]. The global, prospective AWARE (A World-wide Antihistamine-Refractory chronic urticaria patient Evaluation) study revealed that 78% of European patients with CU who were refractory to at least one dose of antihistamines had uncontrolled disease (defined as an UCT score < 12) [43].
Untreated or poorly managed CSU has a substantial negative impact on patients’ quality of life (QoL) [2, 7], affecting their emotional well-being, daily activities, sleep quality, and work performance [7]. In a cross-sectional study of 75 patients with CSU, sleep quality impairment was associated with worse disease control, greater pruritus and swelling, higher rates of anxiety and depression, and female sexual dysfunction [44]. Furthermore, a small cohort study (N = 60) has shown that CSU disease activity is higher in patients with comorbid depression compared with those without depression [45], and another showed that patients with CSU react with excessive anger and experience more passive aggressive interpersonal relationships than healthy controls [46].
CU is associated with a wide range of comorbidities, including thyroid disease, psychiatric disorders, atopic diseases, rheumatic diseases, inflammatory diseases, osteoporosis, and cancer [47]. A retrospective observational study showed that a longer duration of CSU was associated with a higher frequency of comorbidities (N = 173) [48]. Furthermore, an observational study in Israel that compared patients with (n = 12,778) and without CU (n = 10,714), showed that CU is associated with an increased risk of developing autoimmune disease, particularly in women, with conditions mostly diagnosed during the 10 years after CU diagnosis [49].
CSU is also associated with substantial direct and indirect costs, including healthcare visits and absence from and/or reduced productivity at work [7]. In a prospective, cross-sectional study in patients with CSU (N = 72), 40% (n = 29) required emergency room visit(s) for symptom control [15].
Diagnosis and Management
Diagnosis
Diagnosis of urticaria is typically straightforward, regardless of subtype or endotype, and symptom presentation is usually similar across age groups, ethnicities, and genders [1]. The recently published international EAACI/GA2LEN/EuroGuiDerm/APAAACI urticaria guideline provides an evidence-based diagnostic algorithm for CSU (Fig. 1b) [5]. In it, the essential steps in the diagnostic and prognostic workup of patients with suspected CSU comprise: (1) a comprehensive medical history; (2) physical examination; (3) basic tests, i.e., differential blood count, C-reactive protein (CRP) and/or erythrocyte sedimentation rate (ESR), and, for patients under specialist care, total IgE and IgG anti-TPO; and (4) assessment of disease activity, impact on QoL, and disease control, utilizing patient-reported outcome measures such as the weekly urticaria activity score (UAS7; used to determine disease activity and response to treatment), the angioedema activity score (AAS; used to assess disease activity), the chronic urticaria quality of life questionnaire (CU-Q2oL; used to determine QoL impairment in patients with wheals), the angioedema quality of life questionnaire (AE-QoL; used to determine QoL impairment in patients with angioedema, with/without wheals), the Urticaria Control Test (UCT; used to determine disease control in patients with wheals with/without angioedema), and the angioedema control test (AECT; used to determine disease control in patients with angioedema, with/without wheals) [5]. Although this guideline recommends basic laboratory testing in all patients, this is not always done in clinical practice. There are seven components of the diagnostic workup for CSU, which include confirming the diagnosis and excluding differential diagnoses, identifying markers of autoimmune CSU, and checking for comorbidities (Fig. 1b) [5].
History taking should be comprehensive and include the time of symptom onset; duration of specific individual wheals; the shape, size, color, and distribution of wheals; symptom characteristics; and family and personal history. A checklist of items to consider during history taking is included in Table 1 [50]. It is important to obtain sufficient detail from the patient to streamline the diagnostic process and prevent the need for multiple consultations [50, 51]. Important questions to ask patients with suspected CSU, that will help rule out other diagnoses, are included in Table 2.
As part of the physical examination, physicians should review patients’ own pictures of wheals and/or angioedema and any other documentation of signs and symptoms [5]. Scratching signs, such as excoriations, are rarely seen in patients with urticaria since patients typically rub, rather than scratch, their wheals [52]. Identifying signs of scratch can therefore help to differentiate urticaria from atopic dermatitis, which is typically characterized by severe excoriations [52]. Images of skin in patients with CSU are included in Fig. 2.

Images of skin in patients with CSU. Image credit: The Full Spectrum of Dermatology: A Diverse and Inclusive Atlas. CSU chronic spontaneous urticaria
There is a lack of distinguishing diagnostic biomarkers for CSU. As a result, the authors recommend only a small number of additional laboratory tests, not as part of the diagnosis, but to facilitate endotyping and prognostication (Table 3). Total IgE and IgG-anti-TPO should also be assessed in patients with CSU in specialist care for whom type IIb autoimmune CSU is suspected [1], with a high IgG-anti-TPO-to-total IgE ratio currently the most robust surrogate marker of this endotype [5]. In addition, elevated CRP and reduced eosinophil and basophil levels may indicate type IIb autoimmune CSU [5], although this has yet to be firmly established. Additional tests, including extensive food/inhalant allergy testing, are rarely needed and should only be performed if indicated by the patient’s history and physical examination [1, 5]. Although there is no specific guideline-based recommendation to routinely test for antinuclear antibodies (ANA), individual patient factors and clinical judgement can influence the decision to test for ANA or other autoimmune markers [53].
It is important to differentiate urticaria from other conditions that can manifest with wheals and/or angioedema but have distinct pathophysiology and/or clinical presentation [5]. This can be achieved with detailed history taking, supported by basic exploratory tests [5]. In particular, dermographism should also be considered, since this can often co-occur with CSU [5]. Other, albeit often rare, differential diagnoses include urticarial vasculitis, mastocytosis, mast cell activation syndrome, autoinflammatory syndromes (e.g., cryopyrin-associated periodic syndromes or Schnitzler’s syndrome), bradykinin-mediated angioedema, urticarial drug eruption, eczematous urticarial dermatitis, Well’s syndrome, bullous pemphigoid, insect bite reactions, and erythema multiforme [5, 54]. In patients with wheals only, urticarial vasculitis, urticarial pemphigoid, and other autoinflammatory disorders should be ruled out, and in patients with angioedema only, bradykinin-mediated angioedema-like angiotensin-converting enzyme (ACE) inhibitor-induced angioedema and hereditary angioedema should be ruled out [5]. Skin biopsy should only be performed in patients who are suspected to have a condition other than CSU [5].
Adherence to best-practice guidelines is fundamental for improving treatment outcomes in CSU [1, 55]. In a 2017 global survey of 1140 physicians, the majority of whom were allergists/clinical immunologists, almost one-third did not follow a CSU guideline or deviated from guideline recommendations [55]. At least one of the available diagnostic tests was performed by 85% of physicians who followed a guideline, compared with only 70% who did not follow a guideline [55]. Furthermore, physicians who followed a guideline more frequently used sgAHs as first-line therapy (64% vs 46%) and reported higher efficacy with these agents versus those who did not follow a guideline [55]. Another study, from the UK, published in 2015, showed a considerable discrepancy between dermatologists and allergists/immunologists in terms of which guidelines they referred to for patients with CU, as well as differences in investigations performed and use of first-line antihistamine between specialties [56].
Current Treatment Landscape
Treatment of CSU is based on a stepwise approach according to disease severity and treatment response with current standard-of-care therapies targeting histamine and IgE [5]. Despite few therapeutic advances in CSU until recently, many novel compounds are in late-stage clinical development. A treatment algorithm including practical points to consider can be seen in Fig. 3a and a treatment availability timeline is included in Fig. 3b.

Treatment algorithm for CSU (a). Panel adapted under the CC BY 4.0 license from Zuberbier et al. Allergy. 2022;77:734–766 [5]. A timeline of approved and investigational therapies for CSU (b). *300 mg Q4W; †Up to 600 mg Q2W; ‡Up to 5 mg/kg. CSU chronic spontaneous urticaria, Q2W every 2 weeks, Q4W every 4 weeks
Despite being available for the treatment of urticaria since the 1950s, first-generation antihistamines are not recommended because of their pronounced sedative and anticholinergic effects, and interactions with alcohol and other drugs such as analgesics and sedatives [5]. Per the EAACI/GA2LEN/EuroGuiDerm/APAAACI guideline, non-sedating or minimally sedating sgAHs that block the H1 receptor are the standard first-line treatment for CSU, with bilastine, cetirizine, desloratadine, ebastine, fexofenadine, and levocetirizine among the available agents (bilastine and ebastine are not approved in the USA). An increase in dose of sgAHs (up to fourfold) is recommended if the standard dose is not sufficient to control symptoms [5]. Despite the efficacy benefits of sgAHs, approximately 61% of patients with CSU do not respond to standard doses [57] and updosing and/or switching between these agents is not always effective [58]. Available evidence on rates of response to updosed sgAHs in patients whose CSU is refractory to standard doses varies widely: a systematic review and meta-analysis reported remission rates of 63% [57], while other studies have shown that 18–70% of patients continue to have uncontrolled disease [58,59,60]. A retrospective study of patients with CU (N = 657; 556 with CSU) reported that 64% of patients with an inadequate response to standard-dose sgAHs did not benefit from switching to a second sgAH [61]. Furthermore, the AWARE study showed that approximately 42% of patients with uncontrolled CU following updosed sgAHs (n = 44/105) were not offered further treatment escalation to third-line treatment [58].
Omalizumab, a first-in-class anti-IgE monoclonal antibody (mAb), is recommended as an add-on therapy in the second-line setting in adults and adolescents (≥ 12 years old) with CSU who have an inadequate response to H1 antihistamines [62, 63]. The recommended initial dose is 300 mg every 4 weeks [5]. A US Food and Drug Administration (FDA) label change in 2021 permitted the self-administration of omalizumab after at least three physician-guided doses in patients with CSU who have no prior history of anaphylaxis or hypersensitivity reactions [62, 64]. Real-world evidence from the AWARE study in 2727 patients with CSU showed that approximately one-third are treated with omalizumab during the first 2 years after diagnosis [58]. Predictive markers of response to omalizumab have been identified, including higher baseline circulating IgE and basophil FcεRI expression, although findings have been inconsistent to date [65,66,67,68,69]. However, despite robust clinical and real-world evidence supporting the use of omalizumab for CSU [70,71,72,73,74], many patients receiving this agent do not achieve symptom control [75]. Patients also require monitoring for anaphylaxis, for which a black box warning is included in the FDA label [62, 63]. If anaphylaxis occurs, it typically occurs within the first three doses of omalizumab [62, 63]. Anaphylaxis was observed in 0.1% of patients with asthma in clinical trials, and in approximately 0.2% of patients on the basis of post-marketing data [62, 63]. There were no reports of omalizumab-related anaphylaxis in patients with CSU during phase 3 clinical trials [72,73,74].
In patients who are inadequately controlled on sgAHs and omalizumab for 6 months or less, the immunosuppressant cyclosporin at 3.5–5 mg/kg/day is recommended in the third-line setting as add-on to sgAHs [5]. Efficacy of cyclosporin for the treatment of CSU has been demonstrated in clinical and real-world settings [76]. A retrospective analysis (N = 110) showed that features associated with type IIb autoimmune urticaria, namely positive autologous serum skin test, low total IgE, and anti-TPO positivity, were more likely to occur in patients responding to cyclosporin than in those responding to omalizumab [77]. However, the use of cyclosporin for the treatment of CSU is off-label and is only recommended in patients with severe disease refractory to sgAH/omalizumab combination treatment [5].
A short course of systemic corticosteroids is included in the guidelines for rapid control of severe disease flares in patients with CSU; a maximum of 10 days using the lowest effective prednisone-equivalent dose is recommended because of severe side effects associated with long-term steroid treatment [5]. However, the authors advise to avoid treatment with corticosteroids wherever possible. Alternative treatment options that may be valuable in individual patients include azathioprine, hydroxychloroquine, mycophenolate mofetil, methotrexate, dapsone, and narrow-band ultraviolet B; these agents should be used with caution because there is a lack of published evidence [5].
Novel Therapies
Recent advances in the understanding of the pathogenesis of urticaria has led to a rich pipeline of novel targeted therapies that are in late-stage clinical trials for patients with CSU refractory to standard treatment (Fig. 4) [78]. These therapies aim to silence mast cells via inhibitory receptors or reduce mast cell numbers instead of inhibiting mast cell mediators [5].

Pathophysiology and novel treatment targets in CSU. Figure adapted under the CC BY 4.0 license from Mustari AP et al. Indian Dermatol Online J. 2023;14:9–20 and Yosipovitch G et al. Dermatol Ther (Heidelb). 2023;13:1647–1660 [78, 79]. BTK Bruton tyrosine kinase, CRTh2 chemoattractant receptor-homologous molecule expressed on Th2 cells, EP eosinophil peroxide, FcεRI high‑affinity IgE receptor, HR histamine 4 receptor, IgE immunoglobulin E, IL interleukin, JAK-1 Janus kinase 1, MBP major basic protein, MRGPCRX2 Mas‑related G-protein coupled receptor X2, PGD2 prostaglandin D2, R receptor, SYK spleen tyrosine kinase, TH2 T helper 2, TSLP thymic stromal lymphopoietin
BTK is a cornerstone of both FcεRI and B cell receptor signaling, controlling FcεRI-mediated mast cell activation and the production of autoantibodies by B cells, and is therefore a promising therapeutic target in CSU [79,80,81]. In a phase 2b dose-finding study, all doses of remibrutinib elicited significant improvements in UAS7 from baseline to week 4, and a reduced UAS7 score from week 1 until week 12, compared with placebo in patients with CSU that was inadequately controlled with sgAH (N = 311). Most adverse events were mild to moderate in severity [82].
Targeting KIT, a cell surface receptor present on mast cells, has the potential to deplete mast cell numbers and/or prevent mast cell differentiation [79, 83, 84]. Barzolvolimab, an anti-KIT mAb, demonstrated a favorable safety profile and promising clinical activity in a phase 1 multiple ascending dose study in adults with moderate-to-severe CSU refractory to sgAHs [85].
It is believed that inflammatory cytokines, interleukin (IL)-4 and IL-13, play a key role in CSU pathogenesis via activation of mast cells, basophils, and eosinophils. Dupilumab is a monoclonal antibody that blocks IL-4Rα, thus preventing signalling of IL-4 and IL-13, which in turn prevents the activation of the aforementioned immune cells [86].
Other targets include IL-5Rα, Siglec 8, thymic stromal lymphopoietin, cKIT, MRGPRX2, and Janus kinase 1, with clinical trials ongoing.
Conclusion
A substantial proportion of patients with CSU remain undiagnosed, untreated, or uncontrolled on standard-of-care, guideline-recommended therapies. Thus, optimizing the diagnosis and management of CSU is a matter of urgency. We encourage dermatologists to be proactive and adopt straightforward, practical management algorithms to improve the timely diagnosis, personalized care, and lives of patients with CSU.
References
Kolkhir P, Giménez-Arnau AM, Kulthanan K, Peter J, Metz M, Maurer M. Urticaria. Nat Rev Dis Primers. 2022;8(1):61.
Gonçalo M, Giménez-Arnau A, Al-Ahmad M, et al. The global burden of chronic urticaria for the patient and society. Br J Dermatol. 2021;184(2):226–36.
Goldstein S, Eftekhari S, Mitchell L, et al. Perspectives on living with chronic spontaneous urticaria: from onset through diagnosis and disease management in the US. Acta Derm Venereol. 2019;99(12):1091–8.
Toubi E, Giménez-Arnau AM, Maurer M, Vadasz Z. Articular angioedema in patients with chronic spontaneous urticaria is frequently misdiagnosed as arthritis. J Allergy Clin Immunol Pract. 2020;8(9):3232–3.
Zuberbier T, Abdul Latiff AH, Abuzakouk M, et al. The international EAACI/GA2LEN/EuroGuiDerm/APAAACI guideline for the definition, classification, diagnosis, and management of urticaria. Allergy. 2022;77(3):734–66.
Maurer M, Ortonne JP, Zuberbier T. Chronic urticaria: an internet survey of health behaviours, symptom patterns and treatment needs in European adult patients. Br J Dermatol. 2009;160(3):633–41.
Maurer M, Abuzakouk M, Bérard F, et al. The burden of chronic spontaneous urticaria is substantial: real-world evidence from ASSURE-CSU. Allergy. 2017;72(12):2005–16.
Özçeker D, Can PK, Terzi Ö, et al. Differences between adult and pediatric chronic spontaneous urticaria from a cohort of 751 patients: Clinical features, associated conditions and indicators of treatment response. Pediatr Allergy Immunol. 2023;34(2):e13925.
Chang J, Cattelan L, Ben-Shoshan M, Le M, Netchiporouk E. Management of pediatric chronic spontaneous urticaria: a review of current evidence and guidelines. J Asthma Allergy. 2021;14:187–99.
Votto M, Achilli G, De Filippo M, et al. Pediatric chronic spontaneous urticaria: a brief clinician’s guide. Expert Rev Clin Immunol. 2022;18(9):889–99.
Sánchez-Díaz M, Salazar-Nievas MC, Molina-Leyva A, Arias-Santiago S. The burden on cohabitants of patients with chronic spontaneous urticaria: a cross-sectional study. J Clin Med. 2022;11(11):3228.
Doğan N, Çildağ S, Yenisey Ç, Şentürk T. The association between chronic spontaneous urticaria and HLA class I and class II antigen. Turk J Med Sci. 2020;50(5):1231–5.
Plavsic A, Tomic-Spiric V, Arandjelovic S, Miskovic R, Dimitrijevic M, Peric-Popadic A. Biomarkers of disease activity in patients with chronic spontaneous urticaria. Postepy Dermatol Alergol. 2021;38(6):1017–22.
Ye YM, Koh YI, Choi JH, et al. The burden of symptomatic patients with chronic spontaneous urticaria: a real-world study in Korea. Korean J Intern Med. 2022;37(5):1050–60.
Stepaniuk P, Kan M, Kanani A. Natural history, prognostic factors and patient perceived response to treatment in chronic spontaneous urticaria. Allergy Asthma Clin Immunol. 2020;16:63.
Doong JC, Chichester K, Oliver ET, Schwartz LB, Saini SS. Chronic idiopathic urticaria: systemic complaints and their relationship with disease and immune measures. J Allergy Clin Immunol Pract. 2017;5(5):1314–8.
Kolkhir P, Muñoz M, Asero R, et al. Autoimmune chronic spontaneous urticaria. J Allergy Clin Immunol. 2022;149(6):1819–31.
Asero R. Nonsteroidal anti-inflammatory drugs hypersensitivity in chronic spontaneous urticaria in the light of its pathogenesis. Eur Ann Allergy Clin Immunol. 2022;54(4):189–91.
Blanca-Lopez N, Soriano V, Garcia-Martin E, Canto G, Blanca M. NSAID-induced reactions: classification, prevalence, impact, and management strategies. J Asthma Allergy. 2019;12:217–33.
Kocatürk E, Muñoz M, Elieh-Ali-Komi D, et al. How infection and vaccination are linked to acute and chronic urticaria: a special focus on COVID-19. Viruses. 2023;15(7):1585.
Kempuraj D, Selvakumar GP, Ahmed ME, et al. COVID-19, mast cells, cytokine storm, psychological stress, and neuroinflammation. Neuroscientist. 2020;26(5–6):402–14.
Ozdemir O, Dikici U. Safely use of omalizumab during SARS-CoV-2 infection in patients with chronic spontaneous urticaria. J Cosmet Dermatol. 2023;22(1):26–7.
Picone V, Napolitano M, Martora F, Guerriero L, Fabbrocini G, Patruno C. Urticaria relapse after mRNA COVID-19 vaccines in patients affected by chronic spontaneous urticaria and treated with antihistamines plus omalizumab: a single-center experience. Dermatol Ther. 2022;35(11):e15838.
Lee JH, Shin E, Kim HK, et al. Exacerbation of chronic spontaneous urticaria following coronavirus disease 2019 (COVID-19) vaccination in omalizumab-treated patients. J Allergy Clin Immunol Pract. 2023;11(8):2403–10.
Ben-Fredj N, Chahed F, Ben-Fadhel N, et al. Case series of chronic spontaneous urticaria following COVID-19 vaccines: an unusual skin manifestation. Eur J Clin Pharmacol. 2022;78(12):1959–64.
Duperrex O, Tommasini F, Muller YD. Incidence of chronic spontaneous urticaria following receipt of the COVID-19 vaccine booster in Switzerland. JAMA Netw Open. 2023;6(2):e2254298.
Strahan A, Ali R, Freeman EE. Chronic spontaneous urticaria after COVID-19 primary vaccine series and boosters. JAAD Case Rep. 2022;25:63–6.
Balp MM, Halliday AC, Severin T, et al. Clinical remission of chronic spontaneous urticaria (CSU): a targeted literature review. Dermatol Ther (Heidelb). 2022;12(1):15–27.
Garcovich S, Maurelli M, Gisondi P, Peris K, Yosipovitch G, Girolomoni G. Pruritus as a distinctive feature of type 2 inflammation. Vaccines (Basel). 2021;9(3):303.
Moon TC, Befus AD, Kulka M. Mast cell mediators: their differential release and the secretory pathways involved. Front Immunol. 2014;5:569.
Puxeddu I, Pratesi F, Ribatti D, Migliorini P. Mediators of inflammation and angiogenesis in chronic spontaneous urticaria: are they potential biomarkers of the disease? Mediators Inflamm. 2017;2017:4123694.
Sutaria N, Adawi W, Goldberg R, Roh YS, Choi J, Kwatra SG. Itch: pathogenesis and treatment. J Am Acad Dermatol. 2022;86(1):17–34.
Yosipovitch G, Rosen JD, Hashimoto T. Itch: from mechanism to (novel) therapeutic approaches. J Allergy Clin Immunol. 2018;142(5):1375–90.
Jin SY, Wang F. Sensitization mechanisms of chronic itch. Int J Dermatol Venereol. 2019;2(4):211.
Giménez-Arnau AM, DeMontojoye L, Asero R, et al. The pathogenesis of chronic spontaneous urticaria: the role of infiltrating cells. J Allergy Clin Immunol Pract. 2021;9(6):2195–208.
Asero R, Ferrer M, Kocaturk E, Maurer M. Chronic spontaneous urticaria: the role and relevance of autoreactivity, autoimmunity, and autoallergy. J Allergy Clin Immunol Pract. 2023;11(8):2302–8.
Schmetzer O, Lakin E, Topal FA, et al. IL-24 is a common and specific autoantigen of IgE in patients with chronic spontaneous urticaria. J Allergy Clin Immunol. 2018;142(3):876–82.
Altrichter S, Peter HJ, Pisarevskaja D, Metz M, Martus P, Maurer M. IgE mediated autoallergy against thyroid peroxidase—a novel pathomechanism of chronic spontaneous urticaria? PLoS ONE. 2011;6(4):e14794.
Kaplan A, Lebwohl M, Giménez-Arnau AM, Hide M, Armstrong AW, Maurer M. Chronic spontaneous urticaria: focus on pathophysiology to unlock treatment advances. Allergy. 2023;78(2):389–401.
Wedi B, Gehring M, Kapp A. The pseudoallergen receptor MRGPRX2 on peripheral blood basophils and eosinophils: expression and function. Allergy. 2020;75(9):2229–42.
Hoskin B, Ortiz B, Paknis B, Kavati A. Exploring the real-world profile of refractory and non-refractory chronic idiopathic urticaria in the USA: clinical burden and healthcare resource use. Curr Med Res Opin. 2019;35(8):1387–95.
Cappuccio A, Limonta T, Parodi A, et al. Living with chronic spontaneous urticaria in Italy: a narrative medicine project to improve the pathway of patient care. Acta Derm Venereol. 2017;97(1):81–5.
Maurer M, Houghton K, Costa C, et al. Differences in chronic spontaneous urticaria between Europe and Central/South America: results of the multi-center real world AWARE study. World Allergy Organ J. 2018;11(1):32.
Sánchez-Díaz M, Rodríguez-Pozo J, Latorre-Fuentes JM, Salazar-Nievas MC, Alejandro ML, Arias-Santiago S. Sleep quality as a predictor of quality-of-life and emotional status impairment in patients with chronic spontaneous urticaria: a cross-sectional study. Int J Environ Res Public Health. 2023;20(4):3508.
Memet B, Vurgun E, Barlas F, Metz M, Maurer M, Kocatürk E. In chronic spontaneous urticaria, comorbid depression linked to higher disease activity, and substance P levels. Front Psychiatry. 2021;12:667978.
Altınöz AE, Taşkıntuna N, Altınöz ST, Ceran S. A cohort study of the relationship between anger and chronic spontaneous urticaria. Adv Ther. 2014;31(9):1000–7.
Sánchez-Borges M, Ansotegui IJ, Baiardini I, et al. The challenges of chronic urticaria part 1: epidemiology, immunopathogenesis, comorbidities, quality of life, and management. World Allergy Organ J. 2021;14(6):100533.
Agondi RC, Argôlo PN, Mousinho-Fernandes M, Gehlen B, Kalil J, Motta AA. Multiple comorbidities in patients with long-lasting chronic spontaneous urticaria. An Bras Dermatol. 2023;98(1):93–6.
Confino-Cohen R, Chodick G, Shalev V, Leshno M, Kimhi O, Goldberg A. Chronic urticaria and autoimmunity: associations found in a large population study. J Allergy Clin Immunol. 2012;129(5):1307–13.
Cherrez-Ojeda I, Robles-Velasco K, Bedoya-Riofrío P, et al. Checklist for a complete chronic urticaria medical history: an easy tool. World Allergy Organ J. 2017;10(1):34.
Bonnekoh H, Krause K, Kolkhir P. Chronic recurrent wheals—if not chronic spontaneous urticaria, what else? Allergol Select. 2023;7:8–16.
Petkova E, Staevska M. Editor’s pick: managing chronic urticaria: Quo vadis? EMJ. 2020;8(1):66–74.
Arunkajohnsak S, Jiamton S, Tuchinda P, et al. Do antinuclear antibodies influence the clinical features of chronic spontaneous urticaria?: a retrospective cohort study. Biomed Res Int. 2022;2022:7468453.
Schettini N, Corazza M, Schenetti C, Pacetti L, Borghi A. Urticaria: a narrative overview of differential diagnosis. Biomedicines. 2023;11(4):1096.
Kolkhir P, Pogorelov D, Darlenski R, et al. Management of chronic spontaneous urticaria: a worldwide perspective. World Allergy Organ J. 2018;11(1):14.
Wu CH, Ardern-Jones MR, Eren E, Venter C. An observational study of the diagnosis and management of chronic urticaria in the UK. Int Arch Allergy Immunol. 2015;167(1):1–8.
Guillén-Aguinaga S, Jáuregui Presa I, Aguinaga-Ontoso E, Guillén-Grima F, Ferrer M. Updosing nonsedating antihistamines in patients with chronic spontaneous urticaria: a systematic review and meta-analysis. Br J Dermatol. 2016;175(6):1153–65.
Maurer M, Costa C, Gimenez Arnau A, et al. Antihistamine-resistant chronic spontaneous urticaria remains undertreated: 2-year data from the AWARE study. Clin Exp Allergy. 2020;50(10):1166–75.
Wongjirattikarn R, Chaowattanapanit S, Foocharoen C, et al. Factors associated with refractoriness to an up to fourfold dosage of antihistamines in isolated chronic spontaneous urticaria. J Cutan Med Surg. 2022;26(6):593–9.
van den Elzen MT, van Os-Medendorp H, van den Brink I, et al. Effectiveness and safety of antihistamines up to fourfold or higher in treatment of chronic spontaneous urticaria. Clin Transl Allergy. 2017;7:4.
Ayse Ornek S, Orcen C, Church MK, Kocaturk E. An evaluation of remission rates with first and second line treatments and indicators of antihistamine refractoriness in chronic urticaria. Int Immunopharmacol. 2022;112:109198.
Novartis AG. XOLAIR® (omalizumab) prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/103976s5225lbl. Accessed 9 Nov 2023.
Novartis AG. XOLAIR® (omalizumab) summary of product characteristics. https://www.ema.europa.eu/en/documents/product-information/xolair-epar-product-information_en.pdf. Accessed 9 Nov 2023.
XOLAIR® (omalizumab). FDA label update approval letter. 2021 https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2021/103976Orig1s5238ltr.pdf. Accessed 9 Nov 2023.
Marzano AV, Genovese G, Casazza G, et al. Predictors of response to omalizumab and relapse in chronic spontaneous urticaria: a study of 470 patients. J Eur Acad Dermatol Venereol. 2019;33(5):918–24.
Deza G, Bertolin-Colilla M, Pujol RM, et al. Basophil FcεRI expression in chronic spontaneous urticaria: a potential immunological predictor of response to omalizumab therapy. Acta Derm Venereol. 2017;97(6):698–704.
Deza G, Bertolin-Colilla M, Sanchez S, et al. Basophil FcεRI expression is linked to time to omalizumab response in chronic spontaneous urticaria. J Allergy Clin Immunol. 2018;141(6):2313–6.
Deza G, March-Rodríguez A, Sánchez S, et al. Relevance of the basophil high-affinity IgE receptor in chronic urticaria: clinical experience from a tertiary care institution. J Allergy Clin Immunol Pract. 2019;7(5):1619–26.
Gericke J, Metz M, Ohanyan T, et al. Serum autoreactivity predicts time to response to omalizumab therapy in chronic spontaneous urticaria. J Allergy Clin Immunol. 2017;139(3):1059–61.
Manzoor H, Razi F, Rasheed A, et al. Efficacy of different dosing regimens of IgE targeted biologic omalizumab for chronic spontaneous urticaria in adult and pediatric populations: a meta-analysis. Healthcare (Basel). 2022;10(12):2579.
Tharp MD, Bernstein JA, Kavati A, et al. Benefits and harms of omalizumab treatment in adolescent and adult patients with chronic idiopathic (spontaneous) urticaria: a meta-analysis of “real-world” evidence. JAMA Dermatol. 2019;155(1):29–38.
Maurer M, Rosén K, Hsieh HJ, et al. Omalizumab for the treatment of chronic idiopathic or spontaneous urticaria. N Engl J Med. 2013;368(10):924–35.
Kaplan A, Ledford D, Ashby M, et al. Omalizumab in patients with symptomatic chronic idiopathic/spontaneous urticaria despite standard combination therapy. J Allergy Clin Immunol. 2013;132(1):101–9.
Saini SS, Bindslev-Jensen C, Maurer M, et al. Efficacy and safety of omalizumab in patients with chronic idiopathic/spontaneous urticaria who remain symptomatic on H1 antihistamines: a randomized, placebo-controlled study. J Invest Dermatol. 2015;135(1):67–75.
Kaplan A, Ferrer M, Bernstein JA, et al. Timing and duration of omalizumab response in patients with chronic idiopathic/spontaneous urticaria. J Allergy Clin Immunol. 2016;137(2):474–81.
Kulthanan K, Chaweekulrat P, Komoltri C, et al. Cyclosporine for chronic spontaneous urticaria: a meta-analysis and systematic review. J Allergy Clin Immunol Pract. 2018;6(2):586–99.
Kocatürk E, Başkan EB, Küçük ÖS, et al. Omalizumab versus cyclosporin-A for the treatment of chronic spontaneous urticaria: can we define better-responding endotypes? An Bras Dermatol. 2022;97(5):592–600.
Yosipovitch G, Biazus Soares G, Mahmoud O. Current and emerging therapies for chronic spontaneous urticaria: a narrative review. Dermatol Ther (Heidelb). 2023;13(8):1647–60.
Mustari AP, Bishnoi A, Kumaran MS. Biologicals in treatment of chronic urticaria: a narrative review. Indian Dermatol Online J. 2023;14(1):9–20.
Rip J, de Bruijn MJW, Appelman MK, Pal Singh S, Hendriks RW, Corneth OBJ. Toll-like receptor signaling drives Btk-mediated autoimmune disease. Front Immunol. 2019;10:95.
Nyhoff LE, Griffith AS, Clark ES, Thomas JW, Khan WN, Kendall PL. Btk supports autoreactive B cell development and protects against apoptosis but is expendable for antigen presentation. J Immunol. 2021;207(12):2922–32.
Maurer M, Berger W, Giménez-Arnau A, et al. Remibrutinib, a novel BTK inhibitor, demonstrates promising efficacy and safety in chronic spontaneous urticaria. J Allergy Clin Immunol. 2022;150(6):1498–506.e2.
Alvarado D, Maurer M, Gedrich R, et al. Anti-KIT monoclonal antibody CDX-0159 induces profound and durable mast cell suppression in a healthy volunteer study. Allergy. 2022;77(8):2393–403.
Kolkhir P, Elieh-Ali-Komi D, Metz M, Siebenhaar F, Maurer M. Understanding human mast cells: lesson from therapies for allergic and non-allergic diseases. Nat Rev Immunol. 2022;22(5):294–308.
Bernstein J, Metz A, Anderson J, Talreja N, Heath-Chiozzi M, Alvarado D. Effects of multiple dose treatment with an anti-KIT antibody, CDX-0159, in chronic spontaneous urticaria. EAACI 2022. Poster 100097. 2022. https://celldex.com/wp-content/uploads/Effects-of-Multiple-Dose-Treatment-with-an-Anti-KIT-Antibody-CDX-0159-in-Chronic-Spontaneous-Urticaria-EAACI-2022.pdf. Accessed 9 Nov 2023.
Maurer M, Casale TB, Saini SS, et al. Dupilumab in patients with chronic spontaneous urticaria (LIBERTY-CSU CUPID): two randomized, double-blind, placebo-controlled, phase 3 trials. J Allergy Clin Immunol. 2024. https://doi.org/10.1016/j.jaci.2024.01.028.
Giménez-Arnau AM, Podder I. Current perspectives and future directions in the management of chronic spontaneous urticaria and their link to disease pathogenesis and biomarkers. Ital J Dermatol Venerol. 2023;158(4):302–15.
Acknowledgements
Medical Writing Assistance
Medical writing support was provided by Christine Elsner, BOLDSCIENCE®, and was funded by Novartis Pharmaceuticals Corporation. This manuscript was developed in accordance with Good Publication Practice guidelines. Authors had full control of the content and made the final decision on all aspects of this publication.
Funding
Development of this manuscript and the Rapid Service and Open Access Fees for publication were funded by Novartis Pharmaceuticals Corporation, East Hanover, New Jersey, USA.
Author information
Authors and Affiliations
Contributions
All authors contributed equally to the conception, literature search and analysis, and the drafting of the article.
Corresponding author
Ethics declarations
Conflict of Interest
Adam Friedman has participated as an advisory board member for AbbVie, Arcutis, Galderma, Novartis, Pfizer, Pierre Fabre, Regeneron Pharmaceuticals Inc., and Sanofi; has received grants/research funding from Cerave, Eli Lilly, Galderma, Incyte, La Roche-Posay, and Pfizer; and has been a speaker for Eli Lilly, Incyte, Janssen, Regeneron Pharmaceuticals Inc., and Sanofi. Shawn Kwatra is an advisory board member/consultant for AbbVie, Amgen, Arcutis Biotherapeutics, Aslan Pharmaceuticals, Bristol Myers Squibb, Cara Therapeutics, Castle Biosciences, Celldex Therapeutics, Dermavant, Galderma, Genzada Pharmaceuticals, Incyte Corporation, Johnson & Johnson, Leo Pharma, Novartis Pharmaceuticals Corporation, Pfizer, Regeneron Pharmaceuticals, and Sanofi and has served as an investigator for Galderma, Incyte, Pfizer, and Sanofi. Gil Yosipovitch has participated as an advisory board member for AbbVie, Arcutis, Escient Health, Eli Lilly, Galderma, LEO Pharma, Novartis, Pfizer, Pierre Fabre, Regeneron Pharmaceuticals Inc., Sanofi, Vifor, GSK, and Kamari; has received grants/research funding from Eli Lilly, LEO Pharma, Novartis, Pfizer, Galderma, Escient, Sanofi Regeneron, and Celldex; and has been an investigator for Regeneron Pharmaceuticals Inc. and Sanofi.
Ethics Approval
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Friedman, A., Kwatra, S.G. & Yosipovitch, G. A Practical Approach to Diagnosing and Managing Chronic Spontaneous Urticaria. Dermatol Ther (Heidelb) 14, 1371–1387 (2024). https://doi.org/10.1007/s13555-024-01173-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-024-01173-5