
Abstract
Type 2 immunity evolved to combat helminth infections by orchestrating a combined protective response of innate and adaptive immune cells and promotion of parasitic worm destruction or expulsion, wound repair, and barrier function. Aberrant type 2 immune responses are associated with allergic conditions characterized by chronic tissue inflammation, including atopic dermatitis (AD) and asthma. Signature cytokines of type 2 immunity include interleukin (IL)-4, IL-5, IL-9, IL-13, and IL-31, mainly secreted from immune cells, as well as IL-25, IL-33, and thymic stromal lymphopoietin, mainly secreted from tissue cells, particularly epithelial cells. IL-4 and IL-13 are key players mediating the prototypical type 2 response; IL-4 initiates and promotes differentiation and proliferation of naïve T-helper (Th) cells toward a Th2 cell phenotype, whereas IL-13 has a pleiotropic effect on type 2 inflammation, including, together with IL-4, decreased barrier function. Both cytokines are implicated in B-cell isotype class switching to generate immunoglobulin E, tissue fibrosis, and pruritus. IL-5, a key regulator of eosinophils, is responsible for eosinophil growth, differentiation, survival, and mobilization. In AD, IL-4, IL-13, and IL-31 are associated with sensory nerve sensitization and itch, leading to scratching that further exacerbates inflammation and barrier dysfunction. Various strategies have emerged to suppress type 2 inflammation, including biologics targeting cytokines or their receptors, and Janus kinase inhibitors that block intracellular cytokine signaling pathways. Here we review type 2 inflammation, its role in inflammatory diseases, and current and future therapies targeting type 2 pathways, with a focus on AD.
Infographic

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Aberrant type 2 immune responses are associated with allergic conditions characterized by chronic tissue inflammation, including atopic dermatitis. |
Immune dysregulation in such diseases is often highly complex and involves many different cell types and inflammatory mediators. |
Studies of drugs targeting type 2 immune mediators have helped clarify the biologic mechanisms underlying type 2 immunity while also providing tremendous therapeutic advances for diseases involving type 2 inflammation. |
Digital features
This article is published with digital features, including an infographic, to facilitate understanding of the article. To view digital features for this article, go to https://doi.org/10.6084/m9.figshare.19609980.
Introduction
Immune-mediated diseases involve aberrant responses to self-antigens and/or inappropriate activation of cellular and cytokine responses leading to chronic inflammation and tissue damage [1]. Type 2 immunity originally evolved to protect hosts from parasitic helminths or toxins [2,3,4,5], but aberrant type 2 inflammation underlies numerous chronic inflammatory pathologies, including atopic dermatitis (AD) and asthma. The pathways underlying type 2 inflammatory diseases are diverse and may vary by age and ethnicity but often share common mediators that can be therapeutically targeted. Mechanistic characterization of these drug targets will improve our understanding of the efficacy and safety of therapeutic immunomodulation. Here, we provide an overview of type 2 inflammation, its role in inflammatory diseases, and therapeutic approaches to disease control. This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Immune Response and Associated Inflammation
Immune Pathways and Their Role in Homeostasis
The immune system consists of the innate system, which recognizes conserved pathogenic features and provides initial defense mechanisms, and the adaptive system, which acts with specificity and provides long-term immune memory through effector T cells and antibody production by B cells [1, 6]. These two branches are intrinsically coordinated. Antigen-presenting cells (APCs), part of the innate immune system, process and present antigen on major histocompatibility complex (MHC) molecules to activated lymphocytes, members of the adaptive immune system [7]. APCs also produce cytokines and display costimulatory signals facilitating lymphocyte activation and differentiation, usually along one of three main differentiation pathways: types 1, 2, and 3 (Fig. 1) [8,9,10,11].

Overview of types 1, 2, and 3 inflammation. This diagram represents immune concepts that do not occur in complete isolation. IFNγ interferon-γ, IL interleukin, ILC innate lymphoid cell, NK natural killer, Th1/2 T helper type 1/2 cell, TNF tumor necrosis factor
Type 1 immunity evolved to protect against intracellular pathogens such as bacteria, protozoa, and viruses and is characterized by group 1 innate lymphoid cells (ILC1s), T-helper (Th) 1 cells, and T-cytotoxic (Tc) 1 cells. Intercellular communication facilitates the elimination of infected cells through the release of proinflammatory cytokines including interferon (IFN)-γ, interleukin (IL)-1β, IL-12, and tumor necrosis factor (TNF), promoting phagocytic activation and cytolytic activity. ILC1s also induce B cells to produce immunoglobulin (Ig) G, IgM, and IgA (but not IgE) [7, 12].
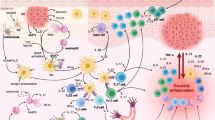
Type 2 immunity evolved to ensure epithelial barrier integrity and protection against helminth parasites and noxious environmental substances [2,3,4,5]. Its main cellular components are group 2 ILCs (ILC2s), Th2 cells, and Tc2 cells that release signature cytokines, including IL-4, IL-5, IL-9, and IL-13 [5]. These type 2 mediators contribute to the recruitment of innate effector cells such as eosinophils and mast cells and promote B-cell isotype class switching to IgE production [2, 7, 10, 12, 13]. Type 2 immune mechanisms, together with mediators such as histamine and prostaglandins, induce a coordinated response of epithelia, sensory neurons, and smooth muscle cells to expel parasites by triggering an itch/scratch reflex followed by wound repair [4, 5, 14]. Unlike most microorganisms, helminths do not “outrun” the immune system through rapid replication but instead rely on downregulating host immunity, leading to chronic infection [3]. Moreover, helminths tend to be endemic, and repeat exposure is common, increasing the need for a robust memory response.
Type 3 immunity is driven by the group 3 ILCs (ILC3s), Th17, and Tc17 cells. These cells are characterized by production of the IL-17 and IL-22 families of cytokines and coordinated neutrophilic activation that protects against extracellular bacteria and fungi through tissue inflammation, phagocytosis, and pathogen lysis [7, 15, 16].
Role of Type 2 Inflammation in Disease
Immune dysregulation can lead to inappropriate activation of inflammatory pathways upon microbial and nonmicrobial stimuli. Aberrant type 2 responses have been associated with allergic conditions, including AD, asthma, allergic rhinitis, and food allergy. Type 2 inflammatory pathways have also been implicated in other disorders characterized by elevated levels of eosinophils, mast cells, and/or IgE, such as chronic rhinosinusitis with nasal polyposis (CRSwNP), eosinophilic esophagitis, chronic urticaria, bullous pemphigoid, and prurigo nodularis [17,18,19].
Key Players in Type 2 Inflammation
In the absence of helminth infection, inappropriate stimulation of type 2 immunity is triggered by allergens, irritants, noxious microenvironmental cues (i.e., temperature, pH), bacteria, viruses, and endogenous host molecules and lacks a downmodulating effect mediated by helminths [3, 20]. Allergens presented by APCs induce naïve Th0 cell polarization to the Th2 phenotype, a process orchestrated by IL-4 and leading to the production of type 2 inflammatory cytokines [10, 20, 21]. Because the type 2 immune response evolved to combat helminths transmitted through the skin and mucosal surfaces, aberrant reactions to antigens are typically found at the epidermal and epithelial barriers. Epidermal or epithelial barrier dysfunction often occurs in inflammatory diseases, including AD, asthma, and chronic rhinitis. Compromised barrier integrity facilitates entry of allergens, irritants, and infectious agents, and damaged or stressed epidermal and epithelial cells release thymic stromal lymphopoietin (TSLP), IL-25 (also known as IL-17E), and IL-33 (alarmins), triggering type 2 inflammatory cytokine production by ILC2s and Th2 cells [13, 22, 23].
Type 2 cytokines: Signature cytokines of type 2 immunity include IL-4, IL-5, IL-9, IL-13, and IL-31, mainly secreted from immune cells, and IL-25, IL-33, and TSLP, mainly secreted from nonimmune cells (Table 1) [13, 24,25,26,27].
IL-4 is produced by many cell types, including activated Th2 cells, ILC2s, basophils, eosinophils, mast cells, natural killer T cells, and macrophages [40,41,42,43,44,45,46]. It plays a central role in Th2 differentiation and drives isotype class switching of B cells, mainly to IgE and IgG4 (human) or IgG1 (mouse) [47,48,49,50]. IL-4 also promotes alternative activation of macrophages, eosinophil migration, and production of other type 2 cytokines, indicating that it is a key upstream immunomodulator and driver of type 2 inflammation [51,52,53]. IL-4 facilitates two major axes for type 2 inflammatory cell recruitment to the skin. First, vasopressin-activated calcium-mobilizing receptor (VCAM) 1 upregulation of endothelial cells leads to perivascular infiltration of very late antigen-4-expressing granulocytes (eosinophils, basophils, and mast cells, but not neutrophils). Th2 cell CCR4 expression, binding to C–C motif chemokine ligand 17 (CCL17, also known as thymus and activation-regulated chemokine [TARC]) and CCL22 (also known as macrophage-derived chemokine [MDC]) on endothelial cells, skin-resident dendritic cells (DCs), and Langerhans cells facilitates inflammatory cell skin-homing [54, 55]. IL-4 drives these processes at low picomolar concentrations given its high affinity for its receptor [28, 56].
IL-13, which shares a receptor subunit with IL-4, is also implicated in type 2 inflammation. Some overlap in the inflammatory effects of IL-13 and IL-4 occurs, and both have been implicated in fibrosis and wound healing [57,58,59,60,61]. In aberrant inflammatory states, both cytokines contribute to barrier disruption by inhibiting keratinocyte differentiation, promoting epidermal hyperplasia [62], and reducing expression of epidermal structural proteins and antimicrobial peptides [62,63,64,65,66,67,68]. IL-13 also directly stimulates tissue remodeling and production of mucus in the respiratory epithelial barrier [39]. In vivo differences between IL-13 and IL-4 derive from their relative productions and receptor expression in different cell types. Notably, because they lack IL-13Rα1 expression, naïve T cells respond to IL-4 but not IL-13, highlighting the unique role of IL-4 in Th2 cell development [69].
IL-5 is the most potent mediator of eosinophil function. Produced by Th2 cells, ILC2s, and mast cells, its receptor, IL-5Rα, is selectively expressed on eosinophils, basophils, and some mast cells [31, 45, 46, 70]. IL-5 controls eosinophil growth, differentiation, migration, and survival. IL-4 and IL-13 promote eosinophil trafficking to the site of inflammation, in part by mediating production of IL-5 and chemokines, such as CCL26 (also known as eotaxin-3) and CCL17, and induction of adhesion molecules such as VCAM-1 in endothelial cells [56, 71,72,73].
IL-9 is produced primarily by T cells but also by mast cells, eosinophils, and ILC2s. It promotes T-cell growth, proliferation, and survival, B-cell IgE production, and mast cell proliferation and differentiation; it also stimulates secretion of mucus by mucosal epithelial cells [74]. IL-9 plays a critical role in type 2 immunity against parasitic worm infection and has been implicated in allergic inflammation [32, 74].
IL-31 is primarily produced by Th2 cells, implying the requirement for the upstream presence of IL-4. Some IL-31 production has been observed by CD8+ T cells, monocytes/macrophages, DCs, keratinocytes, eosinophils, basophils, mast cells, and fibroblasts [25]. This cytokine plays a critical role in histamine-independent pruritus [37] and can alter epidermal barrier function in vitro [75].
Type 2 alarmins: Type 2 alarmins IL-25, IL-33, and TSLP are released by epidermal and epithelial cells in response to external insults and enhance innate and adaptive immune responses by triggering downstream signaling pathways.
IL-25 plays a prominent role in enhancing production of type 2 cytokines (IL-4, IL-5, IL-13, IL-33, and TSLP) from Th2 cells and ILC2s [76]. It may play an important role in AD and asthma as levels of IL-25 are elevated in tissues biopsied from these patients [77].
IL-33 plays a role in the development of allergic diseases by acting on innate cell types such as ILC2s, basophils, and mast cells, and is elevated in patients with allergic airway diseases [78]. Its receptor is found on memory Th2 cells, suggesting a contribution of IL-33 to activation of Th2 cells [79].
TSLP promotes cytokine production from Th2 cells and ILC2s and plays a role in polarizing DCs to induce a Th2 response [80,81,82]. TSLP-activated DCs induce an inflammatory Th2 response through expression of the OX40 ligand (also known as CD252 or tumor necrosis factor receptor superfamily member 4 [TNFSF4]) [83]. TSLP expression is high in acute and chronic AD lesions, and expression of TSLP in skin is correlated with AD severity [81, 84]. TSLP release in keratinocytes can also trigger itch by acting on TRPA1-positive sensory neurons [85].
Signaling Mechanisms Driving Type 2 Inflammation
The polarization of naïve Th0 cells toward a Th2 phenotype is predominantly regulated by the cytokine milieu. IL-4 causes Th0 cells in the lymph nodes to bind (via T-cell receptors) to antigens presented by MHC class II molecules on APCs. The polarized and activated Th2 cells release IL-4, IL-5, IL-9, IL-13, and IL-31 and migrate to germinal centers. In the germinal centers, in the presence of IL-4 and IL-13, follicular helper T cells situate in near B cells to induce isotype class switching and differentiation to antigen-specific, IgE-producing plasma cells. IgE travels via circulation and binds to its antigen and FcεRI receptors on basophils and mast cells [86]. IL-4 and IL-13 can directly upregulate FcεRI expression at the cell surface, which may prime FcεRI-expressing innate cells for activation [56]. Bound IgE on basophils and mast cells confers a selective response to the target antigen [86]. When activated, basophils and mast cells degranulate and release histamine, proteoglycans (e.g., heparin), proteolytic enzymes, leukotrienes, and cytokines to further drive vasodilation and inflammation [87].
The IL-4Rα receptor chain is involved in both IL-4 and IL-13 signaling and is critical to the type 2 response. It can form two types of IL-4R complexes. When IL-4 binds to IL-4Rα, it recruits either the common gamma chain (γc) to form the type I receptor or IL-13Rα1 to form the type II receptor. IL-13 binds to IL-13Rα1 and recruits IL-4Rα, forming the type II receptor [39, 88]. Myeloid cells express both types of receptors; nonhematopoietic cells predominantly express type II receptors, and lymphocytes mainly express type I receptors [39]. IL-4Rα is also expressed on sensory neurons, and activation of neuronal IL-4Rα plays a critical role in chronic itch [89]. IL-4 can activate human sensory neurons, and sensory neuron-specific genetic deletion of IL-4rα in mice attenuates both itch and skin inflammation, further suggesting that IL-4Rα critically mediates chronic itch. Moreover, activation of neuronal IL-4Rα sensitizes sensory neurons to multiple other pruritogens. IL-4 was shown to significantly amplify scratching behavior to low doses of pruritogens such as histamine [90]. These data suggest that type 2 cytokines may act as master regulators of chronic itch by intensifying itch responses to multiple pruritogens present in inflamed skin [89]. Once IL-4 and IL-13 bind to the receptor, they initiate an intracellular signaling cascade involving mainly signal transducer and activator of transcription (STAT) 6, which translocates to the nucleus and induces transcription of type 2 response genes [39].
A key type 2 cytokine signaling pathway is the Janus kinase (JAK)-STAT pathway. Dimerization of cytokine receptors upon binding with cytokines induces auto- and trans-phosphorylation of the noncovalently associated intracellular JAKs. There are four JAK molecules (JAK1, JAK2, JAK3, and tyrosine kinase-2 [TYK2]), and the type of JAK involved depends on the components of the receptor complex (in the case of IL-4 and IL-13 signaling: JAK1 for IL-4Rα, JAK3 for γc, and JAK2 or TYK2 for IL-13Rα1). Their redundant nature accounts for the role of JAKs in a wide variety of biologic processes, including immunity, inflammation, cell division, development, tumor formation, and cell death [91].
Atopic Dermatitis
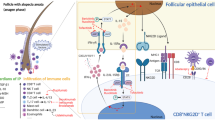
AD is a chronic, relapsing, pruritic, systemic inflammatory disease affecting up to 20% of children and 2–8% of adults worldwide [92, 93]. It often begins in early childhood [93, 94]. Recent research has expanded our understanding of the pathogenesis of AD, which is thought to involve genetic and environmental factors that predispose an individual to epidermal barrier dysfunction and type 2 inflammation [95, 96]. There is considerable heterogeneity within AD [29, 97]; however, type 2 inflammation is the principal driving mechanism (Fig. 2) [98,99,100]. Type 2 inflammation leads to defects in the epithelial barrier, allowing entry of allergens/pathogens, where they are taken up by Langerhans cells and other APCs [101]. Disruption to the epithelial barrier also stimulates the release of alarmins (e.g., IL-25, IL-33, TSLP) from keratinocytes. Alarmins induce production of type 2 mediators such as IL-4 and IL-13 by innate immune cells, thereby shaping the immune response toward the type 2 pathway, and the subsequent recruitment of mast cells, basophils, eosinophils, alternatively activated macrophages, and allergen-specific IgE into the dermal layers [100, 102, 103]. Continued activation of type 2 immunity leads to chronic inflammation with emergence of memory Th2 cells, sensitization of sensory neurons to a range of pruritogens, an amplified itch–scratch cycle, and skin barrier disruption resulting in a clinical picture characterized by erythema, induration, papulation, excoriation, and in more chronic stages, lichenification [19]. The T-cell receptor OX40 (also known as CD134 or tumor necrosis factor receptor superfamily member 4 [TNFRSF4]) and the OX40 ligand contribute to Th2 memory cell formation and are thought to play a role in AD pathogenesis [104, 105]. Th2 cells are increased in young children with AD, suggesting that systemic components of inflammation may predispose nonlesional skin to a pathologic response upon allergen exposure [106]. Genetic risk factors may predispose patients to AD; many patients have a loss-of-function mutation in FLG, but this alone is insufficient to develop disease [93, 107].

Pathologic inflammation in AD (A), and IL-4 as a key driver of Th2 differentiation and activation (B). CCL17 C–C motif chemokine ligand 17, DC dendritic cell, Ig immunoglobin, IL interleukin, ILC innate lymphoid cell, LTC4 leukotriene C4, MHC-II major histocompatibility complex II, TCR T-cell receptor, TSLP thymic stromal lymphopoietin, VCAM-1 vasopressin-activated calcium-mobilizing receptor 1
Barrier protein expression is regulated by both external and internal stimuli, including IL-4, IL-13, and IL-31. In AD, excessive IL-4, IL-13, and IL-31 levels downregulate the expression of several skin barrier proteins, including filaggrin (FLG) [64, 108], loricrin [66, 109], and involucrin [65, 67, 68]. Dysfunction of the epidermal barrier can itself further promote type 2 inflammation through the production of alarmins. Alarmins IL‐33 and IL‐25 are believed to trigger ILC2 activation and overexpression of IL‐13 in the skin; this leads to the recruitment of activated Th2 cells via induction of Th2 cell-recruiting chemokines such as CCL17 and further increases IL‐4/IL‐13 expression within the skin. This pathway is believed to play a critical role in chronic inflammation in AD [110]. IL-4 and IL-13 also induce the production of periostin, a matricellular protein that plays a role in chronic inflammation, skin fibrosis, and pruritus [111]. Periostin promotes the production of inflammatory cytokines by keratinocytes, amplifying chronic inflammation [112].
Lesional skin in AD exhibits significantly more Th2 cells, ILC2s, and basophils than healthy skin [113, 114], and serum IgE levels are often increased in patients with AD. Basophils in AD skin are a key producer of peripheral IL-4 [115,116,117]. The basophil response has been shown to precede the ILC2 response, and the IL-4 produced by basophils induces the proliferation and activation of cutaneous ILC2s in a manner independent of IgE or the Th2 induction that occurs in the lymph nodes, underscoring the role of peripheral IL-4 in AD pathogenesis [115].
Recent genomic studies support widespread dysregulation of immune and barrier function genes in AD [118,119,120,121]. Transcriptome profiling from tape-stripped skin samples indicate upregulation of genes associated with the Th2 response, including IL-4 and IL-13, and downregulation of genes associated with barrier function in lesional versus nonlesional skin [119,120,121]. In children with early-onset AD, the genes encoding the IL-4 receptor and the chemokine CCL17 were upregulated compared with healthy skin [121].
One of the cardinal symptoms of AD is debilitating itch, which can lead to scratching and excoriation that disrupt the epithelial barrier, perpetuate the inflammatory response, and can lead to epidermal thickening and lichenification [19, 122]. Pruritus is regulated by a range of immune mediators and leads to neurocutaneous anatomical alterations. In patients with AD, lesions have increased sensory nerve density in the epidermis and dermis [123], and altered expression of neuropeptides, neurotrophins, and neurotransmitters is seen in the skin and nerves of the dermis. Increased expression of PGP9.5, amphiregulin, nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and substance P have been observed, with a contrasting decrease in semaphorin 3A expression [124]; these mediators are associated with nerve fiber penetration of the epidermis in patients with AD.
Basophils and mast cells release inflammatory mediators, including histamine and tryptase, that contribute to itch [102, 123]. Histamine elicits a range of proinflammatory and pruritic effects, primarily via histamine 1 and histamine 4 receptors. Histamine regulates production of NGF and semaphorin 3A in keratinocytes, promotes IL-31 release from Th2 cells, and directly activates sensory neurons [125]. Recent studies also reveal a critical role for basophils in acute itch flares in AD. Following activation by allergen-specific IgE, basophils produce IL-4 and promote itch through mast cell-independent leukotriene C4-CysLTR2 signaling [126, 127]. Chronic itch in AD appears to be a distinct entity from acute itch, with signaling via a variety of nonhistaminergic nerve fiber pathways, including IL-4Ra and IL-31R [89, 128].
IL-31 is a key mediator of pruritus and is increased in lesional skin in patients with AD. IL-31 not only stimulates sensory nerves but also promotes sensory nerve fiber elongation and branching [129]. It regulates brain-derived natriuretic peptide, upregulating its release in the skin and acting as a neuroimmune link to drive the sensory pathways that result in itch. IL-31 receptor signaling involves JAK1 and JAK2 [25, 130, 131]. The primary source of IL-31 is Th2 cells. IL-4, but not IL-13, upregulates the histaminic H4 receptor (H4R) on CD4+ Th2 cells, and, in turn, H4R activation on T cells upregulates IL-31 production [132]. More recently, IL-4 and IL-13 were shown to directly stimulate sensory neurons and promote itch [89, 133]. The gene for IL-4Rα is widely expressed across neuronal subsets, and IL-4 enhances responsiveness to pruritogens, including IL-31, histamine, and chloroquine [89]. By promoting neural hypersensitivity, IL-4Rα may therefore play a role in the prolonged or outsized reactions to pruritogens seen in patients with AD.
Although the pathogenesis of AD is predominantly a type 2 immune response, types 1 and 3 immune responses also contribute to disease, especially in chronic stages. The type 3 cytokines IL-17 and IL-22 are upregulated in the skin of patients with AD, mainly in patients with relatively low IgE levels [97]. IL-17 reduces expression of FLG and involucrin [134]. Th1 cell activation appears to occur more in the chronic phase of AD after prolonged inflammation at the epithelial barrier. Early-onset AD in children is characterized by relatively strong activation of the Th2 axis and increased Th17 cytokine levels compared with adult-onset AD [106, 135]. Asian patients typically have a higher expression of Th17 and Th22 cytokines and lower activation of Th1 cytokines in AD skin lesions than European–American patients; however, Th2 cytokines are upregulated consistently regardless of ethnicity or disease stage [136, 137].
Inhibition of Type 2 Inflammation
Overview
Targeted immune-modulating therapies provide a specific, effective treatment approach that is less prone to adverse events (AEs) than broad-spectrum immunosuppressant therapy. Biologic therapies have been developed for conditions mediated by type 1 and/or type 3 immunity, such as rheumatoid arthritis (RA), psoriasis, Crohn disease, hidradenitis suppurativa, and ankylosing spondylitis. Therapies for conditions mediated by type 2 inflammation have been similarly developed, with therapies for asthma and AD leading the way. Licensed biologics or biologics in advanced clinical stages targeting type 2 cytokines in AD include lebrikizumab (IL-13), tralokinumab (IL-13), mepolizumab (IL-5), and reslizumab (IL-5); those targeting type 2 cytokine receptors include dupilumab (IL-4Rα), benralizumab (IL-5Rα), and nemolizumab (IL-31Rα); and IgE-targeting therapies include omalizumab and ligelizumab (Fig. 3). The clinical efficacy of these biologics has significantly advanced our understanding of the pathogenic role of type 2 cytokines in diseases such as AD, asthma, and CRSwNP, and has spurred the development of biologics targeting other mediators associated with type 2 inflammation, such as alarmins; for example, IL-33 (astegolimab, etokimab, itepekimab, MEDI-3506), IL-33R (melrilimab), and TSLP (tezepelumab), which are at various stages of development.

Biologics that inhibit type 2 molecules. AD atopic dermatitis, ADCC antibody-dependent cellular cytotoxicity, ABPA allergic bronchopulmonary aspergillosis, AFR allergic fungal rhinosinusitis, BP bullous pemphigoid, CCL17 C–C motif chemokine ligand 17, COPD chronic obstructive pulmonary disorder, CRSsNP chronic rhinosinusitis without nasal polyposis, CRSwNP chronic rhinosinusitis with nasal polyposis, CIU chronic idiopathic urticaria, CSU chronic spontaneous urticaria, EGPA eosinophilic granulomatosis with polyangiitis, EoE eosinophilic esophagitis, FeNO fractional exhaled nitric oxide, HES hypereosinophilic syndrome, IFNγ interferon-gamma, IgE immunoglobulin E, IL interleukin, MCP-4 monocyte chemoattractant protein-4, NP nasal polyps, OSMRβ oncostatin-M specific receptor subunit β, PC20 provocative concentration causing a 20% drop in FEV1 from baseline, PN prurigo nodularis, TSLP thymic stromal lymphopoietin, TSLPR thymic stromal lymphopoietin receptor
Some biologics, including those that target TNF, IL-17, and IL-12/-23, have been associated with an increased risk of opportunistic and/or serious bacterial, fungal, or viral infections [138]. This finding may be attributed to the role of type 1 or type 3 immunity and IFNγ (which has antimicrobial properties and signals via JAK1 and JAK2) in preventing bacterial, fungal, and viral infections [139]. In theory, targeting type 2 inflammation could increase the risk of helminth infections, although no evidence currently exists.
Biologics demonstrating lack of efficacy may also be useful for understanding the precise mechanisms underlying type 2 inflammatory diseases and their treatment. For example, although peripheral blood eosinophil numbers are typically elevated in patients with AD, and eosinophil numbers appear to correlate with disease activity [140], reducing peripheral blood eosinophil numbers through IL-5 inhibition (mepolizumab) failed to yield clinically significant improvements in AD [141, 142]. These findings suggest that IL-5-mediated elevations in eosinophil levels are not likely a primary source of the inflammatory cascade in AD. Similarly, although biologics targeting IgE (omalizumab and ligelizumab) have shown some clinical efficacy in AD, treatment efficacy has been inconsistent, suggesting that elevated IgE is not a predominant clinical factor in AD [143].
Biologic Therapy
Dupilumab: The central and pleiotropic role of IL-4 and IL-13 in type 2 inflammatory conditions make these cytokines attractive therapeutic targets, with beneficial downstream effects for patients [30, 144]. Dupilumab is a fully human monoclonal antibody against IL-4Rα, which inhibits both IL-4 and IL-13 signaling and is approved for patients with type 2 inflammatory diseases, including AD, asthma, and CRSwNP [145, 146]. In multiple, randomized, placebo-controlled phase 3 clinical trials, dupilumab improved signs, symptoms, and quality of life (QoL) in adults and adolescents with moderate-to-severe AD and in children (aged ≥ 6 years) with severe disease [147,148,149,150,151,152]. Dupilumab also demonstrated acceptable safety and long-term efficacy, with continued improvements in AD signs and symptoms for up to 4 years in adults with AD [153, 154]. The most common AEs associated with dupilumab are listed in Table 2, including conjunctivitis and injection-site reactions.
Previous analyses have shown that treatment with dupilumab normalizes the AD transcriptome by downregulating markers of type 2 inflammation (IL-13, IL-31, CCL13, CCL17, CCL18 [also known as pulmonary and activation-regulated chemokine, PARC], CCL22, CCL26, periostin) and Th17/Th22 activity (IL-17A, IL-22) while normalizing gene products associated with barrier function, such as FLG [183,184,185]. Another study suggests that dupilumab might suppress ILC2 and Th2 cell populations [186].
In phase 3 trials in asthma, dupilumab reduced the rate of severe exacerbations and improved lung function as evidenced by increased forced expiratory volume in the first second compared with control in patients with uncontrolled, moderate-to-severe asthma and glucocorticoid-dependent severe asthma [187, 188]. In two phase 3 trials in adults with CRSwNP that was uncontrolled despite prior treatment with systemic corticosteroids, surgery, or both, dupilumab treatment significantly reduced polyp size, roentgenographic sinus opacification, and severity of symptoms compared with controls [189]. These findings suggest the potential for dupilumab to curtail AD progression in children and adolescents, although further evaluation is required [190, 191].
Two different analyses of multiple, randomized, placebo-controlled trials of dupilumab in adults with moderate-to-severe AD demonstrated that dupilumab reduced the risk of serious and severe infections as well as nonherpetic skin infections, and did not increase the overall infection rates, compared with controls. Clinically important herpes viral infections (eczema herpeticum, herpes zoster) were less common with dupilumab than placebo [192,193,194]. Dupilumab also reduced overall infections and skin infections in children and adolescents with moderate-to-severe AD [195], and it reduced upper and lower respiratory infections and anti-infective medication use in adults and adolescents with moderate-to-severe asthma and adults with severe CRSwNP [196]. Dupilumab laboratory safety data also support the long-term use of dupilumab in children, adolescents, and adults without the need for routine laboratory monitoring [164, 197, 198].
The efficacy of dupilumab in AD, asthma, and CRSwNP supports the central roles of IL-4 and IL-13 in inflammatory disease pathology [30, 199]. Trials are ongoing in other conditions sharing elements of type 2 immune dysregulation (Fig. 3).
Tralokinumab and lebrikizumab: Tralokinumab is an anti–IL-13 recombinant humanized IgG4λ monoclonal antibody that binds to IL-13, thereby inhibiting signaling via the IL-13Rα1 and IL-13Rα2 receptors. Tralokinumab is approved in the USA and European Union for the treatment of moderate-to-severe AD in adults [176, 177]. In three phase 3 trials (ECZTRA 1, ECZTRA 2, and ECZTRA 3) in adults with moderate-to-severe AD, tralokinumab improved AD signs and symptoms and QoL and was well tolerated up to 52 weeks [178, 179]. In two phase 3 trials evaluating tralokinumab in patients with severe, uncontrolled asthma (STRATOS 1 and 2), the primary endpoints were not met [200], suggesting that inhibition of IL-13 signaling alone is insufficient to treat patients with severe asthma. Table 2 lists the most common AEs associated with tralokinumab.
Lebrikizumab, a humanized IgG4κ monoclonal antibody, binds to IL-13 at an epitope distinct from that of tralokinumab, preventing heterodimerization of the IL-4Rα and IL-13-IL-13Rα1 complex [30, 166]. Lebrikizumab is currently in phase 3 trials in patients with AD after demonstrating efficacy and an acceptable safety profile in phase 2 trials [166, 167]. In two phase 3 trials (LAVOLTA I and II), lebrikizumab failed to show a consistent reduction in asthma exacerbations in patients with uncontrolled asthma [201]. The most common AEs associated with lebrikizumab are listed in Table 2.
Taken together, the clinical data indicate that inhibiting IL-13 signaling may provide an efficacious treatment option for patients with AD, but treatment failure in asthma underscores the significance of IL-4 in the pathophysiologic nature of type 2 inflammatory diseases.
Nemolizumab: As described above, IL-31 is a key driver of itch in patients with AD. Nemolizumab is a humanized IgG2κ anti-IL-31 receptor A monoclonal antibody, which prevents IL-31 binding and subsequent downstream signaling. In a small phase 3 trial in Japanese patients with AD, nemolizumab in combination with topical agents resulted in clinically significant improvements in pruritus and some improvements in AD signs, QoL, and sleep compared with controls; AD exacerbations were observed both in placebo and in nemolizumab groups [169]. Common AEs associated with nemolizumab are listed in Table 2.
Mepolizumab, reslizumab, and benralizumab: A potent mediator of eosinopoiesis, IL-5 is a logical target for type 2 inflammatory conditions [31]. Mepolizumab, reslizumab, and benralizumab are humanized monoclonal antibodies that block IL-5 signaling. Although mepolizumab and reslizumab directly bind to IL-5, benralizumab binds to its receptor (IL-5Rα), inducing antibody-dependent cell-mediated cytotoxicity. Based on phase 3 trial data, all three agents are approved for use in patients with severe asthma with an eosinophilic phenotype [202,203,204,205,206,207,208,209,210]. Clinically meaningful responses were not observed following reslizumab treatment in patients without elevated eosinophil levels [211], suggesting that the efficacy of IL-5 inhibition is primarily a consequence of reducing eosinophil levels. Although none of these agents are approved for use in AD (mepolizumab failed to report meaningful improvements in AD) [141, 142], and mepolizumab has been licensed for eosinophilic granulomatosis with polyangiitis, investigation of their use in other eosinophil-driven conditions is underway. Table 2 lists the most common AEs associated with mepolizumab, reslizumab, and benralizumab.
Omalizumab and ligelizumab: Omalizumab, a humanized anti‐IgE antibody, binds to serum IgE, inhibiting its downstream effects. It is approved for use in asthma, chronic spontaneous urticaria, nasal polyps, and seasonal allergic rhinitis [170]. Two phase 3 trials support the use of omalizumab as an adjunctive therapy in patients with severe asthma uncontrolled by conventional therapy [212, 213]. Omalizumab has demonstrated some efficacy in a range of other type 2 inflammatory diseases, including AD [171], CRSwNP [214], and food allergies [215, 216]. Omalizumab has also demonstrated efficacy in the treatment of moderate-to-severe chronic spontaneous urticaria, as did recently ligelizumab (another humanized anti-IgE antibody) in controlling symptoms [217]. For the most common AEs associated with omalizumab, see Table 2.
Fezakinumab: Fezakinumab targets IL-22, a Th22 cytokine that plays a role in skin barrier function [165]. Treatment with fezakinumab was shown to affect multiple pathways related to inflammation and barrier function, although the effects were limited to patients with high IL-22 expression at baseline [218], thereby highlighting the population specificity of some biologics [96]. In a small, randomized phase 2 trial, fezakinumab improved signs and symptoms of moderate-to-severe AD with few common AEs (Table 2).
Astegolimab, etokimab, and itepekimab: Astegolimab is a human IgG2 monoclonal antibody that binds to the IL-33 receptor [219]. Phase 2 trials of astegolimab in patients with AD are underway. Another anti-IL-33 monoclonal antibody, itepekimab, is under investigation for chronic obstructive pulmonary disease [220]. Itepekimab was previously under investigation for the treatment of asthma and AD. However, its efficacy in treating asthma was lower than that of dupilumab, and it is no longer being investigated for the treatment of AD due to lack of efficacy [221, 222]. Etokimab, which binds directly to IL-33, is no longer in clinical development [223, 224].
Tezepelumab: TSLP is released from the epithelium following stress or allergen exposure and is an early promoter of polarization toward Th2 phenotype, the production of type 2 cytokines by ILC2s, mast cells, basophils, and eosinophils [26, 225]. Tezepelumab is a monoclonal antibody that binds directly to TSLP to prevent receptor binding. In a phase 2a trial in patients with AD, the addition of tezepelumab to topical corticosteroids did not significantly improve skin lesions compared with placebo and topical corticosteroids [152]. Tezepelumab also failed to show efficacy in a recent phase 2b study in patients with AD [226] and has been discontinued for further development in AD [227].
In patients with moderate-to-severe asthma, tezepelumab significantly reduced asthma exacerbations compared with placebo, regardless of baseline eosinophil count [228] and improved lung function, asthma control, and health-related QoL [229]. However, in adults with oral corticosteroid (OCS)-dependent asthma, tezepelumab did not reduce OCS use without the loss of asthma control [230] but did increase the probability of a reduction in OCS use in patients with high baseline blood eosinophil counts [231].
The inconsistent efficacy of biologics targeting alarmins in treating AD suggests that alarmins may not play a predominant clinical role in eliciting AD pathophysiology but may play a greater role in other type 2 diseases such as asthma.
JAK Inhibitors
More than 50 cytokines signal via intracellular JAK signaling pathways [232] and abnormal JAK–STAT signaling is associated with immune disorders [233], making JAKs a target for treating immune-related diseases. JAK inhibitors have the potential to inhibit signaling from a range of cytokines involved in type 1, type 2, and type 3 pathways (Fig. 4) [234,235,236,237].

Adapted from Schwartz et al. [238]. For simplicity, the depicted receptor subunits do not reflect the extent of differentiation in domain structures of the various JAK cytokine receptors. EPO erythropoietin, G-CSF granulocyte colony-stimulating factor, GH growth hormone, GM-CSF granulocyte macrophage colony-stimulating factor, IFN interferon, IL interleukin, JAK Janus kinase, LIF leukemia inhibitory factor, OSM oncostatin M, TPO thrombopoietin
JAK signaling pathways in type 2 inflammation and JAK inhibitors.
JAK inhibitors were initially approved for use in RA [159, 180, 239] and have been evaluated for the treatment of type 2 immune diseases such as AD. Tofacitinib is a pan-JAK inhibitor that preferentially inhibits JAK1 and JAK3, and is approved for use in RA, psoriatic arthritis, and UC [239, 240]. However, safety concerns that have emerged during trials of tofacitinib for other indications may limit the utility of tofacitinib in treating AD. More selective JAK inhibitors in the treatment of AD have revealed a more acceptable safety profile [160].
Delgocitinib has wide-reaching actions against Th1, Th2, and Th17 responses and inhibitory effects against JAK1, JAK2, JAK3, and TYK2; it is approved in Japan as a topical agent to treat AD [241, 242]. In a phase 3 trial of topical delgocitinib in Japanese patients with moderate or severe AD affecting 10–30% of the body surface area, delgocitinib resulted in greater and more rapid improvements in skin signs and pruritus by week 4 compared with the vehicle. Long-term efficacy was demonstrated in two uncontrolled studies of 28 and 52 weeks’ duration, respectively [162, 163]. Further study in pediatric patients is ongoing. Common AEs associated with delgocitinib are shown in Table 2.
Ruxolitinib is an inhibitor of JAK1 and JAK2. Oral administration is associated with immunosuppression and increased infection risk [243]. Topical ruxolitinib is approved in the USA for the short-term and noncontinuous treatment of mild-to-moderate AD in non-immunocompromised patients aged at least 12 years whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. Approval was based on results from two phase 3 trials (TRuE-AD1 and TRuE-AD2), which investigated topical ruxolitinib cream (0.75% twice daily [BID] and 1.5% BID) in adults and adolescents with mild-to-moderate AD. Both trials met the primary endpoint of Investigator’s Global Assessment Treatment Success for both formulations at week 8 [244]. Additional 52-week data demonstrated that topical ruxolitinib is effective at maintaining treatment effect up to 52 weeks with no AEs suggestive of a relationship to systemic exposure observed, and no meaningful changes or trends in hematologic parameters [245]. Common AEs associated with ruxolitinib are listed in Table 2.
Some serious AEs have been reported with pan-JAK inhibitors, including cytopenias (which may arise due to disrupted JAK-mediated signaling of hematopoietic growth factors), gastrointestinal tract perforation (which may arise due to altered JAK signaling for IL-6, IL-22, IL-10, and IL-9, all of which are involved in intestinal barrier function), and malignancy (which may result from T-cell and NK cell dysregulation) [238]. Although these events are uncommon (generally < 5% incidence), they tend to occur at frequencies larger than those observed with biologics [159, 180, 238, 239, 246, 247].
At the appropriate dose, baricitinib selectively inhibits JAK1- and JAK2-mediated signaling. Baricitinib is approved by the European Union and Japan to treat moderate-to-severe AD in adults [158]. In two phase 3 trials in adults with moderate-to-severe AD (BREEZE-AD 1 and 2), baricitinib showed significant improvements in AD skin signs and symptoms at the higher dose (4 mg once daily) for a 16-week period. Improvements in itch began as early as week 1 at 4 mg and week 2 at 2 mg. Both doses reduced nighttime awakenings and skin pain, and improved QoL. Table 2 lists common AEs associated with baricitinib.
Two next-generation JAK inhibitors, which are believed to be more JAK1 selective at certain lower doses, are upadacitinib and abrocitinib [157, 238, 248,249,250]. JAK1 inhibition blocks signaling pathways for interferons and a range of cytokines, including IL-4, IL-13, IL-31, TSLP, and IFN-γ.
The central antiviral response is mediated by IFNα and IFNβ [251], both of which signal via JAK1 and TYK2. Another central antiviral cytokine, IFN-γ, has antibacterial and antiprotozoan activity and signals via JAK1 and JAK2 [139]. The development and function of natural killer cells capable of killing virus-infected cells depend on IL-15 and IL-7, both of which signal through JAK1- and JAK3-associated receptors [252]. Thus, inhibition of JAK kinases may account for the increased risk of infection, including herpes zoster infection, observed in patients treated with JAK inhibitors [253,254,255,256,257,258,259,260]. Herpes zoster is seen more frequently with JAK inhibitors in patients from Asian regions [257, 259, 260], with crude incidence rates of herpes zoster events (expressed per 100 patient-years) of 9.2 (Japan/Korea), 8.9 (India), 2.7 (Western Europe), and 3.3 (USA/Canada/Australia) in patients treated with tofacitinib [256] and comparable incidence rates in Asian patients treated with baricitinib (Japan: 10.7; Taiwan: 13.0; Korea: 13.1) [258]. This trend has also been noted in patients with plaque psoriasis [261] and in patients with AD treated with upadacitinib [262].
Upadacitinib is approved in the USA, European Union, and Japan for the treatment of AD in adults and adolescents [180, 181]. Table 2 shows the most common AEs associated with upadacitinib. A recent phase 3 trial comparing upadacitinib (30 mg) and dupilumab (300 mg) found that upadacitinib efficacy in reducing AD signs was demonstrated earlier than dupilumab (by week 16) and with greater efficacy compared with dupilumab at week 16, but with no statistical difference at week 24 [263]. Recent data from open-label studies of patients switching from dupilumab to upadacitinib have demonstrated improved efficacy without complete washout of dupilumab, which may suggest enhanced efficacy when these agents are combined [263].
Abrocitinib is approved in the USA for treatment of adults with moderate-to-severe AD [155] and in the UK and Japan for the treatment of moderate-to-severe AD in adults and adolescents [156]. In a phase 3 trial comparing abrocitinib with dupilumab and placebo in adults with moderate-to-severe AD, abrocitinib demonstrated a greater reduction in signs and symptoms of AD compared with placebo at week 12. The 200 mg (but not 100 mg) dose significantly reduced itch by week 2 compared with dupilumab [95]. Moreover, although significantly more patients achieved a 90% improvement in Eczema Area and Severity Index at week 4 and week 16 with abrocitinib compared with dupilumab, no differences were observed at week 26 [264]. In patients with AD aged 12 years and older (JADE MONO 1 and 2), abrocitinib showed rapid and significant improvements by week 12 compared with placebo. The most common AEs associated with abrocitinib are listed in Table 2.
Overall, by broadly impacting cytokine signaling, JAK inhibitors act rapidly to dampen itch and effectively reduce AD lesions, but longer-term head-to-head comparative studies show that a more targeted approach with a biologic such as dupilumab reaches the same goal [95]. Drawing a direct correlation between JAK selectivity and safety profile has proved challenging, which may be because JAK selectivity is tissue selective and dose dependent; at higher doses, selective JAK inhibitors begin to affect other JAKs [265]. The safety profile of semiselective JAK1 inhibitors in patients with RA suggests that they may also inhibit JAK2 to some extent, particularly at higher doses. For example, dose-dependent thrombocytopenia was observed in clinical trials of abrocitinib for AD and psoriasis [249, 266], suggesting that abrocitinib may inhibit JAK2, which is involved in hematopoiesis, at higher doses. Indeed, abrocitinib is expected to inhibit JAK1, JAK2, and TYK2 in vivo [267, 268]. Tofacitinib, which preferentially inhibits JAK1 and JAK3, increased hemoglobin levels at 5 mg, but the effect was diminished at 10 mg [269]. This suggests that tofacitinib may start to inhibit JAK2 at higher doses [270].
To date, studies of JAK inhibitors in type 2 inflammatory disease are mostly limited to AD. Longer studies are required to better understand the role of JAK inhibitors in these diseases. Although selective inhibition of certain JAKs may hypothetically maximize efficacy while minimizing AEs compared with pan-JAK inhibitors, the selectivity of targeted JAK inhibitors demonstrated in vitro may be challenging to maintain in vivo [271].
Discussion and Future Perspectives
Dysregulation of type 2 immunity can lead to a number of chronic diseases such as AD and asthma. Immune dysregulation in these diseases is often highly complex and involves many different cell types and inflammatory mediators. Despite the multifaceted nature of type 2 inflammatory disease pathophysiology, clinical studies of targeted therapies, such as biologic therapies, suggest that only a few components play a clinically significant role. For example, the relative success of tralokinumab in patients with AD, but not asthma, suggests that IL-13 plays a clinically significant role in AD but, by itself, a lesser role in asthma pathophysiology. The clinical efficacy of dupilumab with targeting both IL-4 and IL-13 in multiple type 2 inflammatory diseases suggests that IL-4 and IL-13 are critical target molecules. Similarly, targeting IgE or TSLP is effective in asthma, but modulating these targets has not yielded consistent results in AD [152, 212, 213, 220, 222, 228, 229, 272]. Although pan-JAK inhibitors have demonstrated efficacy in treating some inflammatory diseases, the broad cytokine inhibition elicited by JAK inhibitors is associated with a number of serious AEs that may limit their therapeutic potential in other populations. More recently, JAK inhibitors with greater selectivity (and a more acceptable safety profile) have demonstrated rapid efficacy in AD. Evaluations in larger populations over time in a wider range of type 2 inflammatory diseases are necessary to better understand the impact of JAK inhibition on type 2 inflammatory diseases. Studies of drugs targeting type 2 immune mediators have helped clarify the biological mechanisms underlying type 2 immunity while also providing tremendous therapeutic advances for diseases involving type 2 inflammation.
Many additional targets for modulation of type 2 immunity have been identified, and development of novel agents will no doubt continue to advance our understanding of type 2 inflammatory diseases.
Change history
19 June 2022
A peer-reviewed infographic was retrospectively added to this publication. From digital feature text “video abstract” deleted.
References
Chaplin DD. Overview of the immune response. J Allergy Clin Immunol. 2010;125(2 suppl 2):S3-23.
Palm NW, Rosenstein RK, Medzhitov R. Allergic host defences. Nature. 2012;484(7395):465–72.
Maizels RM. Regulation of immunity and allergy by helminth parasites. Allergy. 2020;75(3):524–34.
Inclan-Rico JM, Siracusa MC. First responders: innate immunity to helminths. Trends Parasitol. 2018;34(10):861–80.
de Kouchkovsky DA, Ghosh S, Rothlin CV. Negative regulation of type 2 immunity. Trends Immunol. 2017;38(3):154–67.
Yatim KM, Lakkis FG. A brief journey through the immune system. Clin J Am Soc Nephrol. 2015;10(7):1274–81.
Sonnenberg GF, Hepworth MR. Functional interactions between innate lymphoid cells and adaptive immunity. Nat Rev Immunol. 2019;19(10):599–613.
Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy Clin Immunol. 2015;135(3):626–35.
Chen L, Deng H, Cui H, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2018;9(6):7204–18.
Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol. 2015;15(5):271–82.
Withers DR. Innate lymphoid cell regulation of adaptive immunity. Immunology. 2016;149(2):123–30.
Mazzurana L, Rao A, Van Acker A, Mjösberg J. The roles for innate lymphoid cells in the human immune system. Semin Immunopathol. 2018;40(4):407–19.
Gandhi NA, Bennett BL, Graham NMH, Pirozzi G, Stahl N, Yancopoulos GD. Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discov. 2016;15(1):35–50.
Makepeace BL, Martin C, Turner JD, Specht S. Granulocytes in helminth infection—who is calling the shots? Curr Med Chem. 2012;19(10):1567–86.
Brockmann L, Giannou AD, Gagliani N, et al. Regulation of TH17 cells and associated cytokines in wound healing, tissue regeneration, and carcinogenesis. Int J Mol Sci. 2017;18(5):1033.
Patel DD, Kuchroo VK. Th17 cell pathway in human immunity: lessons from genetics and therapeutic interventions. Immunity. 2015;43(6):1040–51.
Bachert C, Marple B, Schlosser RJ, et al. Adult chronic rhinosinusitis. Nat Rev Dis Primers. 2020;6(1):1–9.
Ruffner MA, Cianferoni A. Phenotypes and endotypes in eosinophilic esophagitis. Ann Allergy Asthma Immunol. 2020;124(3):233–9.
Garcovich S, Maurelli M, Gisondi P, Peris K, Yosipovitch G, Girolomoni G. Pruritus as a distinctive feature of type 2 inflammation. Vaccines (Basel). 2021;9(3):303.
Lloyd CM, Snelgrove RJ. Type 2 immunity: expanding our view. Sci Immunol. 2018;3(25):1604.
Gieseck RL, Wilson MS, Wynn TA. Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol. 2018;18(1):62–76.
Herbert DR, Douglas B, Zullo K. Group 2 innate lymphoid cells (ILC2): type 2 immunity and helminth immunity. Int J Mol Sci. 2019;20(9):2276.
Zhu J. T helper 2 (Th2) cell differentiation, type 2 innate lymphoid cell (ILC2) development and regulation of interleukin-4 (IL-4) and IL-13 production. Cytokine. 2015;75(1):14–24.
Chakraborty S, Kubatzky KF, Mitra DK. An update on interleukin-9: from its cellular source and signal transduction to its role in immunopathogenesis. Int J Mol Sci. 2019;20(9):2113.
Bağci IS, Ruzicka T. IL-31: a new key player in dermatology and beyond. J Allergy Clin Immunol. 2018;141(3):858–66.
Liu YJ, Soumelis V, Watanabe N, et al. TSLP: an epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu Rev Immunol. 2007;25:193–219.
Saenz SA, Siracusa MC, Perrigoue JG, et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature. 2010;464(7293):1362–6.
Junttila IS, Mizukami K, Dickensheets H, et al. Tuning sensitivity to IL-4 and IL-13: differential expression of IL-4Ralpha, IL-13Ralpha1, and gammac regulates relative cytokine sensitivity. J Exp Med. 2008;205(11):2595–608.
Noda S, Krueger JG, Guttman-Yassky E. The translational revolution and use of biologics in patients with inflammatory skin diseases. J Allergy Clin Immunol. 2015;135(2):324–36.
Moyle M, Cevikbas F, Harden JL, Guttman-Yassky E. Understanding the immune landscape in atopic dermatitis: the era of biologics and emerging therapeutic approaches. Exp Dermatol. 2019;28(7):756–68.
Roufosse F. Targeting the interleukin-5 pathway for treatment of eosinophilic conditions other than asthma. Front Med (Lausanne). 2018;5:49.
Goswami R, Kaplan MH. A brief history of IL-9. J Immunol. 2011;186(6):3283–8.
Salter BM, Oliveria JP, Nusca G, et al. IL-25 and IL-33 induce Type 2 inflammation in basophils from subjects with allergic asthma. Respir Res. 2016;17:5.
Tworek D, Smith SG, Salter BM, et al. Am J Respir Crit Care Med. 2016;193(9):957–64.
Wu L, Zepp JA, Qian W, et al. J Immunol. 2015;194(9):4528–34.
Murdaca G, Greco M, Tonacci A, et al. IL-33/IL-31 axis in immune-mediated and allergic diseases. Int J Mol Sci. 2019;20(23):5856.
Sonkoly E, Muller A, Lauerma AI, et al. IL-31: a new link between T cells and pruritus in atopic skin inflammation. J Allergy Clin Immunol. 2006;117(2):411–7.
Ding W, Zou G-L, Zhang W, Lai XN, Chen HW, Xiong LX. Interleukin-33: its emerging role in allergic disease. Molecules. 2018;23(7):1665.
Junttila IS. Tuning the cytokine responses: an update on interleukin (IL)-4 and IL-13 receptor complexes. Front Immunol. 2018;9:888.
Brown MA, Pierce JH, Watson CJ, Falco J, Ihle JN, Paul WE. B cell stimulatory factor-1/interleukin-4 mRNA is expressed by normal and transformed mast cells. Cell. 1987;50(5):809–18.
Moqbel R, Ying S, Barkans J, et al. Identification of messenger RNA for IL-4 in human eosinophils with granule localization and release of the translated product. J Immunol. 1995;155(10):4939–47.
Yoshimoto T, Paul WE. CD4pos, NK1. 1pos T cells promptly produce interleukin 4 in response to in vivo challenge with anti-CD3. J Exp Med. 1994;179(4):1285–95.
Yoshimoto T, Tsutsui H, Tominaga K, et al. IL-18, although antiallergic when administered with IL-12, stimulates IL-4 and histamine release by basophils. Proc Natl Acad Sci USA. 1999;96(24):13962–6.
La Flamme AC, Kharkrang M, Stone S, Mirmoeini S, Chuluundorj D, Kyle R. Type II-activated murine macrophages produce IL-4. PLoS ONE. 2012;7(10):e46989.
Cherwinski HM, Schumacher JH, Brown KD, Mosmann TR. Two types of mouse helper T cell clone. III. Further differences in lymphokine synthesis between Th1 and Th2 clones revealed by RNA hybridization, functionally monospecific bioassays, and monoclonal antibodies. J Exp Med. 1987;166(5):1229–44.
Pasha MA, Patel G, Hopp R, Yang Q. Role of innate lymphoid cells in allergic diseases. Allergy Asthma Proc. 2019;40(3):138–45.
Swain SL, Weinberg AD, English MI, Huston GA. IL-4 directs the development of Th2-like helper effectors. J Immunol. 1990;145(11):3796–806.
Lebman DA, Coffman RL. Interleukin 4 causes isotype switching to IgE in T cell-stimulated clonal B cell cultures. J Exp Med. 1988;168(3):853–62.
Moon HB, Severinson E, Heusser C, Johansson SG, Möller G, Persson U. Regulation of IgG1 and IgE synthesis by interleukin 4 in mouse B cells. Scand J Immunol. 1989;30(3):355–61.
Gascan H, Gauchat JF, Roncarolo MG, Yssel H, Spits H, de Vries JE. Human B cell clones can be induced to proliferate and to switch to IgE and IgG4 synthesis by interleukin 4 and a signal provided by activated CD4+ T cell clones. J Exp Med. 1991;173(3):747–50.
Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176(1):287–92.
Dubois GR, Bruijnzeel PL. IL-4-induced migration of eosinophils in allergic inflammation. Ann N Y Acad Sci. 1994;725:268–73.
Kopf M, Le Gros G, Bachmann M, Lamers MC, Bluethmann H, Köhler G. Disruption of the murine IL-4 gene blocks Th2 cytokine responses. Nature. 1993;362(6417):245–8.
Gundel R, Lindell D, Harris P, Fournel M, Jesmok G, Gerritsen ME. IL-4 induced leucocyte trafficking in cynomolgus monkeys: correlation with expression of adhesion molecules and chemokine generation. Clin Exp Allergy. 1996;26(6):719–29.
Boone M, Lespagnard L, Renard N, Song M, Rihoux JP. Adhesion molecule profiles in atopic dermatitis vs allergic contact dermatitis: pharmacological modulation by cetirizine. J Eur Acad Dermatol Venereol. 2000;14(4):263–6.
Le Floch A, Allinne J, Martin J, et al. Dupilumab protects from type 2 inflammation by impacting both systemic and local inflammatory events downstream of IL-4/IL-13 signalling. Allergy. 2020;75(5):1188–204.
Borthwick LA, Wynn TA, Fisher AJ. Cytokine mediated tissue fibrosis. Biochim Biophys Acta. 2013;1832(7):1049–60.
Gasparini G, Cozzani E, Parodi A. Interleukin-4 and interleukin-13 as possible therapeutic targets in systemic sclerosis. Cytokine. 2020;125: 154799.
Knipper JA, Willenborg S, Brinckmann J, et al. Interleukin-4 receptor α signaling in myeloid cells controls collagen fibril assembly in skin repair. Immunity. 2015;43(4):803–16.
Nguyen JK, Austin E, Huang A, Mamalis A, Jagdeo J. The IL-4/IL-13 axis in skin fibrosis and scarring: mechanistic concepts and therapeutic targets. Arch Dermatol Res. 2020;312(2):81–92.
Wynn TA. Fibrotic disease and the TH 1/TH 2 paradigm. Nat Rev Immunol. 2004;4(8):583–94.
Danso MO, van Drongelen V, Mulder A, et al. TNF-a and Th2 cytokines induce atopic dermatitis–like features on epidermal differentiation proteins and stratum corneum lipids in human skin equivalents. J Invest Dermatol. 2014;134(7):1941–50.
Howell MD, Gallo RL, Boguniewicz M, et al. Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity. 2006;24(3):341–8.
Howell MD, Kim BE, Gao P, et al. Cytokine modulation of atopic dermatitis filaggrin skin expression. J Allergy Clin Immunol. 2007;120(1):150–5.
Bao L, Alexander JB, Zhang H, Shen K, Chan LS. Interleukin-4 downregulation of involucrin expression in human epidermal keratinocytes involves Stat6 sequestration of the coactivator CREB-binding protein. J Interferon Cytokine Res. 2016;36(6):374–81.
Bao L, Mohan GC, Alexander JB, et al. A molecular mechanism for IL-4 suppression of loricrin transcription in epidermal keratinocytes: implication for atopic dermatitis pathogenesis. Innate Immun. 2017;23(8):641–7.
Kim BE, Leung DYM, Boguniewicz M, Howell MD. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin Immunol. 2008;126(3):332–7.
Furue M. Regulation of filaggrin, loricrin, and involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: pathogenic implications in atopic dermatitis. Int J Mol Sci. 2020;21(15):5382.
Graber P, Gretener D, Herren S, et al. The distribution of IL-13 receptor α1 expression on B cells, T cells and monocytes and its regulation by IL-13 and IL-4. Eur J Immunol. 1998;28(12):4286–98.
Migita M, Yamaguchi N, Mita S, et al. Characterization of the human IL-5 receptors on eosinophils. Cell Immunol. 1991;133(2):484–97.
Yuan Q, Campanella GS, Colvin RA, et al. Membrane-bound eotaxin-3 mediates eosinophil transepithelial migration in IL-4-stimulated epithelial cells. Eur J Immunol. 2006;36(10):2700–14.
Wirnsberger G, Hebenstreit D, Posselt G, Horejs-Hoeck J, Duschl A. IL-4 induces expression of TARC/CCL17 via two STAT6 binding sites. Eur J Immunol. 2006;36(7):1882–91.
Fukuda T, Fukushima Y, Numao T, et al. Role of interleukin-4 and vascular cell adhesion molecule-1 in selective eosinophil migration into the airways in allergic asthma. Am J Respir Cell Mol Biol. 1996;14(1):84–94.
Licona-Limón P, Henao-Mejia J, Temann AU, et al. Th9 cells drive host immunity against gastrointestinal worm infection. Immunity. 2013;39(4):744–57.
Furue M, Yamamura K, Kido-Nakahara M, Nakahara T, Fukui Y. Emerging role of interleukin-31 and interleukin-31 receptor in pruritus in atopic dermatitis. Allergy. 2018;73(1):29–36.
Valizadeh A, Khosravi A, Zadeh LJ, Parizad EG. Role of IL-25 in Immunity. J Clin Diagn Res. 2015;9(4):E01−4.
Wang YH, Angkasekwinai P, Lu N, et al. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC–activated Th2 memory cells. J Exp Med. 2007;204(8):1837–47.
Drake LY, Kita H. IL-33: biological properties, functions, and roles in airway disease. Immunol Rev. 2017;278(1):173–84.
Endo Y, Hirahara K, Iinuma T, et al. The interleukin-33-p38 kinase axis confers memory T helper 2 cell pathogenicity in the airway. Immunity. 2015;42(2):294–308.
Shikotra A, Choy DF, Ohri CM, et al. Increased expression of immunoreactive thymic stromal lymphopoietin in patients with severe asthma. J Allergy Clin Immunol. 2012;129(1):104−11:E1−9.
Soumelis V, Reche PA, Kanzler H, et al. Human epithelial cells trigger dendritic cell-mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3(7):673–80.
Tatsuno K, Fujiyama T, Yamaguchi H, Waki M, Tokura Y. TSLP directly interacts with skin-homing Th2 cells highly expressing its receptor to enhance IL-4 production in atopic dermatitis. J Invest Dermatol. 2015;135(12):3017–24.
Ito T, Wang YH, Duramad O, et al. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J Exp Med. 2005;202(9):1213–23.
Sano Y, Masuda K, Tamagawa-Mineoka R, et al. Thymic stromal lymphopoietin expression is increased in the horny layer of patients with atopic dermatitis. Clin Exp Immunol. 2013;171(3):330–7.
Wilson SR, Thé L, Batia LM, et al. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell. 2013;155(2):285–95.
Stone KD, Prussin C, Metcalfe DD. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol. 2010;125(2 Suppl 2):S73-80.
Bochner BS. Systemic activation of basophils and eosinophils: markers and consequences. J Allergy Clin Immunol. 2000;106(5 Suppl):S292-302.
Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol. 1999;17:701–38.
Oetjen LK, Mack MR, Feng J, et al. Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell. 2017;171(1):217-28.e13.
Miake S, Tsuji G, Takemura M, et al. IL-4 augments IL-31/IL-31 receptor alpha interaction leading to enhanced Ccl 17 and Ccl 22 production in dendritic cells: implications for atopic dermatitis. Int J Mol Sci. 2019;20(16):4053.
Harrison DA. The Jak/STAT pathway. Cold Spring Harb Perspect Biol. 2012;4(3): a011205.
Barbarot S, Auziere S, Gadkari A, et al. Epidemiology of atopic dermatitis in adults: results from an international survey. Allergy. 2018;73(6):1284–93.
Nutten S. Atopic dermatitis: global epidemiology and risk factors. Ann Nutr Metab. 2015;66(Suppl 1):8–16.
Hill DA, Spergel JM. The atopic march: critical evidence and clinical relevance. Ann Allergy Asthma Immunol. 2018;120(2):131–7.
Bieber T, Simpson EL, Silverberg JI, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med. 2021;384(12):1101–12.
Guttman-Yassky E, Krueger JG, Lebwohl MG. Systemic immune mechanisms in atopic dermatitis and psoriasis with implications for treatment. Exp Dermatol. 2018;27(4):409–17.
Grobe W, Bieber T, Novak N. Pathophysiology of atopic dermatitis. JDDG J Dtsch Dermatol Ges. 2019;17(4):433–40.
Fiset PO, Leung DYM, Hamid Q. Immunopathology of atopic dermatitis. J Allergy Clin Immunol. 2006;118(1):287–90.
Bieber T. Atopic dermatitis. Ann Dermatol. 2010;22(2):125–37.
Furue M, Chiba T, Tsuji G, et al. Atopic dermatitis: immune deviation, barrier dysfunction, IgE autoreactivity and new therapies. Allergol Int. 2017;66(3):398–403.
Stingl G, Maurer D. IgE-mediated allergen presentation via fc epsilon rl on antigen-presenting cells. Int Arch Allergy Immunol. 1997;113(1–3):24–9.
Guttman-Yassky E, Nograles KE, Krueger JG. Contrasting pathogenesis of atopic dermatitis and psoriasis—part II: immune cell subsets and therapeutic concepts. J Allergy Clin Immunol. 2011;127(6):1420–32.
Honda T, Kabashima K. Reconciling innate and acquired immunity in atopic dermatitis. J Allergy Clin Immunol. 2020;145(4):1136–7.
Elsner JS, Carlsson M, Stougaard JK, et al. The OX40 axis is associated with both systemic and local involvement in atopic dermatitis. Acta Derm Venereol. 2020;100(6):99.
Furue M, Furue M. OX40L–OX40 signaling in atopic dermatitis. J Clin Med. 2021;10(12):2578.
Czarnowicki T, He H, Cancer T, et al. Evolution of pathologic T-cell subsets in atopic dermatitis from infancy to adulthood. J Allergy Clin Immunol. 2020;145(1):215–28.
Irvine AD, McLean WHI, Leung DYM. Filaggrin mutations associated with skin and allergic diseases. N Engl J Med. 2011;365(14):1315–27.
Cornelissen C, Marquardt Y, Czaja K, et al. IL-31 regulates differentiation and filaggrin expression in human organotypic skin models. J Allergy Clin Immunol. 2012;129(2):426–33.
Beck LA, Cork MJ, Amagai M, et al. Type 2 Inflammation contributes to skin barrier dysfunction in atopic dermatitis. JID Innovations 2022 [In press].
Bieber T. Interleukin-13: Targeting an underestimated cytokine in atopic dermatitis. Allergy. 2020;75(1):54–62.
Hashimoto T, Mishra SK, Olivry T, Yosipovitch G. Periostin, an emerging player in itch sensation. J Invest Dermatol. 2021;141(10):2338–43.
Yamaguchi Y. Periostin in skin tissue skin-related diseases. Allergol Int. 2014;63(2):161–70.
Mashiko S, Mehtaa H, Bissonnette R, Sarfati M. Increased frequencies of basophils, type 2 innate lymphoid cells and Th2 cells in skin of patients with atopic dermatitis but not psoriasis. J Dermatol Sci. 2017;88(2):167–74.
Kim BS, Siracusa MC, Saenz SA, et al. TSLP elicits IL-33–independent innate lymphoid cell responses to promote skin inflammation. Sci Transl Med. 2013;5(170):170ra16.
Kim BS, Wang K, Siracusa MC, et al. Basophils promote innate lymphoid cell responses in inflamed skin. J Immunol. 2014;193(7):3717–25.
Imai Y, Yasuda K, Nagai M, et al. IL-33-induced atopic dermatitis-like inflammation in mice is mediated by group 2 innate lymphoid cells in concert with basophils. J Invest Dermatol. 2019;139(10):2185-94.e3.
Yamanishi Y, Mogi K, Takahashi K, Miyake K, Yoshikawa S, Karasuyama H. Skin-infiltrating basophils promote atopic dermatitis-like inflammation via IL-4 production in mice. Allergy. 2020;75(10):2613–22.
Ewald DA, Malajian D, Krueger JG, et al. Meta-analysis derived atopic dermatitis (MADAD) transcriptome defines a robust AD signature highlighting the involvement of atherosclerosis and lipid metabolism pathways. BMC Med Genomics. 2015;8:60.
He H, Bissonnette R, Wu J, et al. Tape strips detect distinct immune and barrier profiles in atopic dermatitis and psoriasis. J Allergy Clin Dermatol. 2021;147(1):199–212.
Guttman-Yassky E, Diaz A, Pavel AB, et al. Use of tape strips to detect immune and barrier abnormalities in the skin of children with early-onset atopic dermatitis. JAMA Dermatol. 2019;155(12):1358–70.
Pavel AB, Renert-Yuval Y, Wu J, et al. Tape strips from early-onset pediatric atopic dermatitis highlight disease abnormalities in nonlesional skin. Allergy. 2021;76(1):314–25.
Weidinger S, Novak N. Atopic dermatitis. Lancet. 2016;387(10023):1109–22.
Gupta K, Harvima IT. Mast cell-neural interactions contribute to pain and itch. Immunol Rev. 2018;282(1):168–87.
Kubanov AA, Katunina OR, Chikin VV. Expression of neuropeptides, neurotrophins, and neurotransmitters in the skin of patients with atopic dermatitis and psoriasis. Bull Exp Biol Med. 2015;159(3):318–22.
Ohsawa Y, Hirasawa N. The role of histamine H1 and H4 receptors in atopic dermatitis: from basic research to clinical study. Allergol Int. 2014;63(4):533–42.
Wang F, Trier AM, Li F, et al. A basophil-neuronal axis promotes itch. Cell. 2021;184(2):422-40.e17.
Brunner T, Heusser CH, Dahinden CA. Human peripheral blood basophils primed by interleukin 3 (IL-3) produce IL-4 in response to immunoglobulin E receptor stimulation. J Exp Med. 1993;177(3):605–11.
Mollanazar NK, Smith PK, Yosipovitch G. Mediators of chronic pruritus in atopic dermatitis: getting the itch out? Clin Rev Allergy Immunol. 2016;51(3):263–92.
Feld M, Garcia R, Buddenkotte J, et al. The pruritus-and TH2-associated cytokine IL-31 promotes growth of sensory nerves. J Allergy Clin Immunol. 2016;138(2):500–8.
Furue M, Ulzii D, Vu YH, Tsuji G, Kido-Nakahara M, Nakahara T. Pathogenesis of atopic dermatitis: current paradigm. Iran J Immunol. 2019;16(2):97–107.
Meng J, Moriyama M, Feld M, et al. New mechanism underlying IL-31-induced atopic dermatitis. J Allergy Clin Immunol. 2018;141(5):1677-89.e8.
Gutzmer R, Mommert S, Gschwandtner M, Zwingmann K, Stark H, Werfel T. The histamine H4 receptor is functionally expressed on T(H)2 cells. J Allergy Clin Immunol. 2009;123(3):619–25.
Campion M, Smith L, Gatault S, Métais C, Buddenkotte J, Steinhoff M. Interleukin-4 and interleukin-13 evoke scratching behavior in mice. Exp Dermatol. 2019;28(12):1501–4.
Kim J, Kim BE, Leung DYM. Pathophysiology of atopic dermatitis: clinical implications. Allergy Asthma Proc. 2019;40(2):84–92.
Esaki H, Brunner PM, Renert-Yuval Y, et al. Early-onset pediatric atopic dermatitis is TH2 but also TH17 polarized in skin. J Allergy Clin Immunol. 2016;138(6):1639–51.
Noda S, Suárez-Fariñas M, Ungar B, et al. The Asian atopic dermatitis phenotype combines features of atopic dermatitis and psoriasis with increased TH17 polarization. J Allergy Clin Immunol. 2015;136(5):1254–64.
Sugaya M. The role of th17-related cytokines in atopic dermatitis. Int J Mol Sci. 2020;21(4):1314.
Henrickson SE, Ruffner MA, Kwan M. Unintended immunological consequences of biologic therapy. Curr Allergy Asthma Rep. 2016;16(6):46.
Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–45.
Liu FT, Goodarzi H, Chen HY. IgE, mast cells, and eosinophils in atopic dermatitis. Clin Rev Allergy Immunol. 2011;41(3):298–310.
Oldhoff JM, Darsow U, Werfel T, et al. Anti-IL-5 recombinant humanized monoclonal antibody (mepolizumab) for the treatment of atopic dermatitis. Allergy. 2005;60(5):693–6.
Kang EG, Narayana PK, Pouliquen IJ, Lopez MC, Ferreira-Cornwell MC, Getsy JA. Efficacy and safety of mepolizumab administered subcutaneously for moderate to severe atopic dermatitis. Allergy. 2020;75(4):950–3.
Howell MD, Parker ML, Mustelin T, Ranade K. Past, present, and future for biologic intervention in atopic dermatitis. Allergy. 2015;70(8):887–96.
Deleanu D, Nedelea I. Biological therapies for atopic dermatitis: an update. Exp Ther Med. 2019;17(2):1061–7.
US Food and Drug Administration. DUPIXENT® (dupilumab). Highlights of prescribing information. https://www.regeneron.com/sites/default/files/Dupixent_FPI.pdf. Accessed 13 Dec 2021.
European Medicines Agency. DUPIXENT® (dupilumab). Summary of product characteristics. https://www.ema.europa.eu/en/documents/product-information/dupixent-epar-product-information_en.pdf. Accessed 13 Dec 2021.
Simpson EL, Bieber T, Guttman-Yassky E, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375(24):2335–48.
Blauvelt A, de Bruin-Weller M, Gooderham M, et al. Long-term management of moderate-to-severe atopic dermatitis with dupilumab and concomitant topical corticosteroids (LIBERTY AD CHRONOS): a 1-year, randomised, double-blinded, placebo-controlled, phase 3 trial. Lancet. 2017;389(10086):2287–303.
de Bruin-Weller M, Thaçi D, Smith CH, et al. Dupilumab with concomitant topical corticosteroid treatment in adults with atopic dermatitis with an inadequate response or intolerance to ciclosporin A or when this treatment is medically inadvisable: a placebo-controlled, randomized phase III clinical trial (LIBERTY AD CAFÉ). Br J Dermatol. 2018;178(5):1083–101.
Worm M, Simpson EL, Thaçi D, et al. Efficacy and safety of multiple dupilumab dose regimens after initial successful treatment in patients with atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156(2):131–43.
Simpson EL, Parnes JR, She D, et al. Tezepelumab, an anti-thymic stromal lymphopoietin monoclonal antibody, in the treatment of moderate to severe atopic dermatitis: a randomized phase 2a clinical trial. J Am Acad Dermatol. 2019;80(4):1013–21.
Simpson EL, Paller AS, Siegfried EC, et al. Efficacy and safety of dupilumab in adolescents with uncontrolled moderate to severe atopic dermatitis: a phase 3 randomized clinical trial. JAMA Dermatol. 2020;156(1):44–56.
Beck LA, Thaçi D, Deleuran M, et al. Dupilumab provides favorable safety and sustained efficacy for up to 3 years in an open-label study of adults with moderate-to-severe atopic dermatitis. Am J Clin Dermatol. 2020;21(4):567–77.
Thyssen JP, Blauvelt A, Lockshin B, et al. Dupilumab provides long-term efficacy for up to 4 years in an open-label extension study of adults with moderate-to-severe atopic dermatitis. Poster presented at the 3rd Annual Conference of Revolutionizing Atopic Dermatitis (RAD); Virtual Conference; December 11–13, 2021.
US Food and Drug Administration. Abrocitinib PI (US). CIBINQO (Abrocitinib). Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/213871s000lbl.pdf. Accessed 13 May 2022.
Abrocitinib PI (EMA). CIBINQO (abrocitinib). Summary of product characteristics. https://www.medicines.org.uk/emc/product/12873/smpc. Accessed 13 Dec 2021.
Simpson EL, Sinclair R, Forman S, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet. 2020;396(10246):255–66.
European Medicines Agency. Baricitinib PI (EMA). OLUMIANT (baricitinib). Summary of Product Characteristics. https://www.ema.europa.eu/en/documents/product-information/olumiant-epar-product-information_en.pdf. Accessed 13 Dec 2021.
US Food and Drug Administration. Baricitinib PI (US). OLUMIANT (baricitinib). Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/207924s000lbl.pdf. Accessed 13 Dec 2021.
Simpson EL, Lacour J-P, Spelman L, et al. Baricitinib in patients with moderate-to-severe atopic dermatitis and inadequate response to topical corticosteroids: results from two randomized monotherapy phase III trials. Br J Dermatol. 2020;183(2):242–55.
US Food and Drug Administration. Benralizumab PI (USA). FASENRA (benralizumab). Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761070s000lbl.pdf. Accessed 20 Jan 2022.
Nakagawa H, Nemoto O, Igarashi A, Saeki H, Kaino H, Nagata T. Delgocitinib ointment, a topical Janus kinase inhibitor, in adult patients with moderate to severe atopic dermatitis: a phase 3, randomized, double-blind, vehicle-controlled study and an open-label, long-term extension study. J Am Acad Dermatol. 2020;82(4):823–31.
Nakagawa H, Nemoto O, Igarashi A, et al. Long-term safety and efficacy of delgocitinib ointment, a topical Janus kinase inhibitor, in adult patients with atopic dermatitis. J Dermatol. 2020;47(2):114–20.
Paller AS, Wollenberg A, Siegfried E, et al. Laboratory safety of dupilumab in patients aged 6–11 years with severe atopic dermatitis: results from a phase III clinical trial. Pediatr Drugs. 2021;23(5):515–27.
Guttman-Yassky E, Brunner PM, Neumann AU, et al. Efficacy and safety of fezakinumab (an IL-22 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by conventional treatments: a randomized, double-blind, phase 2a trial. J Am Acad Dermatol. 2018;78(5):872–81.
Simpson EL, Flohr C, Eichenfield LF, et al. Efficacy and safety of lebrikizumab (an anti-IL-13 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical corticosteroids: a randomized, placebo-controlled phase II trial (TREBLE). J Am Acad Dermatol. 2018;78(5):863-71.e11.
Guttman-Yassky E, Blauvelt A, Eichenfield LF, et al. Efficacy and safety of lebrikizumab, a high-affinity interleukin 13 inhibitor, in adults with moderate to severe atopic dermatitis. JAMA Dermatol. 2020;156(4):411–20.
US Food and Drug Administration. Mepolizumab PI (USA). NUCALA (mepolizumab). Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/125526Orig1s000Lbl.pdf. Accessed 14 Dec 2021.
Kabashima K, Matsumura T, Komazaki H, Kawashima M, Nemolizumab-JP01 Study Group. Trial of nemolizumab and topical agents for atopic dermatitis with pruritus. N Engl J Med. 2020;383(2):141–50.
US Food and Drug Administration. Omalizumab PI (US): XOLAIR® (omalizumab) Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/103976s5225lbl.pdf. Accessed 14 Dec 2021.
Chan S, Cornelius V, Cro S, Harper JI, Lack G. Treatment effect of omalizumab on severe pediatric atopic dermatitis the ADAPT randomized clinical trial. JAMA Pediatr. 2020;174(1):29–37.
US Food and Drug Administration. Reslizumab PI (USA). CINQAIR (reslizumab). Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/761033lbl.pdf. Accessed 14 Dec 2021.
US Food and Drug Administration. Ruxolitinib PI (USA). JAKAFI™ (ruxolitinib). Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202192lbl.pdf. Accessed 14 Dec 2021.
Kim BS, Howell MD, Sun K, et al. Treatment of atopic dermatitis with ruxolitinib cream (JAK1/JAK2 inhibitor) or triamcinolone cream. J Allergy Clin Immunol. 2020;145(2):572–82.
US Food and Drug Administration. Tofacitinib PI (US). ZELJANZ (tofacitinib). Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/203214s028,208246s013,213082s003lbl.pdf. Accessed 20 Jan 2022.
European Medicines Agency. Tralokinumab PI (EMA). Summary of product characteristics. https://www.ema.europa.eu/documents/product-information/adtralza-epar-product-information_en.pdf. Accessed 15 Dec 2021.
US Food and Drug Administration. Tralokinumab PI (USA). Adbry (Tralokinumab). Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2022/761180Orig1s000lbl.pdf. Accessed 13 May 2022.
Silverberg JI, Toth D, Bieber T, et al. Tralokinumab plus topical corticosteroids for the treatment of moderate-to-severe atopic dermatitis: results from the double-blind, randomized, multicentre, placebo-controlled phase III ECZTRA 3 trial. Br J Dermatol. 2021;184(3):450–63.
Wollenberg A, Blauvelt A, Guttman-Yassky E, et al. Tralokinumab for moderate-to-severe atopic dermatitis: results from two 52-week, randomized, double-blind, multicentre, placebo-controlled phase III trials (ECZTRA 1 and ECZTRA 2). Br J Dermatol. 2021;184(3):437–49.
US Food and Drug Administration. Upadacitinib PI (US). RINVOQ™ (upadacitinib). Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/211675s004lbl.pdf. Accessed 20 Jan 2022.
Upadacitinib PI (EMA). RINVOQ (upadacitinib). Summary of product characteristics. https://www.ema.europa.eu/en/documents/product-information/rinvoq-epar-product-information_en.pdf. Accessed 15 Dec 2021.
Guttman-Yassky E, Teixeira HD, Simpson EL, et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet. 2021;397(10290):2151–68.
Guttman-Yassky E, Bissonnette R, Ungar B, et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J Allergy Clin Immunol. 2019;143(1):155–72.
Möbus L, Rodriguez E, Harder I, et al. Atopic dermatitis displays stable and dynamic skin transcriptome signatures. J Allergy Clin Immunol. 2021;147(1):213–23.
Rohner MH, Thormann K, Cazzaniga S, et al. Dupilumab reduces inflammation and restores the skin barrier in patients with atopic dermatitis. Allergy. 2021;76(4):1268–70.
Imai Y, Kusakabe M, Nagai M, Yasuda K, Yamanishi K. Dupilumab effects on innate lymphoid cell and helper T cell populations in patients with atopic dermatitis. JID Innovations. 2021;1(1): 100003.
Busse WW, Maspero JF, Rabe KF, et al. Liberty asthma QUEST: phase 3 randomized, double-blind, placebo-controlled, parallel-group study to evaluate dupilumab efficacy/safety in patients with uncontrolled, moderate-to-severe asthma. Adv Ther. 2018;35(5):737–48.
Rabe KF, Nair P, Brusselle G, et al. Efficacy and safety of dupilumab in glucocorticoid-dependent severe asthma. N Engl J Med. 2018;378(26):2475–85.
Bachert C, Han JK, Desrosiers M, et al. Efficacy and safety of dupilumab in patients with severe chronic rhinosinusitis with nasal polyps (LIBERTY NP SINUS-24 and LIBERTY NP SINUS-52): results from two multicentre, randomised, double-blind, placebo-controlled, parallel-group phase 3 trials. Lancet. 2019;394(10209):1638–50.
Rial MJ, Barroso B, Sastre J. Dupilumab for treatment of food allergy. J Allergy Clin Immunol Pract. 2019;7(2):673–4.
Bawany F, Franco AI, Beck LA. Dupilumab: One therapy to treat multiple atopic diseases. JAAD Case Rep. 2020;6(11):1150–2.
Eichenfield LF, Bieber T, Beck LA, et al. Infections in dupilumab clinical trials in atopic dermatitis: a comprehensive pooled analysis. Am J Clin Dermatol. 2019;20(3):443–56.
Ou Z, Chen C, Chen A, Yang Y, Zhou W. Adverse events of dupilumab in adults with moderate-to-severe atopic dermatitis: a meta-analysis. Int Immunopharmacol. 2018;54:303–10.
Blauvelt A, Wollenberg A, Eichenfield L, et al. Infections in adults with moderate-to-severe atopic dermatitis treated with dupilumab: long-term data from an open-label extension (OLE) study. J Am Acad Dermatol. 2021;85(3 Suppl):143.
Paller AS, Beck LA, Blauvelt A, et al. Infections in dupilumab pediatric clinical trials in atopic dermatitis—a pooled analysis. Pediatr Dermatol: In press; 2022.
Geng B, Bachert C, Busse WW, et al. Respiratory infections and anti-infective medication use from phase 3 dupilumab respiratory studies. J Allergy Clin Immunol Pract. 2021;S2213–2198(21)01374-X.
Wollenberg A, Howell MD, Guttman-Yassky E, et al. Treatment of atopic dermatitis with tralokinumab, an anti-IL-13 mAb. J Allergy Clin Immunol. 2019;143(1):135–41.
Siegfried EC, Bieber T, Simpson EL, et al. Effect of dupilumab on laboratory parameters in adolescents with atopic dermatitis: results from a randomized, placebo-controlled, phase 3 clinical trial. Am J Clin Dermatol. 2021;22(2):243–55.
Sastre J, Dávila I. Dupilumab: a new paradigm for the treatment of allergic diseases. J Investig Allergol Clin Immunol. 2018;28(3):139–50.
Panettieri RA, Sjöbring U, Péterffy A, et al. Tralokinumab for severe, uncontrolled asthma (STRATOS 1 and STRATOS 2): two randomised, double-blind, placebo-controlled, phase 3 clinical trials. Lancet Respir Med. 2018;6(7):511–25.
Hanania NA, Korenblat P, Chapman KR, et al. Efficacy and safety of lebrikizumab in patients with uncontrolled asthma (LAVOLTA I and LAVOLTA II): replicate, phase 3, randomised, double-blind, placebo-controlled trials. Lancet Respir Med. 2016;4(10):781–96.
Bleecker ER, FitzGerald JM, Chanez P, et al. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting β 2-agonists (SIROCCO): a randomised, multicentre, placebo-controlled phase 3 trial. Lancet. 2016;388(10056):2115–27.
Busse WW, Bleecker ER, FitzGerald JM, et al. Long-term safety and efficacy of benralizumab in patients with severe, uncontrolled asthma: 1-year results from the BORA phase 3 extension trial. Lancet Respir Med. 2019;7(1):46–59.
FitzGerald JM, Bleecker ER, Nair P, et al. Benralizumab, an anti-interleukin-5 receptor α monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2016;388(10056):2128–41.
Bel EH, Wenzel SE, Thompson PJ, et al. Oral glucocorticoid-sparing effect of mepolizumab in eosinophilic asthma. N Engl J Med. 2014;371(13):1189–97.
Chupp GL, Bradford ES, Albers FC, et al. Efficacy of mepolizumab add-on therapy on health-related quality of life and markers of asthma control in severe eosinophilic asthma (MUSCA): a randomised, double-blind, placebo-controlled, parallel-group, multicentre, phase 3b trial. Lancet Respir Med. 2017;5(5):390–400.
Lugogo N, Domingo C, Chanez P, et al. Long-term efficacy and safety of mepolizumab in patients with severe eosinophilic asthma: a multi-center, open-label, phase IIIb study. Clin Ther. 2016;38(9):2058-70.e1.
Ortega HG, Liu MC, Pavord ID, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371(13):1198–207.
Pavord ID, Korn S, Howarth P, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet. 2012;380(9842):651–9.
Castro M, Zangrilli J, Wechsler ME, et al. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir Med. 2015;3(5):355–66.
Corren J, Weinstein S, Janka L, Zangrilli J, Garin M. Phase 3 study of reslizumab in patients with poorly controlled asthma: effects across a broad range of eosinophil counts. Chest. 2016;150(4):799–810.
Busse W, Corren J, Lanier BQ, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol. 2001;108(2):184–90.
Solèr M, Matz J, Townley R, et al. The anti-IgE antibody omalizumab reduces exacerbations and steroid requirement in allergic asthmatics. Eur Respir J. 2001;18(2):254–61.
Gevaert P, Omachi TA, Corren J, et al. Efficacy and safety of omalizumab in nasal polyposis: 2 randomized phase 3 trials. J Allergy Clin Immunol. 2020;146(3):595–605.
Brandström J, Vetander M, Sundqvist AC, et al. Individually dosed omalizumab facilitates peanut oral immunotherapy in peanut allergic adolescents. Clin Exp Allergy. 2019;49(10):1328–41.
Fiocchi A, Artesani MC, Riccardi C, et al. Impact of omalizumab on food allergy in patients treated for asthma: a real-life study. J Allergy Clin Immunol Pract. 2019;7(6):1901-9.e5.
Maurer M, Giménez-Arnau AM, Sussman G, et al. Ligelizumab for chronic spontaneous urticaria. N Engl J Med. 2019;381(14):1321–32.
Brunner PM, Pavel AB, Khattri S, et al. Baseline IL-22 expression in patients with atopic dermatitis stratifies tissue responses to fezakinumab. J Allergy Clin Immunol. 2019;143(1):142–54.
Metz M, Maurer M. Use of biologics in chronic spontaneous urticaria–beyond omalizumab therapy? Allergol Select. 2021;5:89–95.
ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). Identifier NCT04701983, Study to assess the efficacy, safety, and tolerability of SAR440340/REGN3500/Itepekimab in Chronic Obstructive Pulmonary Disease (COPD) (AERIFY-1). https://clinicaltrials.gov/ct2/show/NCT04701983. Accessed 13 Dec 2021.
Adis Insight Drug Profile. Itepekimab - Regeneron Pharmaceuticals/Sanofi. https://adisinsight.springer.com/drugs/800048225. Accessed 13 Dec 2021.
Wechsler ME, Ruddy MK, Pavord ID, et al. Efficacy and safety of itepekimab in patients with moderate-to-severe asthma. N Engl J Med. 2021;385(18):1656–68.
Puar N, Chovatiya R, Paller AS. New treatments in atopic dermatitis. Ann Allergy Asthma Immunol. 2021;126(1):21–31.
AnaptysBio. AnaptysBio reports etokimab ATLAS phase 2b clinical trial in moderate-to-severe atopic dermatitis fails to meet primary endpoint [press release]. 2019. https://ir.anaptysbio.com/news-releases/news-release-details/anaptysbio-reports-etokimab-atlas-phase-2b-clinical-trial. Accessed 13 Dec 2021.
West EE, Kashyap M, Leonard WJ. TSLP: a key regulator of asthma pathogenesis. Drug Discov Today Dis Mech. 2012;9(3–4):10.1016.
ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). Identifier NCT03809663, A dose ranging placebo-controlled double-blind study to evaluate the safety and efficacy of tezepelumab in atopic dermatitis. https://clinicaltrials.gov/ct2/show/NCT03809663. Accessed 13 Dec 2021.
Adis Insight Drug Profile. Tezepelumab - Amgen/AstraZeneca. https://adisinsight.springer.com/drugs/800031599. Accessed 15 Dec 2021.
Corren J, Parnes JR, Wang L, et al. Tezepelumab in adults with uncontrolled asthma. N Engl J Med. 2017;377(10):936–46.
Menzies-Gow A, Corren J, Bourdin A, et al. Tezepelumab in adults and adolescents with severe, uncontrolled asthma. N Engl J Med. 2021;384(19):1800–9.
AstraZeneca. Update on SOURCE phase III trial for tezepelumab in patients with severe, oral corticosteroid-dependent asthma [Press Release]. 2020. https://www.astrazeneca.com/media-centre/press-releases/2020/update-on-source-phase-iii-trial-for-tezepelumab-in-patients-with-severe-oral-corticosteroid-dependent-asthma.html. Accessed 13 Dec 2021.
Wechsler M, Menzies Gow A, Brightling CE, et al. Oral corticosteroid-sparing effect of tezepelumab in adults with severe asthma. Am J Respir Crit Care Med. 2021;203:A1197.
Morris R, Kershaw NJ, Babon JJ. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018;27(12):1984–2009.
Shuai K, Liu B. Regulation of JAK–STAT signalling in the immune system. Nat Rev Immunol. 2003;3(11):900–11.
Gadina M, Le MT, Schwartz DM, et al. Janus kinases to jakinibs: from basic insights to clinical practice. Rheumatology (Oxford). 2019;58(Suppl 1):i4-16.
Virtanen AT, Haikarainen T, Raivola J, Silvennoinen O. Selective JAKinibs: prospects in inflammatory and autoimmune diseases. BioDrugs. 2019;33(1):15–32.
Welsch K, Holstein J, Laurence A, Ghoreschi K. Targeting JAK/STAT signalling in inflammatory skin diseases with small molecule inhibitors. Eur J Immunol. 2017;47(7):1096–107.
Yamaoka K, Saharinen P, Pesu M, Holt VE, Silvennoinen O, O’Shea JJ. The Janus kinases (Jaks). Genome Biol. 2004;5(12):253.
Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;16(12):843–62.
US Food and Drug Administration. Tofacitinib PI (US). XELJANZ® (tofacitinib). Highlights of prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/203214s018lbl.pdf. Accessed 15 Dec 2021.
European Medicines Agency. Tofacitinib PI (EMA). Summary of product characteristics. https://www.ema.europa.eu/en/documents/product-information/xeljanz-epar-product-information_en.pdf. Accessed 15 Dec 2021.
Tanimoto A, Ogawa Y, Oki C, et al. Pharmacological properties of JTE-052: a novel potent JAK inhibitor that suppresses various inflammatory responses in vitro and in vivo. Inflamm Res. 2015;64(1):41–51.
Tanimoto A, Shinozaki Y, Yamamoto Y, et al. A novel JAK inhibitor JTE-052 reduces skin inflammation and ameliorates chronic dermatitis in rodent models: comparison with conventional therapeutic agents. Exp Dermatol. 2018;27(1):22–9.
Lussana F, Cattaneo M, Rambaldi A, Squizzato A. Ruxolitinib-associated infections: a systematic review and meta-analysis. Am J Hematol. 2018;93(3):339–47.
Papp K, Szepietowski JC, Kircik L, et al. Efficacy and safety of ruxolitinib cream for the treatment of atopic dermatitis: results from two phase 3, randomized, double-blind studies. J Am Acad Dermatol. 2021;85(4):863–72.
Papp K, Szepietowski J, Kircik L, et al. Long-term safety and disease control with ruxolitinib cream in atopic dermatitis: results from two phase 3 studies. Presented at the Annual Revolutionizing Atopic Dermatitis (RAD) Conference; Virtual Conference; June 13, 2021.
Curtis JR, Lee EB, Kaplan IV, et al. Tofacitinib, an oral Janus kinase inhibitor: analysis of malignancies across the rheumatoid arthritis clinical development programme. Ann Rheum Dis. 2016;75(5):831–41.
King B, Maari C, Lain E, et al. Extended safety analysis of baricitinib 2 mg in adult patients with atopic dermatitis: an integrated analysis from eight randomized clinical trials. Am J Clin Dermatol. 2021;22(3):395–405.
Guttman-Yassky E, Thaçi D, Pangan AL, et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J Allergy Clin Immunol. 2020;145(3):877–84.
Silverberg JI, Simpson EL, Thyssen JP, et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156(8):863–73.
Gooderham MJ, Forman SB, Bissonnette R, et al. Efficacy and safety of oral Janus kinase 1 inhibitor abrocitinib for patients with atopic dermatitis: a phase 2 randomized clinical trial. JAMA Dermatol. 2019;155(12):1371–9.
Schoggins JW, Rice CM. Interferon-stimulated genes and their antiviral effector functions. Curr Opin Virol. 2011;1(6):519–25.
Vian L, Le MT, Gazaniga N, et al. JAK inhibition differentially affects NK cell and ILC1 homeostasis. Front Immunol. 2019;10:2972.
Colombel JF. Herpes zoster in patients receiving JAK inhibitors for ulcerative colitis: mechanism, epidemiology, management, and prevention. Inflamm Bowel Dis. 2018;24(10):2173–82.
Sunzini F, McInnes I, Siebert S. JAK inhibitors and infections risk: focus on herpes zoster. Ther Adv Musculoskelet Dis. 2020;12:1759720X20936059.
Winthrop KL, Melmed GY, Vermeire S, et al. Herpes zoster infection in patients with ulcerative colitis receiving tofacitinib. Inflamm Bowel Dis. 2018;24(10):2258–65.
Winthrop KL, Yamanaka H, Valdez H, et al. Herpes zoster and tofacitinib therapy in patients with rheumatoid arthritis. Arthritis Rheumatol. 2014;66(10):2675–84.
Winthrop KL, Harigai M, Genovese MC, et al. Infections in baricitinib clinical trials for patients with active rheumatoid arthritis. Ann Rheum Dis. 2020;79(10):1290–7.
Chen YC, Yoo DH, Lee CK, et al. Safety of baricitinib in East Asian patients with moderate-to-severe active rheumatoid arthritis: an integrated analysis from clinical trials. Int J Rheum Dis. 2020;23(1):65–73.
Harigai M, Takeuchi T, Smolen JS, et al. Safety profile of baricitinib in Japanese patients with active rheumatoid arthritis with over 1.6 years median time in treatment: an integrated analysis of phases 2 and 3 trials. Mod Rheumatol. 2020;30(1):36–43.
Kameda H, Takeuchi T, Yamaoka K, et al. Efficacy and safety of upadacitinib in Japanese patients with rheumatoid arthritis (SELECT-SUNRISE): a placebo-controlled phase IIb/III study. Rheumatology (Oxford). 2020;59(11):3303–13.
Zhang J, Tsai TF, Lee MG, et al. The efficacy and safety of tofacitinib in Asian patients with moderate to severe chronic plaque psoriasis: a phase 3, randomized, double-blind, placebo-controlled study. J Dermatol Sci. 2017;88(1):36–45.
PMDA review report on upadacitinib. https://www.pmda.go.jp/drugs/2021/P20210831002/112130000_30200AMX00027_A100_1.pdf. Accessed 12 Dec 2021.
Blauvelt A, Teixeira HD, Simpson EL, et al. PT29: Upadacitinib versus dupilumab in adults with moderate-to-severe atopic dermatitis: Analysis of the HEADS UP phase 3 trial. Presented at the Annual Meeting of the International Society of Atopic Dermatitis (ISAD 2021); April 19, 2021. Seoul, South Korea.
Reich K, Thyssen J, Blauvelt A, et al. Efficacy and safety of abrocitinib versus dupilumab in adults with moderate-to-severe atopic dermatitis who received background topical therapy in a 26-week, randomized, head-to-head trial. EADV 30TH Congress 2021 (EADV 2021); Sept 29, 2021. Virtual.
Winthrop KL. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat Rev Rheumatol. 2017;13(4):234–44.
Schmieder GJ, Draelos ZD, Pariser DM, et al. Efficacy and safety of the Janus kinase 1 inhibitor PF-04965842 in patients with moderate-to-severe psoriasis: phase II, randomized, double-blind, placebo-controlled study. Br J Dermatol. 2018;179(1):54–62.
Vazquez ML, Kaila N, Strohbach JW, et al. Identification of N-{cis-3-[Methyl (7 H-pyrrolo [2, 3-d] pyrimidin-4-yl) amino] cyclobutyl} propane-1-sulfonamide (PF-04965842): a selective JAK1 clinical candidate for the treatment of autoimmune diseases. J Med Chem. 2018;61(3):1130–52.
Peeva E, Hodge MR, Kieras E, et al. Evaluation of a Janus kinase 1 inhibitor, PF-04965842, in healthy subjects: A phase 1, randomized, placebo-controlled, dose-escalation study. Br J Clin Pharmacol. 2018;84(8):1776–88.
Schulze-Koops H, Strand V, Nduaka C, et al. Analysis of haematological changes in tofacitinib-treated patients with rheumatoid arthritis across phase 3 and long-term extension studies. Rheumatology (Oxford). 2017;56(1):46–57.
Choy EH. Clinical significance of Janus Kinase inhibitor selectivity. Rheumatology (Oxford). 2019;58(6):953–62.
Danese S, Argollo M, Le Berre C, Peyrin-Biroulet L. JAK selectivity for inflammatory bowel disease treatment: does it clinically matter? Gut. 2019;68(10):1893–9.
Holm JG, Thomsen SF. Omalizumab for atopic dermatitis: evidence for and against its use. G Ital Dermatol Venereol. 2019;154(4):480–7.
Acknowledgements
Funding
This research was sponsored by Sanofi and Regeneron Pharmaceuticals, Inc. The journal’s Rapid Service Fee was sponsored by Sanofi and Regeneron Pharmaceuticals, Inc.
Authorship
All named authors meet the International Committee of Medical Journal Editors criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
El-Bdaoui Haddad, Kazuhiko Arima, Robert McDonald, and Sonya Cyr contributed to manuscript concept and design. All authors interpreted the data, provided critical feedback on the manuscript, approved the final manuscript for submission, and are accountable for the accuracy and integrity of the manuscript.
Medical Writing, Editorial, and Other Assistance
The authors thank Alexandra Hicks, PhD, of Sanofi for her critical review of the manuscript. Medical writing and editorial assistance were provided by Jamie Casswell, PhD, and Carolyn Ellenberger, PhD, of Excerpta Medica, funded by Sanofi and Regeneron Pharmaceuticals, Inc., according to the Good Publication Practice guideline.
Disclosures
Kazuhiko Arima, El-Bdaoui Haddad, Robert McDonald, and Frank O. Nestle are employees of Sanofi and may hold stock and/or stock options in the company. Sonya Cyr and Noah Levit are employees and shareholders of Regeneron Pharmaceuticals, Inc.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Haddad, EB., Cyr, S.L., Arima, K. et al. Current and Emerging Strategies to Inhibit Type 2 Inflammation in Atopic Dermatitis. Dermatol Ther (Heidelb) 12, 1501–1533 (2022). https://doi.org/10.1007/s13555-022-00737-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-022-00737-7