
Abstract
Maturation of metabolomics has brought a deeper appreciation for the importance of isomeric identity of lipids to their biological role, mirroring that for proteoforms in proteomics. However, full characterization of the lipid isomerism has been thwarted by paucity of rapid and effective analytical tools. A novel approach is ion mobility spectrometry (IMS) and particularly differential or field asymmetric waveform IMS (FAIMS) at high electric fields, which is more orthogonal to mass spectrometry. Here we broadly explore the power of FAIMS to separate lipid isomers, and find a ~75% success rate across the four major types of glycero- and phospho- lipids (sn, chain length, double bond position, and cis/trans). The resolved isomers were identified using standards, and (for the first two types) tandem mass spectrometry. These results demonstrate the general merit of incorporating high-resolution FAIMS into lipidomic analyses.

ᅟ
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
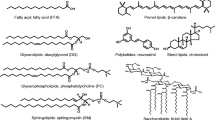
Proteomics and metabolomics measure the complement of proteins and metabolites in living tissues presumed to reflect and predict the health and disease states [1, 2]. Expression of metabolites (unlike proteins) is not coded in the genome, creating an open-ended number of options that incorporates massive isomeric complexity. For instance, a common lipid subclass is glycerolipids made by esterifying fatty acid (FA) chains to one or more of the three glycerol hydroxyls. These feature four major isomer types (Figure 1): (i) acyl chain substitution or stereonumbering (sn-) that results from exchange of FA chains (transacylation), (ii) chain length from swapping pieces between FAs, and, for unsaturated lipids, (iii) double bond (DB) positional isomers and (iv) cis/trans (Z/E) geometry at each DB [3,4,5,6,7,8,9,10,11,12]. Permuting these variations can yield hundreds of isomers with often distinct biological activities, many present in vivo [3,4,5,6,7,8,9,10,11,12,13]. As is widely known, trans-fats differ in uptake rates from the cis- analogs and are implicated in health issues, including cardiovascular and psychiatric diseases and slow infant growth [8,9,10,11]. In another example, the nutritional absorption and bioavailability of constituent FAs differs across sn- isomers [4, 14]. Such isomers with distinct FAs exchanged between sn1 and sn3 positions are enantiomers [15, 16], and proper chirality also matters. Accordingly, characterizing the lipidome to individual isomer level should be inherent to biomedical analyses [17].

Scheme of four major lipid isomer types: (i) sn-, (ii) chain length, (iii) DB position, (iv) cis/trans
The prevailing “shotgun” lipidomics approach [18] based on tandem mass spectrometry (MS) via collision-induced dissociation (CID) effectively profiles lipid stoichiometries, but the CID spectra of isomers typically differ only in the fragment ratios that are sensitive to the excitation energy and collision cross-section and thus depend on the ionization source, gas pressure, and details of the instrument design and operation that control the ion temperatures and product mass discrimination [12, 13, 19, 20]. Thus relying on those ratios requires thorough calibration using pure standards for each conceivable isomer and is difficult for mixtures. In a new ozone-induced dissociation (OzID) mode, ozone mixed into the CID collision gas selectively severs the DBs in unsaturated lipids, revealing their position [21,21,23]. One can also subject CID products to OzID [15, 24]. Unfortunately, OzID is blind to other isomer types.
This situation calls for isomer separations before the MS/MS step. This has been traditionally pursued with liquid chromatography (LC) [16,25,26,27,28,29,, 25–31], using both the usual reversed phase and tailored non-aqueous reversed phase [27], silver-ion normal phase that exploits Ag ions binding to DBs [28,28,30], and their combination [31]. Despite dramatic advances in HPLC of lipids, many isomers could be separated only partly (if at all) despite lengthy gradients [28]. Other protocols involve complicated and laborious prior derivatization [28, 29]. Hence, the assessment that “the inability to distinguish isomeric lipids is the major limitation of existing lipidomics weaponry that seriously impedes the understanding of lipid biochemistry” [22].
An alternative or complement to condensed-phase methods is rapid post-ionization ion mobility spectrometry (IMS) in gases that has unique selectivity [32]. Linear IMS devices based on absolute mobility (K) comprise the original drift-tube (DT) IMS with constant electric field and dynamic-field systems such as traveling-wave (TW) IMS implemented in the Synapt platforms (Waters, Milford, MA). These can be “nested” between LC and MS stages, with the peaks eluting from LC dispersed by IMS and analyzed by (time-of-flight) MS without slowing the LC step [33]. The resulting domains in the IMS/MS palette differ for major biomolecular classes (e.g., peptides, lipids, carbohydrates, oligonucleotides), for some enough to classify by the peak position [34, 35]. However, finer categorization is obstructed by tight correlation between the ion mass (m) and K for homologous compounds that have analogous elemental composition and global morphology [34, 35]. This especially holds for lipids that normally form +1 (−1) ions and therefore one trend line in the IMS/MS space (unlike peptides where multiply-charged ions distribute over several lines with unequal slopes) [33]. Further, the mean and top deviations of those +1 ions from the central trend are just 2.6% and ~5% (versus 7.3% and ~30% for peptides and 8.8% and ~35% for carbohydrates) [36], which stringently constrains the power of linear IMS in isomer separations. Hence, while linear IMS can clean up lipid mixtures from isobaric and matrix interferences for superior component elucidation [36,36,37,38,39,40,42] and tissue imaging [41] and largely separate some subclasses (e.g., sphingolipids and phospholipids [36] or ceramides and lysophospholipids [41]), isomer resolution has been rare: most isomers resolved by LC were not separated by IMS in LC/IMS experiments [43]. Poor orthogonality between IMS and MS dimensions can be mitigated by high IMS resolving power (R), and the latest DT IMS systems [44] operated at >1 Atm to attain R > 100 distinguish some isomers such as phosphocholines (PC) 18:1 with DB in positions 9 and 6. However, some separations claimed in that work were elicited by sophisticated peak fitting.
A newer FAIMS approach exploits nonlinear ion motion in electric fields of high intensity (E) to sort ions by the average derivative of K(E) function over a certain range [45, 46]. This quantity is extracted by a periodic asymmetric field applied across a gap between two electrodes, through which ions are carried by the gas flow. Ions injected into the gap are pushed toward either electrode, but a given species can be balanced (and thus passed and detected) by superposing a particular compensation voltage (CV) on the waveform. As the K(E) derivative is correlated to m substantially weaker than the mobility itself, FAIMS is far more orthogonal to MS than linear IMS [46,46,47,48,49,51]. The specific advantage depends on the analyte nature and is about 4-fold for lipids [48]. Hence, FAIMS tends to resolve isomers much better than linear IMS with equal R metric.
The resolving power maximizes in planar gaps where homogeneous electric field equilibrates only one species at any CV, although finite resolution may allow similar species to pass [46, 52]. The resolution is sensitive to the carrier gas, and is generally augmented by addition of He or H2 to N2 because of higher ion mobilities in lighter gases and pronounced non-Blanc effect in mixtures of molecules with disparate masses [46,53,54,, 53–56]. The latter is theoretically maximized at ~80% He, in line with the findings in FAIMS chips (where microscopic gaps permit extreme fields by the Paschen law) [56, 57]. However, the resolving power of these chips is low, and the electrical breakdown in macroscopic devices employed here [53, 54] constrains the He fraction to ~50%–75%.
High-resolution FAIMS has been deployed to separate various biomolecular isomers, including the peptide sequence inversions and variants differing in the localization of post-translational modifications [49], protein conformers [50], and isotopomers [51]. There were three such studies for lipids: a survey mapping subclasses in the FAIMS/MS space and demonstrating first resolution of isomers (sn- for diacylglycerols) [48], targeted separation of two phospholipid isomers (requiring Ag+ cationization, as the usual protonated species could not be pulled apart) [58], and resolution of sn-isomers for triacylglycerols using alcohol vapors doped into the gas [59]. Fractionation of complex samples into subclasses (e.g., based on the head group) without isomer separation was also documented [60, 61].
Hence, FAIMS has been shown to separate sn- but not the other three isomer types. Here, we explore them all using the system with highest resolving power provided by He/N2 buffers and maximum feasible voltages. The results prove broad utility of FAIMS for lipid isomer separations.
Experimental
We employed a planar (p-) FAIMS unit with the gap width (g) of 1.88 mm [51]. The high-definition bisinusoidal waveform [62] with adjustable profile and frequency is put out by a supply protected from electrical shorts that enables operation near the arc breakdown threshold without risking equipment damage [51]. Here we adopted the 2:1 harmonic ratio, 1 MHz frequency, and zero-to-peak amplitude (dispersion voltage, DV) of 4 kV, leading to the dispersion field (E D) of 21.3 kV/cm. The compensation voltage (CV) was scanned at 0.3–1 V/min. Ions are pulled along the gap by flow formulated by digital meters (MKS Instruments, Andover, MA, USA) from UHP He and N2 with up to ~70% (v/v) He, the breakdown limit. The total flow rate Q was varied from 2 to 0.7 L/min to control the resolution/sensitivity trade-off by adjusting the filtering time (roughly proportional to 1/Q) from t = 0.2 to 0.5 s [55, 62]. The inflow is split, with one part desolvating incoming ions in a curtain plate/orifice inlet and other carrying ions through the device [52]. The optimum rate depends on the ion nature and gas composition, increasing to offset faster diffusion for smaller ions or lighter gases.
The FAIMS stage is coupled to an LTQ XL mass spectrometer (Thermo Fisher Scientific, Waltham, MA) by an electrodynamic ion funnel interface [51, 52, 62]. The inlet features a multi-hole slit of ~6 mm length that fits the rectangular shape of ion beams leaving the FAIMS gap while matching the conductance of regular cylindrical capillary [51]. The funnel assembled from metalized plastic electrodes has the length of 80 mm, a 25-mm entrance orifice, and 2-mm exit aperture, and operates with axial DC voltage of ~100 V, confining rf voltage of ~1 MHz frequency, and ~110 V amplitude. This funnel effectively captures a supersonic jet expanding from the inlet capillary and focuses it into the first quadrupole of the LTQ.
Ions are generated by electrospray ionization (ESI) source biased at ~2.5 kV versus the FAIMS curtain plate (with subsequent ~1 kV drop to the orifice) from samples infused at ~0.5 μL/min. We focused on ubiquitous glycerolipids and phospholipids in the 500–900 Da range, represented by 32 species (Table 1): 10 diacylglycerols (DG), 11 triacylglycerols (TG), and 11 phosphocholines (PC) that form 19 isomer pairs (5 for DGs, 7 for PCs, and 7 for TGs). These were procured from Larodan (Solna, Sweden) or Avanti (Alabaster, AL, USA) and dissolved to 10 μM in 30:70 chloroform/methanol (Fisher HPLC grade) salinized by ammonium acetate (Fisher ACS grade) at 2% by weight. We adopt the lipid nomenclature outlined by Liebisch [63], Holcapek [27, 59], and Blanksby [15] with the subclass (DG, TG, or PC) followed by giving FAs (or entering 0 if none) at sn1, sn2, and (for DG and TG) sn3 sites. The FAs are identified by the number of carbons, number of DBs, and (if any) location and symmetry of DBs. They are abbreviated as L (lauric, 12:0), M (myristic, 14:0), P (palmitic, 16:0), S (stearic, 18:0), A (arachidic, 20:0), and B (behenic, 22:0) for saturated FAs and Pe (palmitelaidic, 16:1[9E]), Po (palmitoleic, 16:1[9Z]), E (elaidic, 18:1[9E]), O (oleic, 18:1[9Z]), Ps (petroselenic, 18:1[6Z]), Pd (petroselaidic, 18:1[6E]), Lo (linoleic, 18:2[9Z,12Z]), and Le (linoelaidic, 18:2[9E,12E]) for unsaturated FAs. For example, the DG 16:0/0/18:0 is P0S and PC 18:0/18:1(9Z) is SO.
Comparisons of FAIMS spectra acquired not concurrently are complicated by shifts due to variations of DV, ambient pressure, and temperature. Like with MS, the increasing resolving power of IMS and FAIMS renders such shifts more prominent. As with “lock mass” in MS [64], this problem in FAIMS of peptides was addressed using internal calibrants exhibiting consistent CV shifts as a function of experimental parameters [48]. Good calibrants are homologous to the analyte(s) with close (but not overlapping) mass and CV to (1) maximize the correlation between the CV dependences on instrumental parameters for the two species, and (2) minimize the time between the appearances of those species and, thus, the parameter drift during it. A calibrant must be easily ionized to yield strong peaks from traces in the sample, yet not materially suppress the analyte ionization. It should also produce one or few well-defined CV peaks and not complex with the analyte, which could cause artifacts for the analyte and/or calibrant upon the fragmentation of complexes during or after the FAIMS step. By these criteria, we selected lipids similar but not isomeric to targeted analytes: LeLeLe for EEE and its isomers, PdPdPd for other TGs and MS/SM pair, P0A for M0E, and M0O, M0E for other DGs, and MS for other PCs (with the E C scale anchored by PdPdPd and others being secondary calibrants). Spectra were aligned by linear CV scaling to match the calibrant peaks. In some cases, two calibrants (straddling the analyte peak) were employed for utmost accuracy. For transferability of findings across FAIMS devices with unequal gaps, CV is converted to the compensation field (E C) deemed positive for type C ions (where ∂K/dE < 0).
We first inspected the positive-mode mass spectra for all lipids to clarify the stoichiometry of ions from ESI. As expected, those were singly ammoniated and protonated species for glycerides and PC, respectively [48]. We then obtained the E C spectra for individual isomers over a range of gas compositions and flow rates, and applied CID at the apexes of peaks of interest or during E C scans. The calibrated spectra for isomers were superposed to optimize the resolution (E C spread divided by the mean peak width at half maximum, w) and signal at sufficient resolution. Those results were validated by FAIMS/MS/(MS) analyses for isomeric mixtures, in some instances with several component ratios.
Results
As expected, all lipid ions in all He/N2 compositions are type C [46, 48]. Typical behaviors are seen in the separation of type (iii) isomers trielaidin and tripetroselaidin (TGs comprising FAs with single trans-DB in the w9 or w12 position). The E C values go up upon He addition (Figure 2a), while peaks narrow (Figure 2 b–d) as the ion mobility increases [48, 65]. These observations track those for peptides, with E C and peak widths somewhat exceeding those for 1+ ions of similar mass under identical conditions [53, 54]. For example, at 65% He we find w ~2–2.5 V/cm versus 1.6 V/cm on average for 1+ peptides [53] around 900 Da (at Q = 2 L/min). Broader peaks may reflect larger cross-sections and, thus, lower K for lipids compared with isobaric peptides [66] and/or more diverse conformations. Hence, the resolving power for lipids approximates that for 1+ peptides [53], increasing from ~10 in N2 to ~40 at the maximum He content.

Separations of ammoniated trielaidin and tripetroselaidin (m/z = 902.8) at Q = 2 L/min: (a) E C for EEE as a function of He content (circles - measurements, line - quadratic regression), and (b)–(d) spectra for both with peak widths listed. The data measured for 1:1 mixture are added in (d)
The E C difference between EEE and PdPdPd likewise expands at higher He content (Figure 2b–d). Combined with narrower peaks, this elevates the resolution from none in N2 and marginal at 40% He to fair at 65% He. The improved resolving power also pulls away two minor features a1 and a2 on the low-E C sides of dominant peaks b. These species with intensities sensitive to ESI conditions may be protomer-like [67] isomers with distinct ammoniation sites. (Probable attachment of NH4 + to DB may place it on any FA, although that would not engender three peaks as FA1 and FA3 are equivalent). Although these features confound the sought separation in principle, at the highest He fractions they lie too far from the base peaks to interfere (Figure 2d). Such secondary features potentially enhance the isomer resolution, although here their difference (if any) is less than that for the base peaks.
The peak widths in p-FAIMS devices scale as t 1/2 fundamentally and actually faster in view of fringe effects [65, 68]; hence, halving Q to ~1 L/min should decrease w by >1.4 times. The actual narrowing of ~1.7 times delivers peak resolution at half-maximum and perfect filtering of either isomer at its apex (Figure 3 a, b), at a sensitivity penalty of ~5× (a) and ~30× (b). Yet higher resolution (near the detection limit) is achievable at Q = 0.7 L/min. Spectral annotations were confirmed by varying the isomer ratios in a mixture, e.g., 2:1 and 1:2 (Figure 3c). This illustrates the method for realistic samples, where those ratios are generally unequal.

(a), (b) FAIMS spectra for triglyceride isomer pairs (flow rates Q marked for each trace) with widths w (fwhm, V/cm) shown for all peaks. Panel (c) presents the spectra for 2:1 and 1:2 EEE/PdPdPd mixtures. Resolution was improved using Q = 0.9 L/min (e) and 0.7 L/min (h)
Given these trends, we settled on 65% He, throttling the gas as needed for isomer resolution. With peptides, one commonly has to lower the He fraction (often to ~30%–40%) to maintain reasonable ion counts even at the “standard” Q = 2 L/min [49]. This is not the case for lipids because of (1) lower K values compared with multiply charged peptides generated by ESI [43], and perhaps (2) lack of “self-cleaning”, which eliminates ions that underwent significant E C change upon isomerization (unfolding) induced by field heating inside the FAIMS gap [69, 70].
Another isomer of EEE is OOO, comprising the oleic FA with cis-DB in the same place. These formed essentially coincident peaks at any Q down to 0.7 L/min (Figure 3d), but PsPsPs was thus resolved from OOO as well as from EEE. To gauge the prevalence of two outcomes, we probed other type (iv) TG isomer pairs with one elaidic or oleic FA. The PPE and PPO are fully resolved at reduced flow rates, but PELo and POLo are not (Figure 3e, f). It is tempting to rationalize the trans-to-cis transition (that bends FA chains) as less impactful in an FA in the middle sn2 location than sn1 or sn3, but this fails to explain why EEE and OOO co-elute. Many important lipids are polyunsaturated, allowing isomers with multiple DB shifts and/or cis/trans transitions. Such plural variations should intuitively augment the structural difference between isomers and, thus, their separation. Indeed, trilinoelaidin and trilinolein that feature two DBs with trans- or cis-configuration (in the w6 and w9 positions) in each FA are separated by >4 V/cm, which is more than baseline (Figure 3g) and better than the three cis/trans isomers with one transition. Here, conversely to Figure 3e, E C is higher for the trans- than cis-geometry. Some sn- TG isomers with P, S, or O chains were resolved using vapor dopants [59]. Here, the arachidin-containing PAO and AOP are partly resolved at lower flow rates (Figure 3h).
The DGs have lower mass than TGs (here ~600 versus ~800–900 Da) and higher E C values [48], but exhibit similar isomer separations overall (Figure 4). Some sn-isomers were resolved well: e.g., SL0 and LS0 (Figure 4a) or PL0 and P0L [48]. Others were not: e.g., SP0 and P0S (Figure 4b), where only the lack of customary peak compression for the mixture upon Q decreasing from 2 to 0.9 L/min suggests slight separation. This is not readily interpreted as a consequence of larger length difference between S and L (6 carbons) or P and L (4 carbons) compared with S and P (2 carbons): transacylation changes the chain length in sn1/sn3 and sn2 positions by 6 carbons in the SL0/LS0 pair but 16 carbons in SP0/P0S as P and hydroxyl switch places. The O0O and OO0 isomers with same transposition of O are merged too, although here both exhibit two substantial peaks (Figure 4c). However, the PL0/P0L pair [48] shows that such chain-hydroxyl switch isomers are not necessarily problematic.

FAIMS spectra for diglyceride isomer pairs (notation as in Figure 3)
The type (iv) M0E/M0O pair is separated well, with higher E C for the cis-geometry (Figure 4d). The isomers M0B and P0A are resolved almost baseline, despite B and A differing by the minimal two carbons or <10% of the FA length (Figure 4e). How representative is this single example of type (ii) isomers remains to be determined. As diglyceride ions are smaller and more mobile than triglycerides, their peaks are slightly narrower under identical conditions (e.g., w = 1.8–1.9 versus 2.1–2.4 V/cm at Q = 2 L/min). However, the associated sensitivity drop requires somewhat higher Q for diglycerides and the final peak widths compare. The enantiomers with exchanged FAs are not resolvable by IMS methods unless converted to diastereomers or with a chiral vapor doped into the gas. In this respect, the LC separations are currently indispensable.
Our phospholipids lie between DGs and TGs in mass (~700–800 Da) and E C range. Their FAIMS spectra tend to be cleaner with stronger signal and fewer minor peaks, likely reflecting a more efficient and specific protonation at the unique PC group versus ammoniation of glycero-lipids possible on multiple similarly favorable sites. All three sn-isomer pairs tried are well resolved despite modest differences between the FAs involved: four carbons for MS/SM, two carbons and one DB for OP/PO, and a DB for OS/SO (Figure 5a–c). However, each species features a minor peak coincident with the major one for its isomer at all He fractions, with the height ratios conserved regardless of the ESI conditions. This pattern already encountered for DGs [48] was ascribed to isomeric contaminants resulting from imperfect synthesis and/or transacylation during the storage or ESI process [71]. Their abundance is commonly [38, 71] ~10%–15%, in line with the present range of ~2%–30%. The OO and PsPs (with single cis-DB on each FA in the w9 and w12 position) coincided even at the maximum resolving power (Figure 5d), in contrast to the parallel move involving the trans-DB in TGs (Figure 3a–c). Their isomer EE with trans-DBs was resolved from both (Figure 5e), again in contrast to the co-eluting OOO and EEE (Figure 3d). The other type (iv) pair PoPo/PePe was well-separated too (Figure 5f), also with the trans-geometry at higher E C.

FAIMS spectra for phosphocholine isomer pairs (notation as in Figure 3)
The summary FAIMS/MS palette for lipids studied here (Figure 6) reproduces the main trends found at 50% He [48]: the negative correlation between E C and mass within each subclass, higher E C for phospholipids than glycerolipids of similar mass, and lower E C for unsaturated lipids. Consistency of these trends despite much higher E C here (~80–140 V/cm versus ~40–80 V/cm in [48]) confirms their utility for classifying lipids based on the domains in FAIMS/MS space.

The FAIMS/MS palette for all lipids studied. Domain labels identify the class followed by the number of double bonds. Filled and empty symbols are for saturated and unsaturated lipids, respectively. The lines are 1st-order regressions for glycerides and phosphocholines
Lipids are commonly identified by CID. For example, both sn-isomers PO and OP can eliminate O or P as ketenes, yielding fragments at m/z = 522.6 and 496.6, respectively [38, 72]. However, an FA is easier to sever at sn2 than at sn1 (because the tertiary α-hydrogen forming an H-bond with the carbonyl oxygen hinders the latter process), and the ion counts at 522.6 exceed those at 496.6 by ~3–4 times for OP and vice versa for PO [20, 38, 72]. This was also observed here (Figure 7), confirming the isomer separation found with standards (Figure 5b). The features at 18 units below these fragments (m/z of 504.6 and 478.6) arise from the elimination of same FAs as free acids [72]. These two fragments were previously reported as similarly intense [38, 72], but we see greater losses at the sn1 position. Although this preference appears weaker than that for ketene loss, it can help to confirm the assignments. These trends extend to PCs with saturated FAs, e.g., MS and SM (Figure 7). The chain-length isomers (where two chains differ in length) are trivially disentangled by two FA elimination products: e.g., at m/z of 284 and 398 for M0B versus 314 and 370 for P0A. However, CID cannot identify the DB position and cis/trans isomers, as this information resides in the lost chain. For instance, EE, OO, and PsPs eliminate E, O, and Ps as ketenes and free acids, leaving identical products (m/z = 522.6 and 504.6), and the PC head group (through two 1,2-hydride shifts) [38] to yield the fragments at m/z = 604 (Figure 7).

CID spectral windows for exemplary isomers selected by FAIMS from binary mixtures (at peak apexes): sn-isomers (top panels) and DB position and cis/trans isomers (bottom panels). The FAs lost are marked and underlined for ketenes
Concluding, high-resolution FAIMS has separated ~3/4 of lipid isomer pairs across major types (including one pair from [48]): 7 out of 9 sn- and chain length (i, ii), 3 out of 4 DB position (iii), and 7 out of 9 cis/trans (iv). (The only type (ii) pair was considered together with (i), and the numbers do not total to 15 out of 20 because the two pairs belonging to two types were counted in each. For isomers differing in either the DB position or cis/trans arrangement, 1 out of 2 and 5 out of 7 were separated, respectively.) To address the remaining pairs, we are raising the FAIMS resolving power and investigating cationization by Ag+ and other metal ions that benefited some lipid separations [59]. All species resolved were assigned using standards, with confirmation by CID for most sn- and chain length-isomers. Integration of OzID (in progress) will extend the a priori identification to DB position isomers. Overall, this work showcases FAIMS as a powerful tool for rapid lipid isomer elucidation.
References
Issaq, H.J., Veenstra, T.D.: Proteomic and metabolomic approaches to biomarker discovery. Academic Press, New York (2013)
Jones, O.A.H.: Metabolomics and systems biology in human health and medicine. CABI (2014)
Brockerhoff, H., Ackman, R.G.: Positional distribution of isomers of monoenoic fatty acids in animal glycerolipids. J. Lipid Res. 8, 661–666 (1967)
Weber, N., Richter, I., Mangold, H.K., Mukherjee, K.D.: Positional specificity in the incorporation of isomeric cis- and trans-octadecanoic acids into glycerolipids of cultured soya cells. Planta 145, 479–485 (1979)
Ramirez, M., Amate, L., Gil, A.: Absorption and distribution of dietary fatty acids from different sources. Early Hum. Dev. 65, S95–S101 (2001)
Puppione, D.L., Kuehlthau, C.M., Jandacek, R.J., Costa, D.P.: Positional analyses of triacylglycerol fatty acids in the milk fat of the antarctic fur seal (Arctocephalus gazella). Lipids 27, 637–639 (1992)
Emken, E.A.: Physicochemical properties, intake, and metabolism. Am. J. Clin. Nutr. 62, 659S–669S (1995)
Hu, F.B., Stampfer, M.J., Manson, J.E., Rimm, E., Colditz, G.A., Rosner, B.A., Hennekens, C.H., Willett, W.C.: Dietary fat intake and the risk of coronary heart disease in women. N. Engl. J. Med. 337, 1491–1499 (1997)
Innis, S.M., King, D.J.: Trans fatty acids in human milk are inversely associated with concentrations of essential all-cis N-6 and N-3 fatty acids and determine trans, but not N-6 and N-3, fatty acids in plasma lipids of breast-fed infants. Am. J. Clin. Nutr. 70, 383–390 (1999)
Destaillats, F., Sebé dio, J.L., Dionisi, F., Chardigny, J.M.: Trans fatty acids in human nutrition. Oily Press, Bridgewater (2009)
Golomb, B.A., Evans, M.A., White, H.L., Dimsdale, J.E.: Trans fat consumption and aggression. PLOS ONE 7, e32175 (2012)
McAnoy, A.M., Wu, C.C., Murphy, R.C.: Direct qualitative analysis of triacylglycerols by electrospray mass spectrometry using a linear ion trap. J. Am. Soc. Mass Spectrom. 16, 1498–1509 (2005)
Quehenberger, O., Armando, A.M., Brown, A.H., Milne, S.B., Myers, D.S., Merrill, A.H., Bandyopadhyay, S., Jones, K.N., Kelly, S., Shaner, R.L., Sullards, C.M., Wang, E., Murphy, R.C., Barkley, R.M., Leiker, T.J., Raetz, C.R., Guan, Z., Laird, G.M., Six, D.A., Russell, D.W., McDonald, J.G., Subramaniam, S., Fahy, E., Dennis, E.A.: Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 51, 3299–3305 (2010)
Eichmann, T.O., Kumari, M., Haas, J.T., Farese Jr., R.V., Zimmermann, R., Lass, A., Zechner, R.: Studies on the substrate and stereo/regioselectivity of adipose triglyceride lipase, hormone-sensitive lipase, and diacylglycerol-O-acyltransferases. J. Biol. Chem. 287, 41446–41457 (2012)
Marshal, D.L., Pham, H.T., Bhujel, M., Chin, J.S.R., Yew, J.Y., Mori, K., Mitchell, T.W., Blanksby, S.J.: Sequential collision- and ozone-induced dissociation enables assignment of relative acyl chain position in triacylglycerols. Anal. Chem. 88, 2685–2692 (2016)
Itabashi, Y.: Development and application of chromatographic methods for glycerolipid analysis. Chromatography 32, 59–72 (2011)
Chatgilialoglu, C., Ferreri, C., Melchiorre, M., Sansone, A., Torreggiani, A.: Lipid geometrical isomerism: from chemistry to biology and diagnostics. Chem. Rev. 114, 255–284 (2014)
Han, X., Gross, R.W.: Shotgun lipidomics: electrospray ionization mass spectrometric analysis and quantitation of cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom. Rev. 24, 367–412 (2005)
Fauconnot, L., Hau, J., Aeschlimann, J.M., Fay, L.B., Dionisi, F.: Quantitative analysis of triacylglycerol regioisomers in fats and oils using reversed-phase high-performance liquid chromatography and atmospheric pressure chemical ionization mass spectrometry. Rapid Commun. Mass Spectrom. 18, 218–224 (2004)
Han, X.: Lipidomics: comprehensive mass spectrometry of lipids. Wiley, New York (2016)
Thomas, M.C., Mitchell, T.W., Harman, D.G., Deeley, J.M., Nealon, J.R., Blanksby, S.J.: Ozone-induced dissociation: elucidation of double bond position within mass-selected lipid ions. Anal. Chem. 80, 303–311 (2008)
Mitchell, T.W., Pham, H., Thomas, M.C., Blanksby, S.J.: Identification of double bond position in lipids: from GC to OzID. J. Chromatogr. B 877, 2722–2735 (2009)
Poad, B.L., Pham, H.T., Thomas, M., Hughes, J.R., Campbell, J.L., Mitchell, T.W., Blanksby, S.J.: Ozone-induced dissociation on a modified tandem linear ion-trap: observations of different reactivity for isomeric lipids. J. Am. Soc. Mass Spectrom. 21, 1989–1999 (2010)
Pham, H.T., Maccarone, A.T., Thomas, M.C., Campbell, J.L., Mitchell, T.W., Blanksby, S.J.: Structural characterization of glycerophospholipids by combinations of ozone- and collision-induced dissociation mass spectrometry: the next step towards “top-down” lipidomics. Analyst 139, 204–214 (2014)
Momchilova, S., Tsuji, K., Itabashi, Y., Nikolova-Damyanova, B., Kuksis, A.: Resolution of triacylglycerol positional isomers by reversed phase high-performance liquid chromatography. J. Sep. Sci. 27, 1033–1036 (2004)
Momchilova, S., Itabashi, Y., Nikolova-Damyanova, B., Kuksis, A.: Regioselective separation of isomeric triacylglycerols by reversed phase high-performance liquid chromatography: stationary phase and mobile phase effects. J. Sep. Sci. 29, 2578–2583 (2006)
Lisa, M., Holcapek, M.: Triacylglycerols profiling in plant oils important in food industry, dietetics and cosmetics using high-performance liquid chromatography-atmospheric pressure chemical ionization mass spectrometry. J. Chromatogr. A 1198/1199, 115–130 (2008)
Kuksis, A., Itabashi, Y.: Regio- and stereospecific analysis of glycerolipids. Methods 36, 172–185 (2006)
Lísa, M., Velínská, H., Holčapek, M.: Regioisomeric characterization of triacylglycerols using silver-ion HPLC/MS and randomization synthesis of standards. Anal. Chem. 81, 3903–3910 (2009)
Holcapek, M., Dvorakova, H., Lisa, M., Giron, A., Sandra, P., Cvacka, J.: Regioisomeric analysis of triacylglycerols using silver-ion liquid chromatography-atmospheric pressure chemical ionization mass spectrometry: comparison of five different mass analyzers. J. Chromatogr. A. 1217, 8186–8194 (2010)
van der Klift, E.J., Vivo-Truyols, G., Claassen, F.W., van Holthoon, F.L., van Beek, T.A.: Comprehensive two-dimensional liquid chromatography with ultraviolet, evaporative light scattering, and mass spectrometric detection of triacylglycerols in corn oil. J. Chromatogr. A 1178, 43–55 (2008)
Eiceman, G.A., Karpas, Z., Hill, H.H.: Ion mobility spectrometry. CRC Press, Boca Raton (2013)
Valentine, S.J., Kulchania, M., Srebalus Barnes, C.A., Clemmer, D.E.: Multidimensional separations of complex peptide mixtures: a combined high-performance liquid chromatography/ion mobility/time-of-flight mass spectrometry approach. Int. J. Mass Spectrom. 212, 97–109 (2001)
Fenn, L.S., Kliman, M., Mahsut, A., Zhao, S.R., McLean, J.A.: Characterizing ion mobility-mass spectrometry conformation space for the analysis of complex biological samples. Anal. Bioanal. Chem. 394, 235–244 (2009)
Kaszycki, J.L., Shvartsburg, A.A.: A priori intrinsic PTM size parameters for predicting the ion mobilities of modified peptides. J. Am. Soc. Mass Spectrom. 28, 294–302 (2017)
May, J.C., Goodwin, C.R., Lareau, N.M., Leaptrot, K.L., Morris, C.B., Kurulugama, R.T., Mordehai, A., Klein, C., Barry, W., Darland, E., Overney, G., Imatani, K., Stafford, G.C., Fjeldsted, J.C., McLean, J.A.: Conformational ordering of biomolecules in the gas phase: nitrogen collision cross-sections measured on a prototype high resolution drift tube ion mobility-mass spectrometer. Anal. Chem. 86, 2107–2116 (2014)
Jackson, S.N., Wang, H.Y., Woods, A.S., Ugarov, M., Egan, T., Schultz, J.A.: Direct tissue analysis of phospholipids in rat brain using MALDI-TOFMS and MALDI-ion mobility-TOFMS. J. Am. Soc. Mass Spectrom. 16, 133–138 (2005)
Castro-Perez, J., Roddy, T.P., Nibbering, N.M.M., Shah, V., McLaren, D.G., Previs, S., Attygalle, A.B., Herath, K., Chen, Z., Wang, S.P., Mitnaul, L., Hubbard, B.K., Vreeken, R.J., Johns, D.G., Hankemeier, T.: Localization of fatty acyl and double bond positions in phosphatidylcholines using a dual stage CID fragmentation coupled with ion mobility mass spectrometry. J. Am. Soc. Mass Spectrom. 22, 1552–1567 (2011)
Shah, V., Castro-Perez, J.M., McLaren, D.G., Herath, K.B., Previs, S.F., Roddy, T.P.: Enhanced data-independent analysis of lipids using ion mobility-TOFMSE to unravel quantitative and qualitative information in human plasma. Rapid Commun. Mass Spectrom. 27, 2195–2200 (2013)
Damen, C.W.N., Isaac, G., Langridge, J., Hankemeier, T., Vreeken, R.J.: Enhanced lipid isomer separation in human plasma using reversed-phase UPLC with ion-mobility/high-resolution MS detection. J. Lipid Res. 55, 1772–1783 (2014)
Paglia, G., Kliman, M., Claude, E., Geromanos, S., Astarita, G.: Applications of ion-mobility mass spectrometry for lipid analysis. Anal. Bioanal. Chem. 407, 4995–5007 (2015)
Paglia, G., Angel, P., Williams, J.P., Richardson, K., Olivos, H.J., Thompson, J.W., Menikarachchi, L., Lai, S., Walsh, C., Moseley, A., Plumb, R.S., Grant, G.F., Palsson, B.O., Langridge, J., Geromanos, S., Astarita, G.: Ion mobility-derived collision cross-section as an additional measure for lipid fingerprinting and identification. Anal. Chem. 87, 1137–1144 (2015)
Kyle, J.E., Zhang, X., Weitz, K.K., Monroe, M.E., Ibrahim, Y.M., Moore, R.J., Cha, J., Sun, X., Lovelace, E.S., Wagoner, J., Polyak, S., Metz, T.O., Dey, S.K., Smith, R.D., Burnum-Johnson, K.E., Baker, E.S.: Uncovering biologically significant lipid isomers with liquid chromatography, ion mobility spectrometry, and mass spectrometry. Analyst 141, 1649–1659 (2016)
Groessl, M., Graf, S., Knochenmuss, R.: High resolution ion mobility-mass spectrometry for separation and identification of isomeric lipids. Analyst 140, 6904–6911 (2015)
Guevremont, R.: High-field asymmetric waveform ion mobility spectrometry: a new tool for mass spectrometry. J. Chromatogr. A 1058, 3–19 (2004)
Shvartsburg, A.A.: Differential ion mobility spectrometry. CRC Press, Boca Raton (2008)
Guevremont, R., Barnett, D.A., Purves, R.W., Vandermey, J.: Analysis of a tryptic digest of pig hemoglobin using ESI-FAIMS-MS. Anal. Chem. 72, 4577–4584 (2000)
Shvartsburg, A.A., Isaac, G., Leveque, N., Smith, R.D., Metz, T.O.: Separation and classification of lipids using differential ion mobility spectrometry. J. Am. Soc. Mass Spectrom. 22, 1146–1155 (2011)
Shvartsburg, A.A., Zheng, Y., Smith, R.D., Kelleher, N.L.: Separation of variant methylated histone tails by differential ion mobility. Anal. Chem. 84, 4271–4276 (2012)
Shvartsburg, A.A.: Ultrahigh-resolution differential ion mobility separations of conformers for proteins above 10 kDa: onset of dipole alignment? Anal. Chem. 86, 10608–10615 (2014)
Kaszycki, J.L., Bowman, A.P., Shvartsburg, A.A.: Ion mobility separation of peptide isotopomers. J. Am. Soc. Mass Spectrom. 27, 795–799 (2016)
Shvartsburg, A.A., Li, F., Tang, K., Smith, R.D.: High-resolution field asymmetric waveform ion mobility spectrometry using new planar geometry analyzers. Anal. Chem. 78, 3706–3714 (2006)
Shvartsburg, A.A., Danielson, W.F., Smith, R.D.: High-resolution differential ion mobility separations using helium-rich gases. Anal. Chem. 82, 2456–2462 (2010)
Shvartsburg, A.A., Prior, D.C., Tang, K., Smith, R.D.: High-resolution differential ion mobility separations using planar analyzers at elevated dispersion field. Anal. Chem. 82, 7649–7655 (2010)
Shvartsburg, A.A., Smith, R.D.: Accelerated high-resolution differential ion mobility separations using hydrogen. Anal. Chem. 83, 9159–9166 (2011)
Shvartsburg, A.A., Tang, K., Smith, R.D.: Modeling the resolution and sensitivity of FAIMS analyses. Anal. Chem. 76, 7366–7374 (2004)
Shvartsburg, A.A., Ibrahim, Y., Smith, R.D.: Differential ion mobility separations in up to 100% helium using microchips. J. Am. Soc. Mass Spectrom. 25, 480–489 (2014)
Maccarone, A.T., Duldig, J., Mitchell, T.W., Blanksby, S.J., Duchoslav, E., Campbell, J.L.: Characterization of acyl chain position in unsaturated phosphatidylcholines using differential mobility-mass spectrometry. J. Lipid Res. 55, 1668–1677 (2014)
Sala, M., Lisa, M., Campbell, J.L., Holcapek, M.: Determination of triacylglycerol regioisomers using differential mobility spectrometry. Rapid Commun. Mass Spectrom. 30, 256–264 (2016)
Baker, P.R.S., Armando, A.M., Campbell, J.L., Quehenberger, O., Dennis, E.A.: Three-dimensional enhanced lipidomics analysis combining UPLC, differential ion mobility spectrometry, and mass spectrometric separation strategies. J. Lipid Res. 55, 2432–2442 (2014)
Lintonen, T.P.I., Baker, P.R.S., Suoniemi, M., Ubhi, B.K., Koistinen, K.M., Duchoslav, E., Campbell, J.L., Ekroos, K.: Differential mobility spectrometry-driven shotgun lipidomics. Anal. Chem. 86, 9662–9669 (2014)
Shvartsburg, A.A., Seim, T.A., Danielson, W.F., Norheim, R., Moore, R.J., Anderson, G.A., Smith, R.D.: High-definition differential ion mobility spectrometry with resolving power up to 500. J. Am. Soc. Mass Spectrom. 24, 109–114 (2013)
Liebisch, G., Vizcaíno, J.A., Köfeler, H., Trötzmüller, M., Griffiths, W.J., Schmitz, G., Spener, F., Wakelam, M.J.O.: Shorthand notation for lipid structures derived from mass spectrometry. J. Lipid Res. 54, 1523–1530 (2013)
Williams, D.K., Muddiman, D.C.: Parts-per-billion mass measurement accuracy achieved through the combination of multiple linear regression and automatic gain control in a Fourier transform ion cyclotron resonance mass spectrometer. Anal. Chem. 79, 58–63 (2007)
Shvartsburg, A.A., Smith, R.D.: Scaling of the resolving power and sensitivity for planar FAIMS and mobility-based discrimination in flow- and field- driven analyzers. J. Am. Soc. Mass Spectrom. 18, 1672–1681 (2007)
Fenn, L.S., McLean, J.A.: Biomolecular structural separations by ion mobility-mass spectrometry. Anal. Bioanal. Chem. 391, 905–909 (2008)
Lalli, P.M., Iglesias, B.A., Toma, H.E., de Sa, G.F., Daroda, R.J., Filho, J.C.S., Szulejko, J.E., Araki, K., Eberlin, M.N.: Protomers: formation, separation, and characterization via traveling wave ion mobility mass spectrometry. J. Mass Spectrom. 47, 712–719 (2012)
Shvartsburg, A.A., Smith, R.D.: Ultrahigh-resolution differential ion mobility spectrometry using extended separation times. Anal. Chem. 83, 23–29 (2011)
Shvartsburg, A.A., Li, F., Tang, K., Smith, R.D.: Distortion of ion structures by field asymmetric waveform ion mobility spectrometry. Anal. Chem. 79, 1523–1528 (2007)
Robinson, E.W., Shvartsburg, A.A., Tang, K., Smith, R.D.: Control of ion distortion in field asymmetric waveform ion mobility spectrometry via variation of dispersion field and gas temperature. Anal. Chem. 80, 7508–7515 (2008)
Cossignani, L., Luneia, R., Damiani, P., Simonetti, M.S., Riccieri, R., Tiscornia, E.: Analysis of isomeric diacylglycerolic classes to evaluate the quality of olive oil in relation to storage conditions. Eur. Food Res. Technol. 224, 379–383 (2007)
Hsu, F.F., Turk, J.: Electrospray ionization/tandem quadrupole mass spectrometric studies on phosphatidylcholines: the fragmentation processes. J. Am. Soc. Mass Spectrom. 14, 352–363 (2003)
Acknowledgments
The authors thank Gordon A. Anderson (GAACE) and Dr. Keqi Tang (PNNL) for broad aid with instrument development, Matt Baird and Julia Kaszycki for experimental help, and Professor Stephen Blanksby for numerous insightful discussions. This research was funded by NIH K-INBRE (P20 GM103418), NSF First (EPS-0903806), and NSF CAREER (CHE-1552640). A.S. has interest in Heartland MS that produces FAIMS systems and ion funnel interfaces such as those utilized in this work. A.S. also holds a faculty appointment at the National Research Nuclear University MEPhI (Moscow Engineering Physics Institute), Russia.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bowman, A.P., Abzalimov, R.R. & Shvartsburg, A.A. Broad Separation of Isomeric Lipids by High-Resolution Differential Ion Mobility Spectrometry with Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 28, 1552–1561 (2017). https://doi.org/10.1007/s13361-017-1675-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-017-1675-2