
Abstract
DNA methylation plays a crucial role in the regulation of gene expression, activity of transposable elements, defense against foreign DNA, and inheritance of specific gene expression patterns. The link between stress exposure and sequence-specific changes in DNA methylation was hypothetical until it was shown that stresses can induce changes in the gene expression through hypomethylation or hypermethylation of DNA. To detect changes in DNA methylation under herbicide stress in two local Zea mays inbred lines exhibiting differential susceptibility to Roundup®, the methylation-sensitive amplified polymorphism (MSAP) technique was used. The overall DNA methylation levels were determined at approximately 60% for both tested lines. The most significant changes were observed for the more sensitive Z. mays line, where 6 h after the herbicide application, a large increase in the level of DNA methylation (attributed to the increase in fully methylated bands (18.65%)) was noted. DNA sequencing revealed that changes in DNA methylation profiles occurred in genes encoding heat shock proteins, membrane proteins, transporters, kinases, lipases, methyltransferases, zinc-finger proteins, cytochromes, and transposons. Herbicide stress-induced changes depended on the Z. mays variety, and the large increase in DNA methylation level in the sensitive line resulted in a lower ability to cope with stress conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Herbicides are chemicals used to decrease weed population in crop fields, to protect crops and increase their yield, and to enhance harvest and processing efficiency of the agro-food industry. The most popular weedkillers are non-selective, i.e., they affect not only weed populations but also all plants growing in the sprayed area. Importantly, in the past few years, the amount and diversity of pesticides used in agriculture and horticulture have greatly increased, for example, in 2015, 4.1 Mt of herbicides were applied globally, which constitutes an increase of 35% compared to that used in 2001. By the year 2050, because of the rapid increase in human population and the expected land conversion into arable production, global pesticide applications are likely to increase further (Maggi et al. 2019).
Glyphosate (N-(phosphonomethyl)glycine) is an active compound of Roundup® (a highly effective, broad-spectrum, non-selective herbicide) and many other commercially available herbicides, that targets 5-enolpyruvylshikimate-3-phosphate synthase (EPSPS), an enzyme in the shikimate pathway that mediates the biosynthesis of aromatic amino acids in plants, bacteria, and fungi (Kanissery et al. 2019; Mertens et al. 2018). EPSPS of all higher plants appears to be inhibited by glyphosate, thus making it a non-selective herbicide that is active on a very wide range of plant species. Nevertheless, it remains unclear how glyphosate-induced inhibition of the shikimate pathway actually kills plants (Gomes et al. 2014). Some differences in the effects of glyphosate between plant species and biotypes are consistent with the observed differential susceptibility (Fuchs et al. 2002). It is assumed that insufficient aromatic amino acid production to maintain protein synthesis is the primary effect, and this is consistent with the slow development of symptoms (Duke and Powles 2008). Another possible cause of death, observed in velvetleaf tissues, is the restriction of water availability to the shoots, which is induced by lethal disruption of root processes. Moreover, because of interference with chlorophyll synthesis and its photodestruction caused by glyphosate-induced loss of carotenoids, a gradual inhibition of photosynthesis was also observed (Fuchs et al. 2002; Gomes et al. 2014). There is also evidence that the increased carbon flow to the shikimate pathway (by deregulation of the pathway due to EPSPS inhibition) results in shortages of carbon for other essential pathways (Duke and Powles 2008).
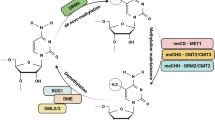
DNA methylation, one of the most important forms of epigenetic modification, has been shown to be involved in gene silencing at both transcriptional and post-transcriptional levels (Gallego-Bartolomé 2020; Agarwal et al. 2020). It plays significant roles in the regulation of gene expression (Meng et al. 2016), plant growth and development (Yang et al. 2014; Han et al. 2019; Agarwal et al. 2020), abiotic and biotic stress responses (Alonso et al. 2019; Kumar et al. 2020; Eriksson et al. 2020; Agarwal et al. 2020), environmental adaptation (Varotto et al. 2020; Gáspár et al. 2019), activity of transposable elements (Dowen et al. 2012; Garg et al. 2015; Lyu et al. 2017; Wang et al. 2018), defense against foreign DNA, and inheritance of specific gene expression patterns (Paszkowski and Whitham 2001; Matzke et al. 2009). Transcriptional gene silencing is associated with hypermethylation of promoter sequences, while post-transcriptional gene silencing is linked with hypermethylation of transcribed or coding sequences (Chinnusamy and Zhu 2009). In the plant genome, methylated cytosine residues (m5C) are found in three nucleotide-sequence contexts: symmetrical CG and two non-CG sites, namely symmetrical CNG and asymmetrical CNN sites (where N is A, T, or C). Moreover, different sequences have different genetic requirements for de novo or maintenance methylation.
The regulation of genomic methylation in plants is complex. Multiple DNA methyltransferases cooperate to establish and maintain methylation in a manner that reflects local sequence features (Matzke et al. 2009; Zhang et al. 2018; Bräutigam and Cronk 2018; Agarwal et al. 2020). Three distinct classes of enzymes are responsible for cytosine methylation. De novo methyltransferases DRM1 and DRM2 (domains rearranged methylase 1 and 2) catalyze new cytosine methylation, while the maintenance of symmetric CG and CHG methylation is mediated by the DNMT1-like enzyme MET1 and the plant-specific enzyme chromomethylase 3 (CMT3), respectively (Matzke et al. 2009; Chinnusamy and Zhu 2009; Zhang et al. 2018; Bräutigam and Cronk 2018; Agarwal et al. 2020). Inhibition of de novo methylation or inability to maintain the parental imprint after DNA replication are two passive ways of a DNA methylation loss, which were shown in met1 mutants (Boyko and Kovalchuk 2008). Apart from that, DNA methylation can be removed enzymatically by the repressor of silencing 1 (ROS1), DEMETER (DME), and DME-like (DML)—proteins that possess DNA glycosylase-lyase activity (Chinnusamy and Zhu 2009; Zhang et al. 2018; Bräutigam and Cronk 2018; Agarwal et al. 2020).
The link between stress exposure and sequence-specific changes in DNA methylation was hypothetical until it was shown that stresses can induce changes in gene expression through hypomethylation or hypermethylation of DNA (Chinnusamy and Zhu 2009). In Zea mays, an immense loss of DNA methylation was reported under nitrogen deficiency; however, phosphate starvation led to much less changes (Mager and Ludewig 2018). It was also reported that in Z. mays under salinity stress, thousands of genes involved in cellular processes, metabolic processes, and signal transduction were associated with differential DNA methylation (Sun et al. 2018).
Cereals occupy the main position in the composition of a human diet, with rice, wheat, and Z. mays being the major staple cereals, having a share in excess of 70% among all food grains (Tyczewska et al. 2018; OECD-FAO Agricultural Outlook 2018–2027). Because of its high productivity, the importance for food and feed production and numerous industrial applications Z. mays, a plant originating from southern Mexico (Yang et al. 2019), is currently cultivated worldwide on a large scale (Langner et al. 2019; Handral et al. 2017). When grown under non-optimal conditions, Z. mays is exposed to environmental stresses, and hence, to minimize the influence of stress on the yield, it is essential to minimize the competition for water, light, and minerals that occurs between Z. mays seedlings and weeds growing in the field (Craine and Dybzinski 2013). The application of herbicides is the easiest and most widely used approach to eradicate weeds (Heap and Duke 2018).
Our aim was to identify the changes in DNA methylation levels and patterns underlying the resistance of Z. mays to the herbicide stress conditions. The two tested lines are inbred lines developed in Poland that naturally differ in susceptibility to Roundup®. In order to observe and analyze the changes in DNA methylation patterns, the methylation-sensitive amplification polymorphism (MSAP) technique was used.
Materials and methods
Plant material
Z. mays lines were chosen based on the results of field tests conducted to verify the response of 25 Z. mays inbred lines to Roundup® stress (K. Adamczewski, data not published). Based on these tests, we chose the S79757 line (sensitive to Roundup®, SL) as it showed the most prominent response to herbicide stress and the S245 line (tolerant to Roundup®, TL) as one of the most resistant (Supplementary Figure 1 in Mahmoud et al. (2020)). When the highest concentration of herbicide (Roundup® 360 SL) was used (300 g, 1.0 l/ha), the level of injuries to the TL variety was 40%, while it reached 85% in the SL 3 weeks after the application of the herbicide (rating is done based on phenotypic analyses of leaf chlorosis and wilting, inhibition of plant growth) (K. Adamczewski, data not published). Seeds from both lines were obtained from a local breeder (HR Smolice, Poland). The same two lines were used in the analyses described in Mahmoud et al. (2020) and Żywicki et al. (2015).
The seedlings were grown in a greenhouse in controlled temperature (22oC), humidity, and light conditions (16 h/8 h—light/dark) (Mahmoud et al. 2020; Żywicki et al. 2015). Uniform seedlings from both lines were selected and divided into two groups: one was sprayed with the herbicide Roundup® (1.0 l/ha, 300 g glyphosate) with adjuvant AS 500 SL (4.0 l/ha) 2 weeks after plants’ emergence (at the stage of 4–5 leaves), and the other (control) group was cultured without the herbicide treatment. To elucidate both early and late responses to stress conditions, for each tested sample, leaves were harvested from 6 plants at specified time points (6 h and 7 days after Roundup® application); leaves were harvested at the same time points for both tested plant groups. After collecting the plant material, the samples were immediately frozen in liquid nitrogen and stored at − 80 °C. Genomic DNA was isolated using the DNeasy Plant Maxi Kit (Qiagen) according to the manufacturer’s protocol. Before the DNA isolation, the plant material, pooled from 6 plants for each sample, was grind to a fine powder using TissueLyser II (Qiagen).
DNA methylation analyses
Methylation-sensitive amplification polymorphism
The protocol was adapted from Yu et al. (2008) with some modifications. It consists of three major steps: digestion and ligation reactions, preamplification and amplification reactions, and detection. The designed adapters and primers for EcoRI and HpaII-MspI are described in Yu et al. (2008) and Xiong et al. (1999) and listed in Table 1.
Digestion and ligation reactions
DNA samples (300 ng) were separately digested using EcoRI-HpaII and EcoRI-MspI restriction enzyme pairs (60 U each, New England Biolabs) at 37 °C in an appropriate reaction buffer. DNA samples were purified by phenol/chloroform and precipitated with 3 M ammonium acetate pH 5.3, glycogen, and ethanol overnight at − 20 °C.
The total amount of digested DNA was used for the ligation reactions with 5 pmoles of 1E and 2E adapters and 50 pmoles of 1HM and 2HM adapters. The ligation was performed in 25 μl of total volume with 2 μl of T4 DNA ligase (10U, Fermentas) for 1 h at 22 °C.
Preamplification and selective amplification
Preamplification PCR reactions were performed in a final volume of 50 μl containing 5 μl of ligation products, 75 ng of E00 and HM00 PreAmp primers (Table 1), and Dream Taq (5 U/μl, Fermentas). DNA fragments were amplified for 25 cycles of 94 °C for 90 s, 56 °C for 30 s, and 72 °C for 1 min, prior to selective amplification. The PCR products were diluted 1:25 (v:v) with ddH2O. Five microliters of the diluted preamplified PCR products was used for selective PCR reactions with Dream Taq (5 U/μl) and 40 ng of a selective primer pair E00+3/HM00+3. Selective primers have additional 3 nucleotides at their 3′ ends (Table 1). The reactions were performed in a total volume of 50 μl. The PCR conditions were as follows: 13 cycles at 94 °C for 30 s, 65oC for 30 s (reduced by 0.7 °C at each cycle), and 72 °C for 1 min followed by 23 cycles: 94 °C for 30 s, 56 °C for 30 s, and 72 °C for 1 min.
Detection assay
The total amount of DNA selective amplification products was mixed with a loading buffer (6×), heated for 5 min at 95 °C, and rapidly chilled on ice. The entire mixtures were loaded on 6% denaturing polyacrylamide gels (50 cm × 33 cm, one for each primer combination). Electrophoresis was performed at a constant power of 700 V for 18 h with cooling with GeneRuler 1kb DNA ladder (Fermentas). The detection of end products was performed using Sybr Gold (Thermo Fisher Scientific).
Isolation, cloning, and sequencing of MSAP fragments
Differentiating DNA fragments were removed from the polyacrylamide gels under UV light by using a razor blade. The fragments were eluted from the gel in 200 μl of 0.3 M ammonium acetate and incubated overnight at 4 °C under shaking. DNA samples were purified by phenol/chloroform and precipitated with 3 M ammonium acetate pH 5.3, glycogen, and ethanol overnight at − 20 °C; the samples were then centrifuged for 30 min at 4 °C, dried, and resuspended in ddH2O. Aliquots (5 μl) were used for reamplification. PCR reactions were performed using the same primer combinations and reaction conditions as those used in selective amplification. The PCR products were then ligated into a vector pCR™2.1-TOPO using a ligation TOPO TA Cloning Kit (Life Technologies). Ligation was performed for 30 min at room temperature in a final volume of 6 μl using 10 ng of a vector and 30 ng of DNA. The ligation products were transformed into an Escherichia coli DH5α strain. The detection of end products was based on the blue/white screening test. Colony PCR was performed for samples taken from white colonies using Clone ID Colony PCR Master Mix (Lucigen) and 12 pmoles each of universal M13 Forward and M13 Reverse primers (Genomed). The PCR conditions were as follows: initial denaturation at 98 °C for 2 min, 29 cycles of 98 °C for 30 s, 54 °C for 30 s, and 72 °C for 3 min, followed by complementary elongation at 72 °C for 10 min. The quality of PCR products was checked by agarose electrophoresis in 1% gel along with GeneRuler 100 bp Plus DNA Ladder (Fermentas). Only full-length DNA samples were sequenced using the Sanger sequencing method at Genomed.
Bioinformatics analysis
The obtained DNA sequences were analyzed using the Basic Local Alignment Search Tool (BLASTn, NCBI, https://blast.ncbi.nlm.nih.gov/Blast.cgi) and Z. mays GDB (B73 filtered gene sets 4a.53 for RefGen_v1, https://www.maizegdb.org/). GO functional classification was performed using Blast2GO (https://www.blast2go.com/) at the default settings. The sequences were blasted using the NCBI Blast service (QBlast) and blastx-fast program. Blast expectation value (E value) was set at 1.0E−3. Next, the GO ontology mapping and InterProScan were performed. GO mapping was performed against extensively curated Gene Ontology annotated proteins, to obtain functional labels. The used data originates from the Gene Ontology Association and Uniprot ID-Mapping. The public EMBL-EBI InterPro web-service was used to scan sequences against InterPro’s signatures at the default settings. GO annotation was performed with Annotation CutOff set at 55, and E value-hit-filter set at 1.0E−6. Enrichment analysis was performed using Fisher’s exact test.
Methylation analysis of CentC in Z. mays
Total Z. mays DNA was isolated using a DNeasy Plant Maxi Kit (Qiagen) according to the manufacturer’s instructions. Target CentC methylation was analyzed with methylation-sensitive restriction enzymes (HpaII and MspI, 60U, New England Biolabs) and Southern blot hybridization, according to previously published protocols (Mette et al. 2000; Aufsatz et al. 2002). A 32P-labeled RNA probe (5′-ATGAGTTTTGGACCTAAAGTAGTGGATTGG-3′) was used for probing the Southern blots.
Results
Extent and pattern of global DNA methylation under control condition and herbicidal stress condition in Z. mays
To evaluate DNA cytosine methylation at 5′-CCGG-3′ sequences in two Z. mays lines that are either tolerant (S245, TL) or sensitive (S79757, SL) to herbicidal stress, 24 primer pairs were used. The enzymes used in the digestion of DNA are sensitive to methylation at either CG and CNG (HpaII: mCmCGG) or only CNG (MspI: mCCGG) sites. To elucidate early and late responses to herbicide stress, the samples were collected 6 h and 7 days after Roundup® treatment. A total of 888 and 826 clear bands were amplified from TL and SL, respectively. For HpaI and MspII restriction enzymes, four distinct restriction patterns can be distinguished (Table 2). A sample photo showing the results of Z. mays DNA amplification for the E41-HM5 primer pair is given in Fig. 1. As can be seen, a highly repetitive banding pattern can be observed for the amplification products for TL (lines 1–4, 9–12) and SL samples (lines 5–8,13–16). The differences in the amplification profiles between the two tested Z. mays lines (TL and SL) are also clearly visible. Additionally, differential bands attributed to the changes in DNA methylation between samples derived from TL or SL under herbicide stress are markedly noticeable (change in intensity, appearance, or disappearance of bands, marked by boxes). The biggest differences in the banding pattern were observed for SL sample 6 h after the application of a herbicide, compared to the control (line 6).

Electrophoregram of DNA samples digested with HpaI and MspII, amplified using E41-HM5 primers. 1–4 and 9–12—TL line; 5–8 and13–16—SL line; 1–8—6 h after herbicide application; 9–16—7 days after the application of herbicide; 1, 3, 5, 7, 9, 11, 13, and 15 (odd numbers)—control samples; 2, 4, 6, 8, 10, 12, 14, and 16 (even numbers)—herbicide-treated samples. M, GeneRuler 1kb DNA ladder 250–10,000 bp (Thermo Fisher Scientific). Some of the differences in the MSAP profiles between samples are marked with white boxes
Under the control conditions, the total methylation level of 5′-CCGG-3′ sequences averaged between 63.11% in TL and 59.38% in SL. The extent of DNA methylation ranged from 62.38% (6 h after treatment) to 63.40% (7 days after treatment) in TL and from 57.87% (6 h after treatment) to 60.90% (7 days after treatment) in SL. Herbicidal stress increased the percentage of total methylated bands in both tested lines 6 h after Roundup® treatment. The change was very slight for the TL (1.13%) compared to SL (18.64%) (Fig. 2). Seven days after herbicide treatment, a decrease in the level of methylation was observed for the TL line (3.94%), and almost no difference was noted for the SL line (0.12%). Importantly, the fully methylated loci were always more abundant than the hemi-methylated ones.

Global DNA methylation changes in TL and SL Z. mays lines resulting from herbicidal stress conditions. T, total methylation level; F, fully methylates sites; H, hemimethylated sites. TL6h-C, TL line 6 h after spraying, control; TL6h-H, TL line 6 h after spraying, herbicide; TL7d-C, TL line 7 days after spraying, control; TL7d-H, TL line 7 days after spraying, herbicide; SL6h-C, SL line 6 h after spraying, control; SL6h-H, SL line 6 h after spraying, herbicide; SL7d-C, SL line 7 days after spraying, control; SL7d-H, TL line 7 days after spraying, herbicide. The biggest changes in DNA methylation in two tested Z. mays lines under herbicide stress conditions were observed in SL line 6 h after herbicide application. These changes are attributed to a large increase (by 18.65%) in the number of fully methylated sites
For TL, a decrease in fully methylated bands (2.59%) and an increase in hemi-methylated bands (3.72%) 6 h after the treatment were observed, while 7 days after treatment, there was a slight increase in fully methylated bands (1.35%) and a decrease in hemi-methylated bands (5.29%). The drastic increase in methylation level in SL line 6 h after herbicide treatment was attributed to the increase in fully methylated bands (18.65%), while the level of hemi-methylated bands stayed almost constant (0.01%). Seven days after treatment, as the total methylation level stayed unchanged in treated vs control plants, there was an increment in fully methylated bands (4.72%) and a decrease in hemi-methylated bands (4.60%) (Table 3, Fig. 2).
Herbicide-induced changes in the level of DNA methylation in Z. mays genotypes that differ in their tolerance to herbicidal stress
To analyze the changes in cytosine methylation patterns under herbicidal stress, all possible banding patterns between control and Roundup® stress in TL and SL lines were calculated and compared (Table 3, Fig. 3). The MSAP analysis revealed 16 banding patterns, and they represent no change in methylation level (A–D), demethylation (E–J), and methylation (K–P) events (Karan R’ DeLeon et al. 2012).

The binding patterns obtained from the MSAP analysis: no change in DNA methylation level (A–D), demethylation (E–J), and methylation (K–P) events in TL and SL Z. mays lines under herbicidal stress conditions, given in percentages. TL6h-C, TL line 6 h after spraying, control; TL6h-H, TL line 6 h after spraying, herbicide; TL7d-C, TL line 7 days after spraying, control; TL7d-H, TL line 7 days after spraying, herbicide; SL6h-C, SL line 6 h after spraying, control; SL6h-H, SL line 6 h after spraying, herbicide; SL7d-C, SL line 7 days after spraying, control; SL7d-H, TL line 7 days after spraying, herbicide
For TL, the methylation of 72.63% and 64.41% of 5′-CCGG-3′ sites remained unchanged under herbicide treatment, 6 h and 7 days after spraying, respectively. We observed a difference in demethylation events at two different time points in TL—13.40% vs 19.25%—and a difference in methylation events—13.96% and 16.32% at 6 h and 7 days after treatment, respectively. For SL, out of 826 bands, 46% and 72.76% of 5′-CCGG-3′ sites remained unchanged under herbicide treatment, 6 h and 7 days after spraying, respectively. There was a decrease in demethylation events at 7 days post-treatment as compared to that at 6 h after treatment (17.32% to 10.52%). The largest difference was observed for the methylation events (19.98%), and it decreased significantly from 36.68% to 16.70% 1 week after spraying. The test of independence between different methylation patterns (total methylation, full methylation, and hemi-methylation) for control and herbicide stress conditions was carried out using the chi-square test (Supplementary Table S1, Supplementary Table S2).
Sequencing and GO analysis of chosen differentially methylated DNA fragments
Selected DNA fragments were isolated from polyacrylamide gels and sequenced at Genomed (Poland). A total of 197 DNA sequences were obtained and subjected to bioinformatics analysis; primer and vector sequences were removed prior to analysis. NCBI and MaizeGDB databases were used for sequence analysis. Of the 197 fragments of 100 to 1200 bp, as many as 151 fragments had sequences that matched with more than 90% accuracy in at least one of the databases searched (Supplementary Table S3). The matched sequences belonged not only to Z. mays but also to the related bicolor sorghum (Sorghum bicolor) and rice (Oryza sativa).
GO functional classification was performed using Blast2GO (https://www.blast2go.com/) at the default settings. Gene Ontology terms were assigned to 87 target genes, which were described by 67 terms in “molecular function,” 58 terms in “biological process,” and 19 terms in “cellular compartment” categories (Supplementary Table S4). Highly represented terms in the “molecular function” category (Fig. 4a) included “magnesium ion binding” (GO:0000287), “transcription regulatory region sequence-specific DNA binding” (GO:0000976), and “RNA polymerase II activity”(GO:0001055); in the “biological process” category (Fig. 4b)—“defense response to oomycetes” (GO:0006468), “mismatch repair” (GO:0006298), and “regulation of transcription, DNA-templated” (GO:0006355); and in the “cellular compartment” category (Fig. 4c)—“extracellular region” (GO:0005576), “nucleus” (GO:0005634), and “nuclear envelope” (GO:0005635). Importantly, GO terms associated with the membranes in the “cellular compartment” category were “plasma membrane” (GO:0005886), “membrane” (GO:0016020), “integral component of membrane” (GO:0016021), “integral component of endoplasmic reticulum membrane” (GO:0030176), and “anchored component of membrane” (GO:0031225). Enrichment analysis (Fisher’s exact test) indicated “hydrolase activity” (GO:0016787) is significant at p value 0.05. Moreover, according to the KEGG analysis (Kanehisa and Goto 2000), 87 target genes were significantly enriched in 20 pathways including “drug metabolism - other enzymes,” “drug metabolism - cytochrome P450,” and “metabolism of xenobiotics by cytochrome P450” (Supplementary Table S4).

A graph showing the percentage of GO enrichment analysis in a “molecular function” category, b “biological process” category, and c “cellular component” category. The values are given in percentages (%)
Among the identified sequences were genes coding for transferases, transporter proteins, methyltransferases, hydrolases, transposons, ribosomal proteins, cytochromes, and proteins participating in transcription processes and involved in stress responses (Supplementary Table S3).
Methylation analysis of repetitive sequences
Southern blot analysis of the centromere-specific satellite repeat CentC (Fig. 5) was performed to analyze whether herbicidal stress influences DNA methylation in such highly methylated genomic regions. In the bioinformatic analysis of sequences with differential DNA methylation, centromeric fragments in Z. mays genomes were identified (Supplementary Table S3). However, it was observed that under herbicide stress conditions, the methylation pattern of the CentC did not change (Fig. 5). The digestion patterns reflected substantial methylation of CGs and non-CGs, as indicated by the increased cleavage with the methylation-insensitive isoschizomer (M) compared to that with the methylation-sensitive isoschizomer (H).

Southern blot analysis of the changes in cytosine DNA methylation in CentC repetitive sequences in Z. mays under herbicide stress conditions. 1, 3, 5, 7, 9, 11, 13, and 15 (odd numbers)—HpaII restriction enzyme digestion; 2, 4, 6, 8, 10, 12, 14, and16 (even numbers)—MspI restriction enzyme digestion; 1 and 2—TL6h-C; 3 and 4—TL6h-H; 5 and 6—TL7d-C; 7 and 8—TL7d-H; 9 and 10—SL6h-C; 11 and 12—SL6h-H; 13 and 14—SL7d-C; 15 and 16—SL7d-H
Discussion
Environmental conditions may sometimes exert a detrimental effect on the development and maturation of organisms. This is particularly true for plants. Because of their sedentary lifestyle, plants require short-term strategies to rapidly and efficiently re-adapt their metabolism and thus have developed unique features in terms of habitat, growth, and reproduction (Boyko and Kovalchuk 2008). A comprehensive understanding of the mechanism of action of glyphosate-based herbicides is important as it affects the growth of plants not only by inhibiting EPSPS but also by altering several crucial plant physiological processes (e.g., photosynthesis, carbon metabolism, mineral nutrition, and oxidative events) (Geiger et al. 1986; Hirayama and Shinozaki 2010; Orcaray et al. 2012; Romero et al. 2011; Ghanizadeh and Harrington 2017; Gomes et al. 2014).
Several authors reported previously changes in DNA methylation in Z. mays and other plant species under abiotic stress conditions (Mager and Ludewig 2018; Sun et al. 2018; Kong et al. 2020; Wang et al. 2014; Wang et al. 2015; Uthup et al. 2011; Tan 2010). Massive loss of DNA methylation in CG and CNG contexts has been observed in Z. mays under nitrogen deficiency. In contrast, phosphorus deficiency caused only a slight change in DNA methylation level, mainly in the CG context (Mager and Ludewig 2018). Salt stress also influenced the DNA methylation level in Z. mays, and it has been concluded that highly methylated CpG islands might participate in the regulation of gene transcription under salt stress. Several other reports showed the influence of stress conditions on DNA methylation in wheat, barley, and rice (Kong et al. 2020; Wang et al. 2014; Wang et al. 2015; Uthup et al. 2011; Tan 2010).
Previously, it was shown that herbicidal stress influences miRNA expression (Żywicki et al. 2015) and that some of the traits related to herbicide resistance may be encoded in the genome (Mahmoud et al. 2020). Here, we focused our efforts on analyzing the influence of herbicidal stress on changes in DNA methylation in the same two Z. mays lines, which are differentially susceptible to the systemic herbicide Roundup®. The results of the analysis of global methylation changes showed a high DNA methylation level in control samples (approximately 63% and 58% in TL in SL, respectively) (Fig. 2). This observation is consistent with a previous report showing that Z. mays is characterized by an overall high level of DNA methylation (Li et al. 2014). This high rate of DNA methylation is attributed to the high content of transposons and repetitive sequences in Z. mays genomes as it has been estimated that the fraction of the genome that appears to be repetitive is 77%, which in general is randomly distributed in the genome (Meyers et al. 2001).
The application of Roundup® to Z. mays plants resulted in only slight changes in total DNA methylation levels in the TL line (Fig. 2)—an increase by 1.13% at 6 h and a decrease of 3.4% at 7 days after the treatment. In contrast, a large increase in the level of DNA methylation (attributed to a large increase in fully methylated context) of 18.64% was observed in the SL line at 6 h after herbicide treatment (Fig. 3, Table 2). Seven days after herbicide spraying, the level of DNA methylation in SL returned to the control level (61%) (Fig. 3, Table 3). The differences in the response to herbicide stress conditions between the tested inbred lines are clearly visible. In Arabidopsis plants subjected to glyphosate stress, 9205 differentially methylated regions have been identified across the genome (Kim et al. 2017). It was shown that the methylation patterns were dose-sensitive and, to a degree, stress-specific. Additionally, two out of seven genes in the shikimate pathway were differentially methylated as a result of the herbicide stress. Herein, we did not detect changes in the methylation pattern in genes encoding shikimate pathway enzymes; however, previous changes in the coding sequences of two shikimate pathways enzymes (bifunctional 3-dehydroquinate dehydratase/shikimate dehydrogenase and chorismate synthase) in the two tested Z. mays lines (TL and SL) were detected (Mahmoud et al. 2020).
Other genes linked with herbicide stress responses are encoding transporter proteins. According to GO analysis, the term highly represented in the “cellular compartment” category was “extracellular region” constituting 37.5%, and the categories associated with membranes constituted 12.5%. A group of transporter proteins (Supplementary Table S3) differentially methylated following herbicide application was identified among the sequences analyzed. Importantly, in rice subjected to atrazine (ATR) stress, and broad bean to glyphosate stress, among differentially methylated genes were those enriched in functions associated with transport activity (Lu et al. 2016; Denis and Delrot 1993). Similarly, enrichment in genes associated with phosphate has been reported in Arabidopsis subjected to glyphosate stress (Kim et al. 2017). Notably, out of 13 miRNAs differentially expressed in the two tested Z. mays lines following glyphosate application, miR444 and miR827 have been shown to regulate the phosphate transport pathways, which seem to be common for Pi and glyphosate uptake (Żywicki et al. 2015). Moreover, recently, single nucleotide polymophisms (SNPs) and indels were detected in genes encoding phosphate transporters in TL and SL Z. mays lines (Mahmoud et al. 2020). This is particularly important, since glyphosate has been shown to be recognized by phosphate transporters (Hetherington et al. 1998). Notably, phosphate transporters 1 and 2 were implicated in the active transport of glyphosate into plant cells (Mahmoud et al. 2020; Denis and Delrot 1993; Hetherington et al. 1998; Morin et al. 1997; Shaner 2009; Gomes et al. 2016).
Out of two types of herbicide resistance, non-target site resistance (NTSR) may occur via enhanced xenobiotics detoxification, herbicide metabolism, or translocation (Markus et al. 2018; González-Torralva et al. 2012). Xenobiotics detoxification is mediated by cytochrome P450 monooxygenases (P450s), glutathione transferases (GSTs), or ATP-dependent (ATP-binding cassette (ABC)) transporters in plants (Jensen and Møller 2010). Cytochrome P450 monooxygenases constitute a group of proteins reported as affected by herbicide stress (Lu et al. 2016; Markus et al. 2018; Jensen and Møller 2010; Samsel and Seneff 2013). Plant P450s are involved in acclimatization to biotic and abiotic stresses and were shown to mediate herbicide metabolism (Siminszky 2006). Moreover, P450s were shown to be involved in the degradation of ATR in rice (Rong Tan et al. 2015). These results are corroborated by KEGG analysis as the enriched pathways included “drug metabolism - other enzymes,” “drug metabolism - cytochrome P450,” and “metabolism of xenobiotics by cytochrome P450.”
The involvement of ABC transporters in glyphosate resistance was reported in Conyza canadensis (Tani et al. 2015; Tani et al. 2016; Moretti et al. 2017), Conyza bonariensis, and C. canadensis (Moretti et al. 2017). Herein, one of the DNA fragments with changed DNA methylation level was identified as the gene encoding the ATP binding protein belonging to the family of ABC transporters and another one as encoding multidrug and toxic compound extrusion (MATE) protein (Supplementary Table S3). ABC transporters are present in all living organisms (Jasinski et al. 2003; Lefèvre and Boutry 2018), and they constitute a very large and diverse family of proteins. The function of ABC transporters is the transport of various compounds across the cell membranes, including lipids, sugars, amino acids, proteins, secondary metabolites, heavy metal ions, and xenobiotics (including herbicides) (Tani et al. 2015). Previously, structural changes in genes encoding the MATE family of proteins have been demonstrated in Z. mays inbred lines differentially resistant to the glyphosate-based herbicide Roundup® (TL and SL) (Mahmoud et al. 2020). Both protein families, ABC and MATE transporters, play significant roles in the transport of xenobiotics and other small molecules and may contribute to glyphosate transport and distribution in plants (Markus et al. 2018). The differences in the DNA methylation level of the protein-coding fragment belonging to the ABC and MATE transporter family in the tested Z. mays lines may suggest their role in the increased adaptation of certain Z. mays lines to herbicidal stress conditions.
Environmental factors may influence DNA methylation either by directly inhibiting enzymes that catalyze DNA methylation or by changing the availability of substrates required for those enzymatic reactions such as the availability and utilization of methyl donor groups (Markus et al. 2018; Munksgaard et al. 1995; Meng et al. 2018). Recently, it was shown that in rice, in response to ATR herbicide, DNA methyltransferases, histone methyltransferases, and DNA demethylases were differentially expressed (Kim et al. 2017). Herein, several methyltransferases (among others SAM-dependent carboxyl methyltransferase, benzoate carboxyl methyltransferase, gamma-tocopherol methyltransferase) had altered the DNA methylation levels in response to glyphosate-based herbicide (Supplementary Table S3) suggesting their possible role in glyphosate-based herbicide stress resistance in Z. mays.
Global analysis of plants such as Arabidopsis and rice suggests that the majority of transposons are inactive, methylated, and targeted by siRNAs (Lisch 2008). Given the large number of potentially active elements in most eukaryotic genomes, global activation could result in an overwhelmingly high level of mutation. Nevertheless, McClintock suggested that all types of stresses could potentially reshape a plant genome via transposon activation (McClintock 1984), a hypothesis that has been confirmed by other authors (Kim et al. 2017; Negi et al. 2016; Galindo-González et al. 2018; Boyko and Kovalchuk 2008; Hashida et al. 2006). Previously, it was shown that transposable elements may contribute to the activation of Z. mays genes in response to abiotic stress (cold, heat, salinity, and UV) as some TE families were associated with stress-responsive expression of nearby genes, and some TE families may act as local enhancers of stress-responsive expression (Makarevitch et al. 2015). Four to nine different TE families were associated with the upregulation of gene expression in each of these stress conditions, thus affecting up to 20% of the genes upregulated in response to abiotic stress, and as many as 33% of the genes that are expressed only in response to stress (Makarevitch et al. 2015). Importantly, changes in the DNA methylation status of TEs have been linked with herbicide stress responses (Markus et al. 2018). Herein, changes in DNA methylation were detected, among others, in miniature inverted-repeat transposable element Hbr22, Mu transposon, retrotransposons, and a helitron (Supplementary Table S3).
Conclusions
The selective pressure exerted by persistent application of herbicides may extort adaptive responses not only in weeds (a phenomenon observed for many years), but also in crop plants. The present analysis showed that herbicide stress, depending on the natural susceptibility of Z. mays varieties to herbicide, caused various changes in the DNA methylation levels and patterns in Z. mays varieties depending on their natural susceptibility to herbicides. This, in turn, may be reflected in the changes in the expression of particular genomic fragments that may result in a heightened or diminished response of organisms for survival under stress conditions. Immense changes in DNA methylation level and profile observed for the SL line 6 h after the herbicide application (18.64%) may lead to the deregulation of gene expression, which as a result ends with the plant’s death. In contrast, the change in DNA methylation level in the TL line following herbicide application was very slight (1.13%). Among the DNA sequences identified based on the changes in DNA methylation were transferases, transporter proteins, methyltransferases, hydrolases, transposons, ribosomal proteins, cytochromes, and proteins participating in transcription processes and involved in stress responses.
It appears that natural resistance of crops to herbicides is much more complicated than just a single-trait change (as observed in genetically modified crops) and is based on many mechanisms and several types of regulation of expression of genetic information (epigenetics, small non-coding RNAs, changes in expression and composition of genic products) that together make up the increased fitness of particular varieties.
References
Agarwal G, Kudapa H, Ramalingam A, Choudhary D, Sinha P, Garg V, Singh VK, Patil GB, Pandey MK, Nguyen HT, Guo B, Sunkar R, Niederhuth CE, Varshney RK (2020) Epigenetics and epigenomics: underlying mechanisms, relevance, and implications in crop improvement. Funct Integr Genomics 20(6):739–761. https://doi.org/10.1007/s10142-020-00756-7
Alonso C, Ramos-Cruz D, Becker C (2019) The role of plant epigenetics in biotic interactions. New Phytol 221(2):731–737. https://doi.org/10.1111/nph.15408
Aufsatz W, Mette MF, van der Winden J, Matzke M, Matzke AJ (2002) HDA6, a putative histone deacetylase needed to enhance DNA methylation induced by double-stranded RNA. EMBO J 21(24):6832–6841. https://doi.org/10.1093/emboj/cdf663
Boyko A, Kovalchuk I (2008) Epigenetic control of plant stress response. Environ Mol Mutagen 49(1):61–72. https://doi.org/10.1002/em.20347
Bräutigam K, Cronk Q (2018) DNA methylation and the evolution of developmental complexity in plants. Front Plant Sci 9:1447. https://doi.org/10.3389/fpls.2018.01447
Chinnusamy V, Zhu JK (2009) Epigenetic regulation of stress responses in plants. Curr Opin Plant Biol 12(2):133–139. https://doi.org/10.1016/j.pbi.2008.12.006
Craine JM, Dybzinski R (2013) Mechanisms of plant competition for nutrients, water and light. Funct Ecol 27:833–840. https://doi.org/10.1111/1365-2435.12081
Denis MH, Delrot S (1993) Carrier-mediated uptake of glyphosate in broad bean (Vicia faba) via a phosphate transporter. Physiol Plant 87:569–575. https://doi.org/10.1111/j.1399-3054.1993.tb02508.x
Dowen RH, Pelizzola M, Schmitz RJ, Lister R, Dowen JM, Nery JR, Dixon JE, Ecker JR (2012) Stress-induced dynamic DNA methylation. PNAS 109(32):E2183–E2191. https://doi.org/10.1073/pnas.1209329109
Duke SO, Powles SB (2008) Glyphosate: a once-in-a-century herbicide. Pest Manag Sci 64(4):319–335. https://doi.org/10.1002/ps.1518
Eriksson MC, Szukala A, Tian B, Paun O (2020) Current research frontiers in plant epigenetics: an introduction to a Virtual Issue. New Phytol 226(2):285–288. https://doi.org/10.1111/nph.16493
Fuchs MA, Geiger DR, Reynolds TL, Bourque JE (2002) Mechanisms of glyphosate toxicity in velvetleaf (Abutilon theophrasti medikus). Pestic Biochem Physiol 74:27–39. https://doi.org/10.1016/S0048-3575(02)00118-9
Galindo-González L, Sarmiento F, Quimbaya MA (2018) Shaping plant adaptability, genome structure and gene expression through transposable element epigenetic control: focus on methylation. Agronomy 8:180. https://doi.org/10.3390/agronomy8090180
Gallego-Bartolomé J (2020) DNA methylation in plants: mechanisms and tools for targeted manipulation. New Phytol 227(1):38–44. https://doi.org/10.1111/nph.16529
Garg R, Narayana Chevala V, Shankar R, Jain M (2015) Divergent DNA methylation patterns associated with gene expression in rice cultivars with contrasting drought and salinity stress response. Sci Rep 5:14922. https://doi.org/10.1038/srep14922
Gáspár B, Bossdorf O, Durka W (2019) Structure, stability and ecological significance of natural epigenetic variation: a large-scale survey in Plantago lanceolata. New Phytol 221(3):1585–1596. https://doi.org/10.1111/nph.15487
Geiger DR, Kapitan SW, Tucci MA (1986) Glyphosate inhibits photosynthesis and allocation of carbon to starch in sugar beet leaves. Plant Physiol 82(2):468–472. https://doi.org/10.1104/pp.82.2.468
Ghanizadeh H, Harrington KC (2017) Non-target site mechanisms of resistance to herbicides. Crit Rev Plant Sci 36(1):24–34. https://doi.org/10.1080/07352689.2017.1316134
Gomes MP, Smedbol E, Chalifour A, Hénault-Ethier L, Labrecque M, Lepage L, Lucotte M, Juneau P (2014) Alteration of plant physiology by glyphosate and its by-product aminomethylphosphonic acid: an overview. J Exp Bot 65(17):4691–4703. https://doi.org/10.1093/jxb/eru269
Gomes MP, Le Manac’h SG, Moingt M, Smedbol E, Paquet S, Labrecque M, Lucotte M, Juneau P (2016) Impact of phosphate on glyphosate uptake and toxicity in willow. J Hazard Mater 304:269–279. https://doi.org/10.1016/j.jhazmat.2015.10.043
González-Torralva F, Rojano-Delgado AM, Luque de Castro MD, Mülleder N, De Prado R (2012) Two non-target mechanisms are involved in glyphosate-resistant horseweed (Conyza canadensis L. Cronq.) biotypes. J Plant Physiol 169(17):1673–1679. https://doi.org/10.1016/j.jplph.2012.06.014
Han Q, Bartels A, Cheng X, Meyer A, An YC, Hsieh TF, Xiao W (2019) Epigenetics regulates reproductive development in plants. Plants (Basel) 8(12):564. https://doi.org/10.3390/plants8120564
Handral AR, Singh A, Singh DR, Suresh A, Jha GK (2017) Scenario of changing dynamics in production and productivity of major cereals in India. Indian J Agric Sci 87:1371–1376
Hashida SN, Uchiyama T, Martin C, Kishima Y, Sano Y, Mikami T (2006) The temperature-dependent change in methylation of the Antirrhinum transposon Tam3 is controlled by the activity of its transposase. Plant Cell 18(1):104–118. https://doi.org/10.1105/tpc.105.037655
Heap I, Duke SO (2018) Overview of glyphosate-resistant weeds worldwide. Pest Manag Sci 74:1040–1049. https://doi.org/10.1002/ps.4760
Hetherington PR, Marshall G, Kirkwood RC, Warner JM (1998) Absorption and efflux of glyphosate by cell suspensions. J Exp Bot 49:527–533. https://doi.org/10.1093/jxb/49.320.527
Hirayama T, Shinozaki K (2010) Research on plant abiotic stress responses in the post-genome era: past, present and future. Plant J 61:1041–1052. https://doi.org/10.1111/j.1365-313X.2010.04124.x
Jasinski M, Ducos E, Martinoia E, Boutry M (2003) The ATP-binding cassette transporters: structure, function, and gene family comparison between rice and Arabidopsis. Plant Physiol 131(3):1169–1177. https://doi.org/10.1104/pp.102.014720
Jensen K, Møller BL (2010) Plant NADPH-cytochrome P450 oxidoreductases. Phytochemistry 71(2-3):132–141. https://doi.org/10.1016/j.phytochem.2009.10.017
Kanehisa M, Goto S (2000) KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res 28(1):27–30. https://doi.org/10.1093/nar/28.1.27
Kanissery R, Gairhe B, Kadyampakeni D, Batuman O, Alferez F (2019) Glyphosate: its environmental persistence and impact on crop health and nutrition. Plants (Basel) 8(11):499. https://doi.org/10.3390/plants8110499
Karan R’ DeLeon T, Biradar H, Subudhi PK (2012) Salt stress induced variation in DNA methylation pattern and its influence on gene expression in contrasting rice genotypes. PLoS One 7(6):e40203. https://doi.org/10.1371/journal.pone.0040203
Kim G, Clarke CR, Larose H, Tran HT, Haak DC, Zhang L, Askew S, Barney J, Westwood JH (2017) Herbicide injury induces DNA methylome alterations in Arabidopsis. PeerJ 5:e3560. https://doi.org/10.7717/peerj.3560
Kong L, Liu Y, Wang X, Chang C (2020) Insight into the role of epigenetic processes in abiotic and biotic stress response in wheat and barley. Int J Mol Sci 21:1480. https://doi.org/10.3390/ijms21041480
Kumar J, Rai KM, Pirseyedi S, Elias EM, Xu S, Dill-Macky R, Kianian SF (2020) Epigenetic regulation of gene expression improves Fusarium head blight resistance in durum wheat. Sci Rep 10(1):17610. https://doi.org/10.1038/s41598-020-73521-2
Langner JA, Zanon AJ, Streck NA, Reiniger LRS, Kaufmann MP, Alves AF (2019) Maize: key agricultural crop in food security and sovereignty in a future with water scarcity. Rev Bras Engenharia Agrícola e Ambienta 23(9):648–654. https://doi.org/10.1590/1807-1929/agriambi.v23n9p648-654
Lefèvre F, Boutry M (2018) Towards identification of the substrates of ATP-Binding Cassette Transporters. Plant Physiol 178(1):18–39. https://doi.org/10.1104/pp.18.00325
Li Q, Eichten SR, Hermanson PJ, Zaunbrecher VM, Song J, Wendt J, Rosenbaum H, Madzima TF, Sloan EA, Huang J, Burgess DL, Richmond TA, McGinnis KM, Meeley RB, Danilevskaya ON, Vaughn MW, Kaeppler SM, Jeddeloh JA, Springer NM (2014) Genetic perturbation of the maize methylome. Plant Cell 26(12):4602–4616. https://doi.org/10.1105/tpc.114.133140
Lisch D (2008) Epigenetic regulation of transposable elements in plants. Annu Rev Plant Biol 60:43–66. https://doi.org/10.1146/annurev.arplant.59.032607.092744
Lu Y, Feng S, Zhang J, Zhang JJ, Luo F, Zhang S, Yang H (2016) Genome-wide identification of DNA methylation provides insights into the association of gene expression in rice exposed to pesticide atrazine. Sci Rep 6:18985. https://doi.org/10.1038/srep18985
Lyu H, He Z, Wu CI, Shi S (2017) Convergent adaptive evolution in marginal environments: unloading transposable elements as a common strategy among mangrove genomes. New Phytol 217:428–438. https://doi.org/10.1111/nph.14784
Mager S, Ludewig U (2018) Massive loss of DNA methylation in nitrogen-, but not in phosphorus-deficient Zea mays roots is poorly correlated with gene expression differences. Front Plant Sci 9:497. https://doi.org/10.3389/fpls.2018.00497
Maggi F, Tang FHM, la Cecilia D, McBratney A (2019) PEST-CHEMGRIDS, global gridded maps of the top 20 crop-specific pesticide application rates from 2015 to 2025. Sci Data 6(170):1–20. https://doi.org/10.1038/s41597-019-0169-4
Mahmoud M, Gracz-Bernaciak J, Żywicki M, Karłowski W, Twardowski T, Tyczewska A (2020) Identification of structural variants in two novel genomes of maize inbred lines possibly related to glyphosate tolerance. Plants 9:523. https://doi.org/10.3390/plants9040523
Makarevitch I, Waters AJ, West PT, Stitzer M, Hirsch CN, Ross-Ibarra J, Springer NM (2015) Transposable elements contribute to activation of maize genes in response to abiotic stress. PLoS Genet 11:1. https://doi.org/10.1371/journal.pgen.1004915
Markus C, Pecinka A, Karan R, Barney JN, Merotto A Jr (2018) Epigenetic regulation - contribution to herbicide resistance in weeds? Pest Manag Sci 74(2):275–281. https://doi.org/10.1002/ps.4727
Matzke M, Kanno T, Daxinger L, Huettel B, Matzke AJM (2009) RNA-mediated chromatin-based silencing in plants. Curr Opin Cell Biol 21(3):367–376. https://doi.org/10.1016/j.ceb.2009.01.025
McClintock B (1984) The significance of responses of the genome to challenge. Science 226(4676):792–801. https://doi.org/10.1126/science.15739260
Meng D, Dubin M, Zhang P, Osborne EJ, Stegle O, Clark RM, Nordborg M (2016) Limited contribution of DNA methylation variation to expression regulation in Arabidopsis thaliana. PLoS Genet 12(7):e1006141. https://doi.org/10.1371/journal.pgen.1006141
Meng J, Wang L, Wang J, Zhao X, Cheng J, Yu W, Jin D, Li Q, Gong Z (2018) Methionine adenozylotransferase4 mediates DNA and histone methylation. Plant Physiol 177(2):652–670. https://doi.org/10.1104/pp.18.00183
Mertens M, Höss S, Neumann G, Afzal J, Reichenbecher W (2018) Glyphosate, a chelating agent-relevant for ecological risk assessment? Environ Sci Pollut Res Int 25(6):5298–5317. https://doi.org/10.1007/s11356-017-1080-1
Mette MF, Aufsatz W, van der Winden J, Matzke MA, Matzke AJM (2000) Transcriptional silencing and promoter methylation triggered by double-stranded RNA. EMBO J 19:5194–5201. https://doi.org/10.1093/emboj/19.19.5194
Meyers BC, Tingey SV, Morgante M (2001) Abundance, distribution, and transcriptional activity of repetitive elements in the maize genome. Genome Res 11(10):1660–1676. https://doi.org/10.1101/gr.188201
Moretti ML, Alárcon-Reverte R, Pearce S, Morran S, Hanson BD (2017) Transcription of putative tonoplast transporters in response to glyphosate and paraquat stress in Conyza bonariensis and Conyza canadensis and selection of reference genes for qRT-PCR. PLoS One 12(7):e0180794. https://doi.org/10.1371/journal.pone.0180794
Morin F, Vera V, Nurit F, Tissut M, Marigo G (1997) Glyphosate uptake in Catharanthus roseus cells: role of a phosphate transporter. Pestic Biochem Physiol 58:13–22. https://doi.org/10.1006/pest.1997.2280
Munksgaard D, Mattsson O, Thyge Okkels F (1995) Somatic embryo development in carrot is associated with an increase in levels of S-adenosylmethionine, S-adenosylhomocysteine and DNA methylation. Physiol Plant 93(1):1–4. https://doi.org/10.1034/j.1399-3054.1995.930102.x
Negi P, Rai AN, Suprasanna P (2016) Moving through the stressed genome: emerging regulatory roles for transposons in plant stress response. Front Plant Sci 7:1448. https://doi.org/10.3389/fpls.2016.01448
OECD-FAO Agricultural outlook 2018-2027, Available online: http://www.agri-outlook.org/commodities/Agricultural-Outlook-2018-Cereals.pdf (accessed 1 July 2020)
Orcaray L, Zulet A, Zabalza A, Royuela M (2012) Impairment of carbon metabolism induced by the herbicide glyphosate. J Plant Physiol 169:27–33. https://doi.org/10.1016/j.jplph.2011.08.009
Paszkowski J, Whitham SA (2001) Gene silencing and DNA methylation processes. Curr Opin Plant Biol 4(2):123–129. https://doi.org/10.1016/S1369-5266(00)00147-3
Romero DM, de Molina MCR, Juarez AB (2011) Oxidative stress induced by a commercial glyphosate formulation in a tolerant strain of Chlorella kessleri. Ecotoxicol Environ Saf 74(4):741–747. https://doi.org/10.1016/j.ecoenv.2010.10.034
Rong Tan L, Chen Lu Y, Jing Zhang J, Luo F, Yang H (2015) A collection of cytochrome P450 monooxygenase genes involved in modification and detoxification of herbicide atrazine in rice (Oryza sativa) plants. Ecotoxicol Environ Saf 119:25–34. https://doi.org/10.1016/j.ecoenv.2015.04.035
Samsel A, Seneff S (2013) Glyphosate’s suppression of cytochrome P450 enzymes and amino acid biosynthesis by the gut microbiome: pathways to modern diseases. Entropy 15:1416–1463. https://doi.org/10.3390/e15041416
Shaner DL (2009) Role of translocation as a mechanism of resistance to glyphosate. Weed Sci 57:118–123. https://doi.org/10.1614/WS-08-050.1
Siminszky B (2006) Plant cytochrome P450-mediated herbicide metabolism. Phytochem Rev 5:445–458. https://doi.org/10.1007/s11101-006-9011-7
Sun L, Miao X, Cui J, Deng J, Wang X, Wang Y, Zhang Y, Gao S, Yang K (2018) Genome-wide high-resolution mapping of DNA methylation identifies epigenetic variation across different salt stress in maize (Zea mays L.). Euphytica 214:25. https://doi.org/10.1007/s10681-017-2076-0
Tan MP (2010) Analysis of DNA methylation of maize in response to osmotic and salt stress based on methylation-sensitive amplified polymorphism. Plant Physiol Biochem 48(1):21–26. https://doi.org/10.1016/j.plaphy.2009.10.005
Tani E, Chachalis D, Travlos IS (2015) A glyphosate resistance mechanism in Conyza canadensis involves synchronization of EPSPS and ABC-transporter genes. Plant Mol Biol Report 33:1721–1730. https://doi.org/10.1007/s11105-015-0868-8
Tani E, Chachalis D, Travlos IS, Bilalis D (2016) Environmental conditions influence induction of key ABC-transporter genes affecting glyphosate resistance mechanism in Conyza canadensis. Int J Mol Sci 17(4):342. https://doi.org/10.3390/ijms17040342
Tyczewska A, Woźniak E, Gracz J, Kuczyński J, Twardowski T (2018) Towards food security: current state and future prospects of agrobiotechnology. Trends Biotechnol 36(12):1219–1229. https://doi.org/10.1016/j.tibtech.2018.07.008
Uthup TK, Ravindran M, Bini K, Thakurdas S (2011) Divergent DNA methylation patterns associated with abiotic stress in Hevea brasiliensis. Mol Plant 4:996–1013. https://doi.org/10.1590/S1415-47572013000400016
Varotto S, Tani E, Abraham E, Krugman T, Kapazoglou A, Melzer R, Radanović A, Miladinović D (2020) Epigenetics: possible applications in climate-smart crop breeding. J Exp Bot 71(17):5223–5236. https://doi.org/10.1093/jxb/eraa188
Wang M, Qin L, Xie C, Li W, Yuan J, Kong L, Yu W, Xia G, Liu S (2014) Induced and constitutive DNA methylation in a salinity-tolerant wheat introgression line. Plant Cell Physiol 55(7):1354–1365. https://doi.org/10.1093/pcp/pcu059
Wang W, Zhao X, Pan Y, Zhu L, Fu B, Li Z (2015) DNA methylation changes detected by methylation-sensitive amplified polymorphism in two contrasting rice genotypes under salt stress. Biophys Res Commun 465(4):790–796. https://doi.org/10.1016/j.jgg.2011.07.006
Wang Y, Liang W, Tang T (2018) Constant conflict between Gypsy LTR retrotransposons and CHH methylation within a stress-adapted mangrove genome. New Phytol 220(3):922–935. https://doi.org/10.1111/nph.15209
Xiong Z, Xu CG, Saghai Maroof MA, Zhang Q (1999) Patterns of cytosine methylation in an elite rice hybrid and its parental lines, detected by a methylation-sensitive amplification polymorphism technique. Mol Gen Genet 261:439–446. https://doi.org/10.1007/s004380050986
Yang H, Chang F, You C, Cui J, Zhu G, Wang L, Zheng Y, Qi J, Ma H (2014) Whole-genome DNA methylation patterns and complex associations with gene structure and expression during flower development in Arabidopsis. Plant J 81:268–281. https://doi.org/10.1111/tpj.12726
Yang CJ, Samayoa LF, Bradbury PJ, Olukolu BA, Xue W, York AM, Tuholski MR, Wang W, Daskalska LL, Neumeyer MA, Sanchez-Gonzalez JJ, Cinta Romay M, Glaubitz JC, Sun Q, Buckler ES, Holland JB, Doebley JB (2019) The genetic architecture of teosinte catalyzed and constrained maize domestication. PNAS 116(12):5643–5652. https://doi.org/10.1073/pnas.1820997116
Yu J, Holland JB, McMullen MD, Buckler ES (2008) Genetic design and statistical power of nested association mapping in maize. Genetics 178:539–551. https://doi.org/10.1534/genetics.107.074245
Zhang H, Lang Z, Zhu JK (2018) Dynamics and function of DNA methylation in plants. Nat Rev Mol Cell Biol 19(8):489–506. https://doi.org/10.1038/s41580-018-0016-z
Żywicki M, Gracz J, Karłowski W, Twardowski T, Tyczewska A (2015) Expression of miRNAs involved in phosphate homeostasis and senescence is altered in glyphosate-treated maize. Acta Physiol Plant 37:265. https://doi.org/10.1007/s11738-015-2022-5
Acknowledgments
We thank prof. K. Adamczewski and prof. J. Adamczyk for providing the plant material.
Funding
This work was supported by grants from the National Science Center Poland: UMO-2011/01/D/NZ9/03631 and UMO-2012/06/A/NZ9/00125. This publication was also supported by the Polish Ministry of Science and Higher Education, under the KNOW program.
Author information
Authors and Affiliations
Contributions
Conceptualization: A.T. Methodology: A.T. and J.G.-B. Experimental work: A.T., J.G.-B., and J.S. Writing—original draft preparation: A.T. Writing—review and editing: A.T., J.G.-B., J.S., and T.T. Supervision: A.T. and T.T. Funding acquisition: A.T. and T.T. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by: Izabela Pawłowicz
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tyczewska, A., Gracz-Bernaciak, J., Szymkowiak, J. et al. Herbicide stress-induced DNA methylation changes in two Zea mays inbred lines differing in Roundup® resistance. J Appl Genetics 62, 235–248 (2021). https://doi.org/10.1007/s13353-021-00609-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-021-00609-4