
Abstract
COVID-19 pandemic has proven to be a dramatic challenge, introducing huge clinical differences that demand extensive investigations. Severe and critical patients may present coagulopathies and microthrombi, which results in varied complications, or acute respiratory distress syndrome that leads to fatality. Although the lung to be the major site of clinical manifestations, COVID-19 has shown extrapulmonary manifestations, especially on the heart and kidney, directly linked to worse disease outcomes. According to the fast-moving of clinical description and scientific publications, the injuries in multiple organs and systemic inflammation appear to be caused by a deregulated immune response, and the NLRP3 inflammasome could be a relevant influencer in this imbalance. However, until now, the precise drivers of the pathophysiology of these injuries remain unknown. In this review, we discuss how inflammasome seems to be directly involved in the clinical profile of patients infected with SARS-CoV-2 and shed light on the mechanisms that lead to fatality.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since the emergence of a novel β-coronavirus (β-CoV) in late December 2019, the entire world is facing an unprecedented crisis. As a reflection of high spread capacity, Coronavirus disease 19 (COVID-19), caused by a virus named SARS-CoV-2, has become pandemic and has caused over 165 million cases and 3 million global deaths [1].
Different from other current strains of coronavirus that causes a simple common cold, in the last decades, other coronavirus (CoVs) have been reported in severe respiratory outbreaks in 2002–2003 and 2012, causing severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome (MERS), respectively. SARS-CoV-2 is responsible for a range of variable symptoms [2] that can progress to critical illness and death [3]. Despite of SARS-CoV-2 exhibit genetic and phylogenetic similarities with SARS-CoV and MERS-CoV, this virus has singularities that made it to be treated as a global emergency [4].
In a genomic perspective, the virus is constituted by non-structural and structural proteins as spike (S), used for binding to angiotensin-converting enzyme 2 (ACE2) on surfaces of host cells; membrane protein (M), linked with virus morphogenesis and assembly; the envelop protein (E), a transmembrane protein important for ionic transportation; and the nucleocapsid (N) where the RNA strand is located [5].
To protect the body against the virus, the innate immune system uses pattern-recognition receptors (PRRs) for the detection of foreign structures called pathogen-associated molecular patterns (PAMPs) [6]. Among the large family of PRRs, the NOD-like receptor (NLR) has notoriety due to its wide recognition of intrinsic or extrinsic stimuli, operating as cytoplasmatic sensors. The primary function of NLR is to participate in a multiprotein complex, known as inflammasome, able to activate the pro-inflammatory cytokines such as IL-1β and IL-18 [7].
Until now, several factors may explain COVID-19 severity, however, dysregulated inflammation and cytokine storm is implicated with tissue damage to the airways [8]. Severe patients show high levels of pro-inflammatory cytokines (such as IL-1β, IL-2, IL-6, IL-18, IFN-γ, TNF-α), and chemokines (CXCL8 CXCL10, macrophage inflammatory protein 1α (MIP1α), MIP1β and MCP1) [9, 10]. Although the importance of strong immune response for pathogens control, these biomolecules, along with others immune effectors, promote unbalanced responses in COVID-19; this mechanism of disease aggravating is also observed in SARS and MERS [11].
The clinical presentations of individuals range from the total absence of symptoms to mild, moderate, severe, or critical disease. The main symptoms reported are fever, fatigue, and dry cough. Laboratory data presented lymphocytopenia (83.2%) thrombocytopenia (36.2%), leucopenia (33.7%), elevated levels of C-reactive protein, alanine aminotransferase (ALT), aspartate aminotransferase (ALT), creatine kinase and also increased d-dimer [9].
Given the high number of patients across the world, understand the pieces in the puzzle of excessive inflammation and clinical outcome is urgently needed. Therefore, insights about the influence of relevant biologic proteins like inflammasome are necessary for design therapies and unravel the mysteries of pathophysiology.
Inflammasome and viral infection
Structurally, inflammasome complex is a scaffold of proteins formed by central domain called the nucleotide-binding and oligomerization domain (NOD), a domain with leucine-rich repeats (LRRs) and an N-terminal protein–protein interaction domain that need to interact with Apoptosis-associated speck-like protein (ASC) which contains a caspase activation and recruitment domain (CARD) to attached pro-caspase-1 and allow its activation. After activation, caspase-1 promote proteolytic cleavage of pro-IL-1β and pro-IL-18 in its mature forms and stimulates its secretion [7].
To develop an efficient response against viral particles, the host immune system uses PRRs as TLR3, TLR7, and TLR8 for the detection of PAMPs. The SARS-CoV-2 RNA is recognized by those receptors and also by RIG-I/MDA5, that work as a cytosolic sensor, promoting a signalling cascade that leads to nuclear factor kappa B (NF-κB) activation. Consequently, this transcription factor is translocated to the nucleus which could induce IFN type I and other pro-inflammatory cytokines [12, 13].
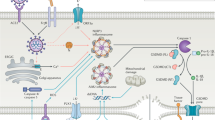
It has been shown that proteins E, ORF3a e ORF8b from SARS-CoV activate NLRP3, the inflammasome more expressed in pulmonary tissue. Viroporin of E protein formed ionic channel that facilitates the leakage of Ca 2+ into the cytosol. Due to the sensitivity to ionic imbalances, inflammasome is activated. On the other hand, ORF3a promotes ubiquitination mediated by a factor-binding domain associated with receptor 3 (TRAF3), as well as expressing the pro-IL-1β gene and the secretion of IL-1β, both of which are necessary for inflammasome activation. Finally, ORF8b activates NLRP3 through direct interaction of the leucine-rich repeat domain, even functioning as ion channels. These cytosolic changes trigger a second step for inflammasome activation [14] (Fig. 1a).

Schematic representation of signaling pathways of pro-inflammatory cytokines involved on COVID-19. a Inflammasome assembly after PAMP and DAMP recognition activate caspase-1, essential for the cleavage of pro-IL-1b and IL-18 into their active form. After externalization, these cytokines can induce pyroptosis and biding their respective receptors to promote the expression of pro-inflammatory molecules. b Signal transduction of IL-6 after binding IL-6 receptor and activates Janus Kinases (JAK) leads to activation of different mediators to promote gene expression. The recognition of TNFa by TNFR2 triggers different cascades which promotes inflammation or gene expression mediated by NFk-B. In parallel, the biding on TNFR1 causes subsequent apoptosis mediated by caspases
Among the other contributors to inflammasome activation, the ROS seems to play a significant role in COVID-19 patients [15,16,17]. Upon SARS-CoV-2 binding ACE2, occur multiple modifications on host homeostasis, including the promotion of inflammatory response and the downregulation of ACE2 receptor. Thus, occur deregulation in the renin-angiotensin system and increase of angiotensin II, resulting in the generation of ROS and raise of oxidative stress. Moreover, ROS can interfere through NF-kB and activate the NLRP3 and increase the expression of pro-IL-1 and pro-IL-18 [18,19,20].
Excessive activation of the inflammasome and release of IL-1β and IL-18 can trigger immune cascades as the discharge of pro-inflammatory cytokines, chemokines, and cell migration, promoting the accumulation of immune cells and lung damage [21]. In COVID-19, there is a complex crosstalk between these components that needs to urgently elucidate especially because when analyzing the patients' serum, high levels of IL-1β and LDH were observed, which are indicative of the inflammasome activation [22,23,24].
As an effort to maintains the proper response, inflammasome activation is tightly regulated. To protect the body, the immune system prevents unnecessary or hyperactivation through different mechanisms, including some proteins and post-translational modifications. CARD-only proteins and PYRIN-only proteins (POP) are small cytoplasmatic proteins that act as regulators with the function in decrease inflammation. Likewise, CARD16, CARD17, and CARD18 inhibit caspase 1 and negatively regulate the inflammasome. Conversely, the target of POP1 and POP2 is ASC—NLRP3 interaction, and as a consequence, the NLRP3 activation is suppressed [25,26,27,28].
Furthermore, the recognition of Trp73 on NLRP3 by FBXL12 and SCF E3 ligase complex leads to ubiquitination and proteasomal degradation. Moreover, TRIM31 appears to be an inflammasome suppressor through feedback positive because induces Lys48-linked ubiquitination. Besides, the Ser295 in NLRP3 is an important site to phosphorylation by protein kinase A (PKA) and subsequent inhibition of inflammasome activation [29, 30].
Immunomodulatory interventions to treat COVID-19 is largely debated and different strategies are applied in clinical trials such as Canakinumab [31], and anti-IL-1β that have NLRP3 inflammasome as a molecular target, Anakinra [32], an IL-1 receptor antagonist, and JAK1/JAK2 inhibitors [33] that interrupt the mediates cytokine signaling pathways. Additionally, some natural compounds as colchicine are frequently used to treat inflammatory diseases due to its ability to inhibit inflammasome, and serving as an example for the wide possibilities of therapeutic strategies [34, 35]. Until now, few clinical trials registered at NIH intend to study anti-inflammasome strategies focus specifically in COVID-19 positive patients.
Neutrophils and pulmonary injury
As the first line of defence against pathogens, neutrophils play an important role against viral infections [36]. However, IL-1β activated by the NLRP3 inflammasome can recruit and activate neutrophils, inducing “NETosis”, which consists of a type of cell death, producing wire-like extracellular structures, called neutrophil extracellular traps (NETs). NETs mainly play a protective role, forming mesh-like structures to trap pathogens, and they consist of DNA, modified histones and cytotoxic proteins, including myeloperoxidase (MPO), and cathepsin [37, 38].
Although there is indeed an important function of kill foreign pathogens, the uncontrolled production of NETs has been strongly implicated as a causal factor associated with acute lung injury (ALI), acute respiratory distress syndrome (ARDS), coagulopathy, multi-organ failure, and autoimmune diseases, besides activating even more inflammasomes [39, 40]. In COVID-19, severely ill patients have a higher neutrophil count in blood plasma and 80% of these cells presented NETs structures [41].
Prior reports suggested that NETs released from neutrophils activated by SARS-CoV-2 promotes lung epithelial cell death [42,43,44]. The increased level of NETs was also reported in post-mortem analyses of lung tissue from COVID-19 patients treated, or not, in intensive care units (ICU), supporting his involvement in mediating tissue injury [45]. It is important to reinforce the relationship of NETs in several inflammatory diseases such as rheumatoid arthritis [46], diabetes [47], and sepsis [48]. According to the results from a recent research, necroinflammation linked with NETs play a central role in the cytokine storm growth, sepsis and multi-organ failure during COVID-19. Furthermore, patients with comorbidities increases neutrophil infiltration sustaining an inflammatory cascade that leads to the migration of more immune cells, the release of cytokines, inflammasome activation, damage to the lungs, increasing disease severity [43]. Other study identified high levels of citrullinated histones H3 (Cit-H3) and MPO-DNA complexes, markers directly associated with NETs, and cell-free DNA, higher in patients on mechanical ventilation [49].
Based on the researches so far, it is evident the impact of NETs on disease outcome, therefore, the inhibition of these extracellular traps represents a potential therapeutic target for SARS-CoV-2.
Influence of inflamassoma on ards
The central role of inflammation in the aggravation of COVID-19 cases is extensively reasoned through different perspectives and the involvement of inflammasome has been carefully presented [50, 51]. Robust evidence demonstrates an association between over-activation of the inflammasome complex with the worsening of clinical features of patients, although it is necessary a detailed investigation about additional mechanisms surrounding this process [52, 53]. Nevertheless, patients in critical stage reveal increased levels of IL-1 and IL-18, which is related to inflammasome [54,55,56] (Fig. 1).
Acute respiratory distress syndrome (ARDS) – the most severe form of acute lung injury—is a dramatic syndrome without specific treatments. Due to the absence of biomarkers, the diagnosis of this severe form is based on a combination of clinical manifestations [57] such as the time for the onset of symptoms, radiographic changes and the existence of bilateral opacity, the severity of the PaO2/FiO2 rate, and the presence of edema [58]. Additionally, it is observed a remarkably increased vascular permeability, raised lung weight, and loss of aerated tissue, which may result in severe hypoxemia and septic shock [59, 60].
Regardless of the new coronavirus, ARDS is, by itself, an extremely complex problem [61]. Known for being an ICU syndrome with a high mortality rate and difficult diagnosis, this disorder becomes even more serious due to the global proportions of SARS-CoV-2 infection. In fact, among symptomatic individuals, it is estimated that 15% develop critical conditions and become susceptible to the emergence of ARDS [62, 63].
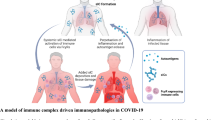
The innate immune response, after the virus entry in permissive cells, is rapid and aggressively triggered to try to eliminate the pathogen. However, in response to this infection, occurs an exacerbated inflammation that becomes responsible for tissue damage more expressive than the viral replication itself. The severity of COVID-19 appears to be directly linked to this inflammatory dysregulation and massive release of cytokines. In some cases this can lead to a state of hypercytokinemia, also known as macrophage release syndrome, capable of triggering ARDS and leading to multi-organ failure [64, 65] (Fig. 2).

The clinical features of COVID-19 patients are resulting from simultaneous events, promoting complex crosstalk between immune components. When SARS-CoV-2 infect host cell through Spike protein binding with ACE2 receptor, a wave of PAMPs and DAMPs ensues, activating pathways that enable inflammasome activation and leads to cleavage of IL-1β and IL-18, creating pro-inflammatory signals and leads to airway inflammation. Consequently, occurs platelet activation, fibrin deposition, and NETs release, which promotes tissue damage. A cascade involving inflammasome assembly and inflammation leads to ARDS development
Among the ARDS hallmarks, the most significant is the exacerbated and unregulated inflammatory response. Simultaneously, several events occur to promote positive feedback of inflammation pathways and, consequently, there is an increase in pro-inflammatory mediators such as chemokines and cytokines, determinant factors for increasing disease severity and mortality [66, 67].
Recent insights into the pathophysiology of ARDS point to inflammasome as one of the main factors responsible for the intense pulmonary inflammatory response that cause tissue injury in affected patients [68,69,70]. The inflammasome has also been correlated with other lung diseases such as chronic obstructive pulmonary disease (COPD), a gradual illness caused by exposure of the airways to irritants, such as cigarette smoke, capable of activating NLRP3, resulting in intense inflammation, mucus production and dyspnea. In the sputum of human patients suffering from this disease, the levels of IL-1β were significantly increased [71], and CPOD modelling using NLRP3 knockout mice observed that gene absence prevented the development of COPD in the animal exposed to cigarette smoke for a long time [72].
The inflammasome is also considered as an important mediator in pulmonary fibrosis characterized by a chronic inflammatory process and scarring that hinders breathing. Although there is no formally established cause for the appearance of this disease, several agents that induce fibrosis are capable of activating Inflammasome [73]. Also, IL-1β secretion induces the production of TGF-β, a pro-fibrotic cytokine, promotes neutrophil chemoattraction, and contribute to epithelial damage [74]. Interestingly, TGF-b was upregulated in SARS and MERS patients, and fibrosis is also related in COVID-19 patients [75]. Although the involvement of IL-1β in worsening ARDS has been known for more than two decades, unravelling the key role of the inflammasome complex in this process allows us to outline assertive clinical management and new treatment strategies.
From the moment of viral infection, several events occur in synergy leading to systemic irregularities perpetuating the inflammatory imbalance [76, 77] (Fig. 1b). In parallel, alveolar macrophages secrete pro-inflammatory cytokines such as TNF-a and IL-1β maintaining an inflammation cascade that results in the release of more PAMPs and DAMPs capable of activating the inflammasome complex. In addition, the presence of ARDS can result in the release of mitochondrial DAMPs that signal and result in increased vascular permeability and activation of polymorphonuclear cells, disseminating inflammation, and possibly increasing patient mortality [78,79,80].
When analysing the peripheral blood of individuals diagnosed with ARDS, it was observed a significant increase in mRNA levels of CASP-1, IL-1β, and IL-18, along with high levels of IL-18 in plasma in comparison with control patients in treatment in ICU. Moreover, severity and mortality was higher among the group with the highest plasma level of IL-18, which in turn is related to lactate levels, a biomarker widely used in the prognosis of critically ill patients [66].
In a murine model of ARDS induced by mechanical ventilation, IL-18 levels is increased in the lung, serum, and bronchoalveolar lavage. In knockout animals for CASP-1 or IL-18, there was less lung injury in response to stress-induced by mechanical ventilation. These findings demonstrate the participation of molecules linked to the inflammasome in the development and severity of ARDS [66].
Coagulopathies, inflammasome and COVID-19
In clinical practice, complications Related to coagulation abnormality was extensively observed in patients with COVID-19. The determination of coagulation markers as D-dimer, fibrin, fibrinogen, prothrombin, and platelets have become usual and fundamental for the determination of coagulopathies [81].
According to the evidence available so far, imbalances in coagulation is one of the most significant variables in determining the COVID-19 disease severity. However, researchers hypothesize that these occurrences, such as pulmonary thromboembolism [82], is not directly caused by virus entry but by the inflammatory response. Nevertheless, indirect events of SARS-CoV-2 infection, like hypoxia, may predispose the patient to develop thrombotic complications [83].
The most common homeostatic abnormality in COVID-19 patients are mild thrombocytopenia [84], high D-dimer levels [85], increased prothrombin time, and disseminated intravascular coagulation (DIC)— more common in critical patients [86].
A key point of interaction between inflammation and coagulation appears to be activation of protease-activated receptor (PARs) by tissue factor (TF), and at the end of this long signalling pathway occurs platelet activation and fibrin deposition [87]. Pro-inflammatory cytokines like IL-1β, IL-6, and TNF can induce TF expression in mononuclear cells, creating coagulation activation feedback. In post-mortem biopsy from the lung of four critically ill patients, was observed platelet aggregation with clot formation and fibrin deposition, validating the suggested hypotheses and the clinical manifestations observed [88,89,90,91].
Platelets play a major role in homeostatic maintenance, however, they have the unusual function of maturing pro-IL-1β from your pre-mRNA [92]. Therefore it is another synthesis pathway for this cytokine which, in addition to participating in coagulation disorders, is a primary effector in different biological processes. Additionally, platelets also express NLRP3 and ASC, being capable of assembly inflammasome and produce IL-1β [93]. This cell line is a fundamental link between immune response and coagulation.
When investigating the association between inflammasome activation and blood clotting, it was found that inflammasome can trigger systemic coagulation, thrombosis, and lead to fatality. In summary, the pyroptosis promoted by inflammasome assembly induces pyroptotic macrophages to release TF. Consequently, this protein promotes disseminated coagulation and systemic disorders [94]. Given the continuous growth of evidence where the Inflammasome plays a role in addition to the immune system, the development of robust studies that explain the unknowns linked to this protein complex is indispensable (Table 1).
Conclusions
Emerging data has presented the variability of players involved in COVID-19 severe or critical illness and until now, evidences clearly demonstrated a major role of hyperinflammation in worse prognosis. Because of this imbalance, there is a feedback release of cytokine, PAMPs, and DAMPs which leads to inflammasome activation and maintenance of pro-inflammatory state promoting platelet activation and coagulopathies, increased NETs release, tissue injury and developing of ARDS. Inflammasome complex has been indicated in pathophysiology of with several symptoms observed in COVID-19 patients, and considering the complex crosstalk involving immune system and others biological components, is crucial elucidate the mechanism behind clinical manifestation. Therefore, the inflammasome has relevance to understand the heterogeneity of clinical features and to develop rational design of therapies.
Change history
21 September 2021
A Correction to this paper has been published: https://doi.org/10.1007/s13337-021-00740-0
References
Dong E, Du H, Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect Dis. 2020;20(5):533–4. https://doi.org/10.1016/S1473-3099(20)30120-1.
Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y, et al. Presumed asymptomatic carrier transmission ofCOVID-19. N Engl J Med. 2020;382(13):1199–207.
Gorbalenya AE, Baker SC, Baric RS, de Groot RJ, Drosten C, Gulyaeva AA, et al. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol. 2020;5(4):536–44.
Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–74.
Khailany RA, Safdar M, Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020. https://doi.org/10.1016/j.genrep.2020.100682.
Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801.
Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20(13):1–24.
Tay MZ, Poh CM, Rénia L, MacAry PA, Ng LFP. The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol. 2020;20(6):363–74.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan. China Lancet. 2020;395(10223):497–506.
Liu J, Li S, Liu J, Liang B, Wang X, Wang H, et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine. 2020. https://doi.org/10.1016/j.ebiom.2020.102763.
Siu KL, Yuen KS, Castano-Rodriguez C, Ye ZW, Yeung ML, Fung SY, et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019;33(8):8865–77.
Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol. 2020;92(10):2105–13.
De Wit E, Van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol. 2016;14(8):523–34.
Nieto-Torres JL, Verdiá-Báguena C, Jimenez-Guardeño JM, Regla-Nava JA, Castaño-Rodriguez C, Fernandez-Delgado R, et al. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology. 2015;485:330–9. https://doi.org/10.1016/j.virol.2015.08.010.
Beltrán-García J, Osca-Verdegal R, Pallardó F V., Ferreres J, Rodríguez M, Mulet S, et al. Oxidative stress and inflammation in COVID-19-Associated sepsis: the potential role of anti-oxidant therapy in avoiding disease progression. antioxidants [Internet]. 2020;9(10):936. Available from: https://www.mdpi.com/2076-3921/9/10/936.
Schönrich G, Raftery MJ, Samstag Y. Devilishly radical NETwork in COVID-19: Oxidative stress, neutrophil extracellular traps (NETs), and T cell suppression. Adv Biol Regul [Internet]. 2020;77(June):100741. Available from: https://linkinghub.elsevier.com/retrieve/pii/S221249262030052X.
Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II–mediated mitochondrial dysfunction. Circ Res [Internet]. 2008;102(4):488–96. https://doi.org/10.1161/CIRCRESAHA.107.162800.
de Cavanagh EMV, Inserra F, Ferder M, Ferder L. From mitochondria to disease: role of the renin-angiotensin system. Am J Nephrol [Internet]. 2007;27(6):545–53. Available from: https://www.karger.com/Article/FullText/107757.
Abais JM, Xia M, Zhang Y, Boini KM, Li P-L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal [Internet]. 2015 May;22(13):1111–29. https://doi.org/10.1089/ars.2014.5994.
Tang Y-S, Zhao Y-H, Zhong Y, Li X-Z, Pu J-X, Luo Y-C, et al. Neferine inhibits LPS-ATP-induced endothelial cell pyroptosis via regulation of ROS/NLRP3/Caspase-1 signaling pathway. Inflamm Res [Internet]. 2019;68(9):727–38. https://doi.org/10.1007/s00011-019-01256-6.
Grailer JJ, Canning BA, Kalbitz M, Haggadone MD, Dhond RM, Andjelkovic AV, et al. Critical role for the NLRP3 inflammasome during acute lung injury. J Immunol. 2014;192(12):5974–83.
Chen IY, Moriyama M, Chang MF, Ichinohe T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front Microbiol. 2019;10:1–9.
Yue Y, Nabar NR, Shi CS, Kamenyeva O, Xiao X, Hwang IY, et al. SARS-coronavirus open reading frame-3a drives multimodal necrotic cell death. Cell Death Dis. 2018. https://doi.org/10.1038/s41419-018-0917-y.
Shao BZ, Xu ZQ, Han BZ, Su DF, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol. 2015;6:1–9.
DeDiego ML, Nieto-Torres JL, Jiménez-Guardeño JM, Regla-Nava JA, Álvarez E, Oliveros JC, et al. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog. 2011. https://doi.org/10.1371/journal.ppat.1002315.
Indramohan M, Stehlik C, Dorfleutner A. COPs and POPs patrol inflammasome activation. J Mol Biol [Internet]. 2018;430(2):153–73. https://doi.org/10.1016/j.jmb.2017.10.004
Jo E-K, Kim JK, Shin D-M, Sasakawa C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol [Internet]. 20169;13(2):148–59. Available from: http://www.nature.com/articles/cmi201595
Madouri F, Guillou N, Fauconnier L, Marchiol T, Rouxel N, Chenuet P, et al. Caspase-1 activation by NLRP3 inflammasome dampens IL-33-dependent house dust mite-induced allergic lung inflammation. J Mol Cell Biol [Internet]. 2015;7(4):351–65. https://doi.org/10.1093/jmcb/mjv012.
McKee CM, Coll RC. NLRP3 inflammasome priming: A riddle wrapped in a mystery inside an enigma. J Leukoc Biol [Internet]. 20203;108(3):937–52. https://doi.org/10.1002/JLB.3MR0720-513R.
Huang P, Liu W, Chen J, Hu Y, Wang Y, Sun J, et al. TRIM31 inhibits NLRP3 inflammasome and pyroptosis of retinal pigment epithelial cells through ubiquitination of NLRP3. Cell Biol Int [Internet]. 2020;44(11):2213–9. https://doi.org/10.1002/cbin.11429.
Pan H, Lin Y, Dou J, Fu Z, Yao Y, Ye S, et al. Wedelolactone facilitates Ser/Thr phosphorylation of NLRP3 dependent on PKA signalling to block inflammasome activation and pyroptosis. Cell Prolif [Internet]. 2020;53(9):1–12. https://doi.org/10.1111/cpr.12868.
Magro G. COVID-19: review on latest available drugs and therapies against SARS-CoV-2. Coagulation and inflammation cross-talking. Virus Res. 2020. https://doi.org/10.1016/j.virusres.2020.198070.
Huet T, Beaussier H, Voisin O, Jouveshomme S, Dauriat G, Lazareth I, et al. Anakinra for severe forms of COVID-19: a cohort study. Lancet Rheumatol. 2020;2(7):e393-400.
Zhang X, Zhang Y, Qiao W, Zhang J, Qi Z. Baricitinib, a drug with potential effect to prevent SARS-COV-2 from entering target cells and control cytokine storm induced by COVID-19. Int Immunopharmacol. 2020. https://doi.org/10.1016/j.intimp.2020.106749.
Deftereos SG, Siasos G, Giannopoulos G, Vrachatis DA, Angelidis C, Giotaki SG, et al. The Greek study in the effects of colchicine in COvid-19 complications prevention (GRECCO-19 study ): Rationale and study design. Hellenic J Cardiol. 2020;61:42–5.
Montealegre-Gómez G, Garavito E, Gómez-López A, Rojas-Villarraga A, Parra-Medina R. Colchicine: A potential therapeutic tool against COVID-19. Experience of 5 patients. Reumatol Clínica. 2020 May;(xx):1–5. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1699258X20301078.
Schönrich G, Raftery MJ. Neutrophil extracellular traps go viral. Front Immunol. 2016;7:11–4. https://doi.org/10.3389/fimmu.2016.00366/full.
Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM, et al. Targeting potential drivers of COVID-19: neutrophil extracellular traps. J Exp Med. 2020;217(6):1–7.
Boeltz S, Amini P, Anders HJ, Andrade F, Bilyy R, Chatfield S, et al. To NET or not to NET: current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019;26(3):395–408.
Tall AR, Westerterp M. Inflammasomes, neutrophil extracellular traps, and cholesterol. J Lipid Res. 2019;60(4):721–7.
Chen KW, Demarco B, Broz P. Beyond inflammasomes: emerging function of gasdermins during apoptosis and NETosis. EMBO J. 2020;39(2):1–11.
Veras FP, Pontelli MC, Silva CM, Toller-Kawahisa JE, de Lima M, Nascimento DC, et al. SARS-CoV-2–triggered neutrophil extracellular traps mediate COVID-19 pathology. J Exp Med. 2020. https://doi.org/10.1084/jem.20201129/152086/SARSCoV2triggered-neutrophil-extracellular-traps.
Tomar B, Anders H-J, Desai J, Mulay SR. Neutrophils and neutrophil extracellular traps drive necroinflammation in COVID-19. Cells. 2020;9(6):1383.
Cicco S, Cicco G, Racanelli V, Vacca A. Neutrophil Extracellular Traps (NETs) and Damage-Associated Molecular Patterns (DAMPs): two potential targets for COVID-19 Treatment. Mediators Inflamm. 2020. https://doi.org/10.1155/2020/7527953.
Schönrich G, Raftery MJ, Samstag Y. Devilishly radical NETwork in COVID-19: oxidative stress, neutrophil extracellular traps (NETs), and T cell suppression. Adv Biol Regul. 2020. https://doi.org/10.1016/j.jbior.2020.100741.
Khandpur R, Carmona-rivera C, Vivekanandan-giri A, Yalavarthi S, Knight JS, Friday S, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. 2013. https://doi.org/10.1126/scitranslmed.3005580.
Med N. Diabetes primes neutrophils to undergo NETosis which severely impairs wound healing. Nat Med. 2015;21(7):815–9.
Colón DF, Wanderley CW, Franchin M, Silva CM, Hiroki CH, Castanheira FVS, et al. Neutrophil extracellular traps (NETs) exacerbate severity of infant sepsis. Crit Care. 2019;23(1):1–13.
Radermecker C, Detrembleur N, Guiot J, Cavalier E, Henket M, d’Emal C, et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J Exp Med. 2020. https://doi.org/10.1084/jem.20201012.
Shah A. Novel coronavirus-induced NLRP3 inflammasome activation: a potential drug target in the treatment of COVID-19. Front Immunol. 2020. https://doi.org/10.3389/fimmu.2020.01021.
Lara PC, Macías-Verde D, Burgos-Burgos J. Age-induced NLRP3 inflammasome over-activation increases lethality of SARS-CoV-2 pneumonia in elderly patients. Aging Dis. 2020;11(4):756.
Yap JKY, Moriyama M, Iwasaki A. Inflammasomes and pyroptosis as therapeutic targets for COVID-19. J Immunol. 2020. https://doi.org/10.4049/jimmunol.2000513.
Freeman TL, Swartz TH. Targeting the NLRP3 inflammasome in severe COVID-19. Front Immunol. 2020;11:1–12.
Liu Y, Zhang C, Huang F, Yang Y, Wang F, Yuan J, et al. Elevated plasma levels of selective cytokines in COVID-19 patients reflect viral load and lung injury. Natl Sci Rev. 2020;7(6):1003–11.
Huang KJ, Su IJ, Theron M, Wu YC, Lai SK, Liu CC, et al. An interferon-γ-related cytokine storm in SARS patients. J Med Virol. 2005;75(2):185–94.
Makabe H, Kojika M, Takahashi G, Matsumoto N, Shibata S, Suzuki Y, et al. Interleukin-18 levels reflect the long-term prognosis of acute lung injury and acute respiratory distress syndrome. J Anesth. 2012;26(5):658–63.
Fan E, Brodie D, Slutsky AS. Acute respiratory distress syndrome advances in diagnosis and treatment. JAMA J Am Med Assoc. 2018;319(7):698–710.
Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. Acute respiratory distress syndrome: the Berlin definition. JAMA J Am Med Assoc. 2012;307(23):2526–33.
Gibelin A, Parrot A, Maitre B, Brun-Buisson C, Mekontso Dessap A, Fartoukh M, et al. Acute respiratory distress syndrome mimickers lacking common risk factors of the Berlin definition. Intensive Care Med. 2016;42(2):164–72.
Kim WY, Hong SB. Sepsis and acute respiratory distress syndrome: recent update. Tuberc Respir Dis (Seoul). 2016;79(2):53–7.
Reiss LK, Schuppert A, Uhlig S. Inflammatory processes during acute respiratory distress syndrome: a complex system. Curr Opin Crit Care. 2018;24(1):1–9.
Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al.: Risk Factors Associated with Acute Respiratory Distress Syndrome and Death in Patients with Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern Med. 2020; 1-10
Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med. 2020;8(5):475–81. https://doi.org/10.1016/S2213-2600(20)30079-5.
Girija ASS, Shankar EM, Larsson M. Could SARS-CoV-2-Induced hyperinflammation magnify the severity of coronavirus disease (CoViD-19) leading to acute respiratory distress syndrome? Front Immunol. 2020;11:1206.
Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20(6):355–62.
Dolinay T, Kim YS, Howrylak J, Hunninghake GM, An CH, Fredenburgh L, et al. Inflammasome-regulated cytokines are critical mediators of acute lung injury. Am J Respir Crit Care Med. 2012;185(11):1225–34.
Rogers AJ, Guan J, Trtchounian A, Hunninghake GM, Kaimal R, Desai M, et al. Association of elevated plasma interleukin-18 level with increased mortality in a clinical trial of statin treatment for acute respiratory distress syndrome∗. Crit Care Med. 2019;47(8):1089–96.
Bortolotti P, Faure E, Kipnis E. Inflammasomes in tissue damages and immune disorders after trauma. Front Immunol. 2018;9:1–17.
Chen J, Wang S, Fu R, Zhou M, Zhang T, Pan W, et al. RIP3 dependent NLRP3 inflammasome activation is implicated in acute lung injury in mice. J Transl Med. 2018;16(1):1–12. https://doi.org/10.1186/s12967-018-1606-4.
Wang S, Zhao J, Wang H, Liang Y, Yang N, Huang Y. Blockage of P2X7 attenuates acute lung injury in mice by inhibiting NLRP3 inflammasome. Int Immunopharmacol. 2015;27(1):38–45. https://doi.org/10.1016/j.intimp.2015.04.035.
Chung KF. Cytokines in chronic obstructive pulmonary disease. Eur Respir J. 2001;18(1):50–9. https://doi.org/10.1183/09031936.01.00229701.
Yang W, Ni H, Wang H, Gu H. NLRP3 inflammasome is essential for the development of chronic obstructive pulmonary disease. Int J Clin Exp Pathol. 2015;8(10):13209–16.
Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest. 2007;117(12):3786–99.
Wilson MS, Madala SK, Ramalingam TR, Gochuico BR, Rosas IO, Cheever AW, et al. Bleomycin and IL-1β–mediated pulmonary fibrosis is IL-17A dependent. J Exp Med. 2010;207(3):535–52.
Lechowicz K, Drożdżal S, Machaj F, Rosik J, Szostak B, Zegan-Barańska M, et al. COVID-19: the potential treatment of pulmonary fibrosis associated with SARS-CoV-2 infection. J Clin Med. 2020;9(6):1917.
Triantafilou K, Triantafilou M. Ion flux in the lung: virus-induced inflammasome activation. Trends Microbiol. 2014;22(10):580–8. https://doi.org/10.1016/j.tim.2014.06.002.
Nieto-Torres JL, DeDiego ML, Verdiá-Báguena C, Jimenez-Guardeño JM, Regla-Nava JA, Fernandez-Delgado R, et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. 2014. https://doi.org/10.1371/journal.ppat.1004077.
Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated Type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. 2016;19(2):181–93. https://doi.org/10.1016/j.chom.2016.01.007.
Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181(5):1036-1045.e9. https://doi.org/10.1016/j.cell.2020.04.026.
Thachil J, Tang N, Gando S, Falanga A, Cattaneo M, Levi M, et al. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J Thromb Haemost. 2020. https://doi.org/10.1111/jth.14810.
Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145–7.
Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;135(23):2033–40.
Lippi G, Plebani M, Henry BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin Chim Acta. 2020;506:145–8. https://doi.org/10.1016/j.cca.2020.03.022.
Lippi G, Favaloro EJ. D-dimer is associated with severity of coronavirus disease 2019: a pooled analysis. Thromb Haemost. 2020;120(05):876–8. https://doi.org/10.1111/jth.14768.
Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18(4):844–7.
Chu AJ. Tissue factor, blood coagulation, and beyond: an overview. Int J Inflam. 2011. https://doi.org/10.4061/2011/367284.
Tian S, Xiong Y, Liu H, Niu L, Guo J, Liao M, et al. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod Pathol. 2020;33(6):1007–14. https://doi.org/10.1038/s41379-020-0536-x.
Menter T, Haslbauer JD, Nienhold R, Savic S, Hopfer H, Deigendesch N, et al. Postmortem examination of COVID-19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings in lungs and other organs suggesting vascular dysfunction. Histopathology. 2020;77(2):198–209.
Carsana L, Sonzogni A, Nasr A, Rossi RS, Pellegrinelli A, Zerbi P, et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect Dis. 2020;20(10):1135–40. https://doi.org/10.1016/S1473-3099(20)30434-5.
Rapkiewicz AV, Mai X, Carsons SE, Pittaluga S, Kleiner DE, Berger JS, et al. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: a case series. EClinicalMedicine. 2020. https://doi.org/10.1016/j.eclinm.2020.100434.
Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, et al. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell. 2005;122(3):379–91.
Hottz ED, Lopes JF, Freitas C, Valls-De-Souza R, Oliveira MF, Bozza MT, et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood. 2013;122(20):3405–14.
Wu C, Lu W, Zhang Y, Zhang G, Shi X, Hisada Y, et al. Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity. 2019;50(6):1401-1411.e4. https://doi.org/10.1016/j.immuni.2019.04.003.
Borthwick LA. The IL-1 cytokine family and its role in inflammation and fibrosis in the lung. Semin Immunopathol. 2016;38(4):517–34. https://doi.org/10.1007/s00281-016-0559-z.
Dinarello CA. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev. 2018;281(1):8–27.
Tahvildari M, Dana R. Low-Dose IL-2 therapy in transplantation, autoimmunity, and inflammatory diseases. J Immunol. 2019;203(11):2749–55. https://doi.org/10.4049/jimmunol.1900733.
Ju S-T, Sharma R, Gaskin F, Fu SM. IL-2 controls trafficking receptor gene expression and Th2 response for skin and lung inflammation. Clin Immunol. 2012;145(1):82–8.
Baran P, Hansen S, Waetzig GH, Akbarzadeh M, Lamertz L, Huber HJ, et al. The balance of interleukin (IL)-6, IL-6soluble IL-6 receptor (sIL-6R), and IL-6sIL-6Rsgp130 complexes allows simultaneous classic and trans-signaling. J Biol Chem. 2018;293(18):6762–75.
Tanaka T, Narazaki M, Kishimoto T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy. 2016;8(8):959–70.
Rex DAB, Agarwal N, Prasad TSK, Kandasamy RK, Subbannayya Y, Pinto SM. A comprehensive pathway map of IL-18-mediated signalling. J Cell Commun Signal. 2020;14(2):257–66.
Fenini G, Contassot E, French LE. Potential of IL-1, IL-18 and inflammasome inhibition for the treatment of inflammatory skin diseases. Front Pharmacol. 2017;8(MAY):1–20.
Mehta AK, Gracias DT, Croft M. TNF activity and T cells. Cytokine. 2018;101(1):14–8.
Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol. 2016;12(1):49–62.
Makris S, Paulsen M, Johansson C, Johansson C. Type i interferons as regulators of lung inflammation. Front Immunol. 2017;8:1–10.
Crow MK, Ronnblom L. Type i interferons in host defence and inflammatory diseases. Lupus Sci Med. 2019;6(1):1–10.
Kopitar-Jerala N. The role of interferons in inflammation and inflammasome activation. Front Immunol. 2017. https://doi.org/10.3389/fimmu.2017.00873.
Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. 2018;176(1):139–48.
Funding
This research was supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Brazil.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Consent to participate
All authors consented with development and participation of this research.
Consent for publication
The authors consented with publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Calado, M.B., da Silva Santana, C.E. & Crovella, S. Do inflammasome impact COVID-19 severity?. VirusDis. 32, 410–420 (2021). https://doi.org/10.1007/s13337-021-00705-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13337-021-00705-3