
Abstract
Monoclonal antibodies have become a mainstay in the treatment of patients with relapsing multiple sclerosis (RMS) and provide some benefit to patients with primary progressive MS. They are highly precise by specifically targeting molecules displayed on cells involved in distinct immune mechanisms of MS pathophysiology. They not only differ in the target antigen they recognize but also by the mode of action that generates their therapeutic effect. Natalizumab, an \(\alpha\)4\(\beta\)1 integrin antagonist, works via binding to cell surface receptors, blocking the interaction with their ligands and, in that way, preventing the migration of leukocytes across the blood–brain barrier. On the other hand, the anti-CD52 monoclonal antibody alemtuzumab and the anti-CD20 monoclonal antibodies rituximab, ocrelizumab, ofatumumab, and ublituximab work via eliminating selected pathogenic cell populations. However, potential adverse effects may be serious and can necessitate treatment discontinuation. Most importantly, those are the risk for (opportunistic) infections, but also secondary autoimmune diseases or malignancies. Monoclonal antibodies also carry the risk of infusion/injection-related reactions, primarily in early phases of treatment. By careful patient selection and monitoring during therapy, the occurrence of these potentially serious adverse effects can be minimized. Monoclonal antibodies are characterized by a relatively long pharmacologic half-life and pharmacodynamic effects, which provides advantages such as permitting infrequent dosing, but also creates disadvantages regarding vaccination and family planning. This review presents an overview of currently available monoclonal antibodies for the treatment of RMS, including their mechanism of action, efficacy and safety profile. Furthermore, we provide practical recommendations for risk management, vaccination, and family planning.
Similar content being viewed by others

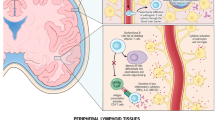
Introduction
Multiple sclerosis (MS) is an autoimmune inflammatory disease of the central nervous system (CNS) that affects about 2.8 million people worldwide [1]. The majority of patients (85%) initially follow a relapsing course (RMS), defined by acute exacerbations and periods of relative clinical stability in between [2]. Disability is accrued associated with relapses but occurs also independent of them. Over the last quarter of a century, an ever-increasing number of disease-modifying treatments (DMTs) has emerged, enabling effective reduction of disease activity, i.e., occurrence of relapses and T2-hyperintense lesions (T2L) contrast-enhancing lesions (CEL) on MRI, and to a lesser degree also disability progression [3]. As the disease course displays a considerable degree of both inter- and intra-individual variation, treatment choices depend on assessing the disease stage and judging the current level of disease activity. Across differing definitions, RMS may be classified as highly active based on the number and severity of relapses in the past 1–2 years, the number of new and/or enlarging T2L and/or Gd-enhancing lesions on MRI, or an insufficient response to treatment with one or more disease-modifying therapy (DMT) for at least one year [4]. Highly active RMS requires highly effective DMT (HET), which is almost exclusively achieved by monoclonal antibodies (mAb).
The advent of mAb has revolutionized treatment of MS due to their targeted mechanism, potent efficacy and favorable risk profile. They were originally developed from mice to prevent organ rejection in 1986; however, reactions to murine mAbs were soon associated with antidrug antibodies which led to the development of chimeric mouse-human mAbs [5, 6]. To minimize risks, particularly the risk of allergic or infusion-related reactions (IRRs), mAb have undergone several engineering generations to humanize their components in the last decades. This renders them less immunogenic and less likely to evoke generation of anti-drug antibodies. Also this increases clearance times [7]. The first-generation biologics were entirely murine in structure, sometimes leading to potentially fatal immune responses. Second-generation biologics were engineered as either chimeric (combining human Fc-regions with murine variable regions) or humanized (the variable region containing relatively more human protein). Third generation biologics are fully human mAb, yet these still appear to induce production of anti-human mAb. The mAb currently licensed for in MS have proven high efficacy in phase 3 studies and are therefore used in patients with high disease activity. Labels given by regulatory agencies in different countries vary. While all mAbs are approved to treat relapsing forms of MS in the USA, none of those are licensed in Europe for use in less active disease, based on weighing benefits vs. risks.
mAbs belong to the immunoglobulin G (IgG) isotype which bind specifically with their fragment antigen-binding (Fab) region to the epitope of the target molecule. The latter can either inhibit a specific function or directly induce an intracellular signaling. The binding of the fragment-crystallizable (Fc) region can lyse a target cell through either antibody-dependent cell-mediated (ADCC) or complement-dependent cytotoxicity (CDC) [8]. However, they differ not only in the target antigen they recognize but also regarding the mechanism by which they exert their therapeutic effect (Fig. 1). Natalizumab, for instance, works via binding to cell surface receptors, blocking interaction with their ligands and, thereby prevents the transition of leukocytes across the blood–brain barrier (BBB). On the other hand, alemtuzumab and the class of anti-CD20 mABs rituximab, ocrelizumab, ofatumumab, and ublituximab work via killing selected cell populations. Potential adverse effects may be serious and can necessitate treatment discontinuation. Such serious adverse events are the risk for (opportunistic) infections, autoimmune diseases or malignancies.

Mechanism of action of mAb in the treatment of multiple sclerosis. Rituximab, ocrelizumab, ofatumumab, and ublituximab target CD20 expressing lymphocytes B causing ADCC and CDC of circulating lymphocytes B. Alemtuzumab targets CD52 expressing lymphocytes, eosinophils, monocytes/macrophages, and dendritic cells, resulting in their rapid depletion. Natalizumab binds to \(\alpha\)4\(\beta\)1 integrin receptor on endothelial cells, preventing interaction between \(\alpha\)4\(\beta\)1 integrin and VCAM-1 and, therefore, inhibiting migration of leukocytes through the BBB into the CNS parenchyma. Created with BioRender.com. ADCC antibody-dependent cell-mediated cytolysis, APC antigen-presenting cell, CD cluster of differentiation, CDC complement-dependent cytolysis, IL interleukin, TGF-\(\beta\) transforming growth factor \(\beta\), TNF-\(\alpha\) tumor necrosis factor \(\alpha\), VCAM-1 vascular cell adhesion molecule 1
mAb are characterized by a relatively long pharmacologic half-life (IgG subclasses up to 30 days) and pharmacodynamic effects [9, 10], which provide advantages such as permitting infrequent dosing, but also create disadvantages regarding vaccination and family planning.
Here, we provide an overview of mAb for RMS treatment with a special focus on potential side effects and risk management, pregnancy and family planning, and vaccinations (Tables 1 and 2).
Natalizumab
Natalizumab is a humanized second-generation mAb that binds to \(\alpha\) 4 integrin receptors on endothelial cells lining blood vessels, disrupting the interaction of \(\alpha\)4\(\beta\)1 integrin (or very late antigen 4, VLA-4) expressed on lymphocytes and monocytes with its ligand vascular cell adhesion molecule 1 (VCAM1) on endothelial cells. Thereby, it inhibits migration of leukocytes through the BBB into the brain and spinal cord. While preventing invasion of autoreactive lymphocytes from peripheral blood into the CNS, cells are not depleted from the circulation. Rather, there is an increase in peripheral lymphocyte and monocyte counts during treatment (natalizumab-induced lymphocytosis, NIL) [11, 12]. Natalizumab is approved at a fixed dose of 300 mg administered intravenously or subcutaneously every 4 weeks (standard interval dosing; SID), allowing natalizumab concentrations to be maintained at levels which ensure continuous maximal \(\alpha\)4\(\beta\)1 integrin receptor saturation [13].
Pharmacology and Pharmacodynamics
The median relative bioavailability following intravenous and subcutaneous administration is 100% and 82.4%, respectively [14]. The median half-life of intravenous application of natalizumab is 27.1 days, with the subcutaneous absorption half-life being estimated to be approximately 2.6 days [14]. After absorption, the elimination phase for subcutaneous and intravenous administration parallels each other, suggesting comparable elimination characteristics [14].
Mean natalizumab serum concentrations are lower for extended interval dosing (EID) compared to SID (18.2 vs. 35.7 µg/ml, respectively); besides, \(\alpha\) 4-integrin receptor occupancy by natalizumab is lower for EID than SID (78.2% vs. 87.4%, respectively) [15]. As \(\alpha\) 4-integrin receptor saturation > 50% correlates with high clinical efficacy of natalizumab, at least 9 natalizumab infusions per year are required to maintain adequate trough saturation and concentrations levels [16]. The pharmacology of natalizumab is mostly affected by body mass index and dosing interval [17].
Clinical Trials
Both AFFIRM (Natalizumab Safety and Efficacy in RRMS) and SENTINEL (Safety and Efficacy of Natalizumab in Combination with Avonex [IFN \(\beta\)-1a] in Patients with RRMS) were phase 3 clinical trials assessing the efficacy of natalizumab in RMS [18, 19]. In the AFFIRM study, natalizumab significantly decreased annualized relapse rate (ARR) by 68% (p < 0.001) and lowered disability progression rates (sustained for 3 months) by 42% (p < 0.001) compared to placebo [18]. Additional analyses showed that over 2 years, natalizumab elicited a 92% and 83% decline in the number of Gd-enhancing lesions and the number of new or enlarging T2L, respectively (both p < 0.001). Besides, natalizumab also reduced the rate of brain atrophy during the second year of treatment [18, 20]. The drug is effective in patients with a more severe disease, and has been shown to have beneficial effects on visual function and several aspects of quality of life [21,22,23].
In the SENTINEL clinical trial, natalizumab plus interferon (IFN) \(\beta\)-1a significantly reduced the cumulative probability of 12-week confirmed disability progression (CDP) by 24% (p = 0.02) and decreased ARR by 55% compared with IFN \(\beta\)-1a alone (p < 0.001) [19].
Long-term data of natalizumab effectiveness from the Austrian MS Treatment Registry show a stable disease course regarding relapse activity and disease progression under natalizumab treatment for more than 7 years, with older age at natalizumab start (> 35 years) being the only significant risk factor for disease progression over long-term [24].
Safety and Adverse Effects
Although natalizumab is generally well-tolerated and safe, progressive multifocal leukoencephalopathy (PML) may occur as a serious, potentially life-threatening adverse effect.
After natalizumab was first approved in 2004, reported cases of PML led to a withdrawal in 2005 and subsequently reintroduction in 2006 with the establishment of an advisory committee that would monitor patients on natalizumab. PML is an acute or subacutely developing demyelinating disease caused by the John Cunningham virus (JCV) that leads to a lytic destruction of oligodendrocytes. Infection is very frequent but most commonly asymptomatic. It usually occurs during childhood and JCV remains latent until a possible reactivation by mutation of the virus, which remains a very rare event. The presence of anti-JCV antibodies as an indirect footprint of infection, duration of natalizumab exposure (particularly beyond 2 years), and immunosuppressant use prior to receiving natalizumab are all risk factors associated with an increased risk of PML [25]. Anti-JCV antibody-negative patients have an estimated PML risk of 0.07/1000 patients, whereas in anti-JCV antibody-positive patients, estimated cumulative PML probability over 6 years is 2.7% and 1.7% in patients with and without previous immunosuppressive therapy, respectively [26].
Monitoring and Screening
A comprehensive and exemplary scientific effort by the MS community yielded a clinically applicable risk stratification model. Special anti-JCV antibody index has been developed to predict the risk of PML with people with an antibody index of ≤ 0.9 having an annual PML risk of 0.6/1000, an index of 0.9–1.5 having a risk of 3.0/1000, and an index of > 1.5 having a risk of 10.0 /1000 in 6 years [26]. Re-testing of anti-JCV antibody negative patients every 6 months is recommended [27]. However, patients should not be tested for anti-JCV antibodies within 2 weeks of plasmapheresis given removal of antibodies from the serum, or within 6 months of IVIg [27]. Frequent MRI monitoring (on a 3–6 monthly basis) is recommended for patients who either have all three risk factors (anti-JCV antibody positive, ≥ 2 years of natalizumab treatment, prior immunosuppressant therapy) or patients with a high anti-JCV antibody index who have received at least 2 years of natalizumab treatment without prior immunosuppressant therapy [27]. The extension of the dosing interval from 4 to 6 weeks (EID) has been associated with lower incidence of PML, and no negative effect on efficacy evidenced by ARR, disability progression and MRI activity [28, 29]. Recently published data show that the proportion of relapse-free patients at 72 weeks (97.6% vs. 96.9%), proportion of patients free of disability worsening (92.0% vs. 90.0%), and proportion of patients with No Evidence of Disease Activity (NEDA) (67.4% vs. 70.0%) do not differ between the intervals of application (4 vs. 6 weeks, respectively) [30,31,32]. Also, real-world evidence indicates equivalent efficacy of SID and EID with EID being safe and well tolerated for over 7 years [33].
Special caution should be exercised when washing out natalizumab in the setting of CNS infection such as PML, since the immune reconstitution inflammatory syndrome (IRIS) in response to viral antigen in the brain may be robust and cause worsening or even death [34].
Pregnancy and Breastfeeding
Natalizumab is classified as a pregnancy category C drug as potential fetal effects have been reported in animal studies [35,36,37]. However, there is a risk of reactivation or even rebound of disease activity after natalizumab cessation, which is of particular importance in the first trimester and during the first 3 months postpartum, where disease activity is not yet or not anymore diminished by the effects of pregnancy itself [38,39,40,41]. The risk of relapse and disability progression during pregnancy is predicted by pre-conception relapse activity, higher EDSS at conception, use of HET and prolonged washout period [42]. Re-initiating natalizumab administration within 4 weeks after delivery in women without a relapse in the year pre-conception on HET is associated with a ninefold decreased risk for relapse and disability progression postpartum [42]. Thus, there is a clear rationale for continuing natalizumab at least until pregnancy occurs, or in patients with higher disease activity even during pregnancy as antibodies, including natalizumab, only minimally cross the placenta during the first trimester [43,44,45].
Even though evidence of safety during natalizumab continuation is limited, various expert guidelines incorporated these recommendations. They suggest to continue natalizumab at least until pregnancy is confirmed and, depending on an individual benefit-risk-assessment even until 32–34 weeks of gestation with EID. Natalizumab administration should be resumed as soon as possible after delivery [46, 47].
With reference to the Tysabri Pregnancy Exposure Registry, 355 pregnancy outcomes were analyzed after exposure to natalizumab 3 months before conceiving or during pregnancy. The rate of birth defects and spontaneous abortions was found to be similar to that of the general population [48]. The same findings were obtained in a retrospective analysis from the Austrian MS Treatment Registry [49]. However, in one case series study, mild to moderate thrombocytopenia and anemia were detected in 10 of 13 newborns when natalizumab was prescribed in the third trimester of gestation [50]. It is, therefore, mandatory to test all exposed newborns for thrombocytopenia and anemia [51].
As natalizumab is excreted in breast milk, the SmPC states that breastfeeding should be discontinued during treatment with natalizumab [27]. However, natalizumab concentrations in breast milk are low and large molecules such as natalizumab are most likely destroyed in the infants` gastrointestinal tract. Thus, treatment with natalizumab can be also considered during breast-feeding [47].
Vaccination
According to EMA, inactivated vaccines can be given to patients receiving natalizumab, whereas live and live-attenuated vaccines have not been studied in those patients and should, therefore, be avoided [27]. There is little evidence on the vaccine response in patients receiving natalizumab. One study confirmed a significant increase in anti-influenza A and B titer after the vaccination in both treated patients and HC, with a lower antibody response to the H3N2 strain [52,53,54]. Another study demonstrated no difference between immunization response to tetanus toxoid in the presence of natalizumab [55]. Currently available data also indicates comparable humoral immune response to SARS-CoV2 vaccines in patients on natalizumab and healthy controls without the need to discontinue the treatment [56,57,58]. Therefore, vaccination in patients treated with natalizumab seems to elicit a sufficient immune response.
Alemtuzumab
Alemtuzumab is a humanized second-generation mAb that binds the CD52 glycoprotein present on lymphocytes, eosinophils, monocytes/macrophages, and dendritic cells but not on hematopoietic progenitors, erythrocytes, or platelets, and elicits rapid depletion of CD52 expressing cells [59, 60]. The function of CD52 is not well understood, but evidence suggests that it may be involved in T cell co-stimulation and migration [61]. The dosing consists of 5 consecutive days of infusions at treatment initiation followed by 3 consecutive days of infusions 12 months later, with optional additional courses per approved local labels [62].
Alemtuzumab was the first monoclonal antibody used for therapeutic purposes. Originally, FDA approved it in 2001 for use in B-cell chronic lymphocytic leukemia. It became FDA-approved for use in RMS in 2014. However, because of the risk of autoimmune disorders and due to rare but severe vascular effects, its use has been recommended to be restricted to patients who have failed at least two other DMT approved for RMS.
Pharmacology and Pharmacodynamics
Following cell surface binding of alemtuzumab to lymphocytes, alemtuzumab results in the depletion of circulating CD52-positive cells in a rapid manner, and the proposed mechanism of lymphocyte depletion includes both antibody-dependent cell-mediated cytolysis (ADCC) and complement-dependent cytolysis (CDC) [60, 63, 64]. As alemtuzumab is administered intravenously, its bioavailability is 100%. It does not cross cell membranes and is expected to distribute between the plasma and interstitial space. Its half-life is approximately 4–5 days and low or undetectable serum concentrations were measured within 30 days after last infusion [62].
Alemtuzumab induces a prolonged lymphopenia, with B-cell counts returning to the lower limits of normal (\(\ge\) 0.1 \(\bullet\) 109/l) within 7 months, CD8 + cell counts (\(\ge\) 0.2 \(\bullet\) 109/l) within 20 months, and CD4 + cell counts (\(\ge\) 0.4 \(\bullet\) 109/l) within 35 months; however, T-cell counts rarely recover to their pretreatment levels [65, 66]. A hyper-repopulation of immature B cell clones to 160–180% of baseline levels is observed at 3–6 months [67]. The peculiar reconstitution of the B-cell compartment has been suggested to be at the root of the development of secondary autoimmunity that is frequently observed in alemtuzumab-treated patients.
However, lymphopenia in absolute number does not seem to be the driving force behind alemtuzumab’s efficacy and safety profile; besides, the rate of lymphocyte count reconstitution seems to be unrelated to relapse risk, infection, or secondary autoimmunity [68, 69]. Moreover, the distinctive pattern of repopulation that begins within weeks and continues over time indicates a possible rebalancing of the immune system, which persists beyond the actual course of treatment. Alemtuzumab treatment results in a relative increase of cells with memory and regulatory phenotypes and a decrease in cells with a proinflammatory signature, and therefore, further promotes an immunoregulatory environment through an impact on other innate immune cells (e.g., dendritic cells) that play a role in MS pathogenesis [70, 71].
Clinical Trials
The efficacy and safety of alemtuzumab compared to that of IFN \(\beta\)-1a was shown in two phase 3 randomized, controlled, clinical trials called CARE-MS I and CARE-MS II. CARE-MS I enrolled only treatment-naïve patients [72, 73]. Alemtuzumab significantly decreased ARR (49.4–54.9%): It was associated with a significant reduction in 6-month CDP in CARE-MS II but not in CARE-MS I [72, 73]. Alemtuzumab was superior to IFN \(\beta\)-1a in reducing the number of Gd-enhancing lesions (9% vs. 23% at year 2, respectively) and new or enlarging T2L (46% vs. 68%, respectively) in both studies [74]. Besides, there were higher proportions of patients free from disease activity during the second year of therapy in the alemtuzumab-treated group in both studies (50% vs. 30–40%). Alemtuzumab also diminished the extent of brain atrophy over 2 years by 40% and 25% in CARE-MS I and CARE-MS II, respectively (p < 0.001 and p = 0.012).
Furthermore, durable efficacy was demonstrated throughout the extension studies, with 62% of patients having NEDA, and the majority of patients (50–68.5%) not requiring retreatment with alemtuzumab or another DMT for 9 years [75,76,77,78]. Imaging data of alemtuzumab-treated patients in exploratory studies have demonstrated potential neuroprotective effects, with increased retinal nerve fiber layer thickness consistent with reduced neurodegeneration, increased myelin water fraction suggestive of remyelination, and stabilized magnetization transfer ratio indicating preserved myelination [79,80,81,82].
Safety and Adverse Effects
In the clinical trials, several adverse effects were reported, with infusion-associated reactions being the most common, occurring in more than 90% of participants [83]. Infusion-associated reaction comprises symptoms like headache, rash, fever, nausea, vomiting, and myalgia, which are part of the so-called cytokine release syndrome and decrease in their occurrence and severity over the course of repeated infusion [84]. They occur within 2–6 h after alemtuzumab infusion. The introduction of high dose methylprednisolone intravenously before alemtuzumab infusion has dramatically reduced infusion-associated reactions [85].
Among side effects, infections were mostly mild or moderate due to the preservation of the innate immune system, with a peak after the first course (66–77%) and declining over time [86,87,88]. The most common infections reported in Clinical trials were upper and lower respiratory tract infections (nasopharyngitis, sinusitis, flu, bronchitis, pneumonia), masticatory and digestive tract infections (oral herpes, dental infections, gastroenteritis, appendicitis), infection of the urinary tract, and superficial fungal infections (oral and vaginal candidiasis) [89].
A rare but serious infection that has been associated with alemtuzumab is listeriosis, an infection with Gram-positive bacteria Listeria monocytogenes, which is usually contracted from unpasteurized dairy products, raw fish and meat, and soft cheeses. Immunocompetent persons rarely develop severe symptoms, whereas defective cellular immunity or pregnancy increase the risk of developing septicemia, meningitis or encephalitis with a mortality rate 20–40% [90, 91]. Furthermore, alemtuzumab administration has been associated with higher rates of HSV infections, sometimes even requiring hospitalization, and VZV infections [72, 73]. Therefore, FDA-approved product label recommends prophylaxis with acyclovir from the start of treatment until CD4 + lymphocytes recover to at least 200 cells/µl, with a minimum duration of prophylaxis of 2 months even if CD4 + lymphopenia resolves earlies [62]. In order to reduce the risk of L. monocytogenes infection, patients are advised to keep a Listeria-free diet at least 2 weeks before, during, and 1 month after each infusion [62]. If prophylactic measures are insufficient or unattainable, antibiotic prophylaxis with trimethoprim/sulfamethoxazole should be considered for the period of 1 month after the last infusion. Although serious opportunistic infections have been observed, they occur very rarely [92].
Development of autoimmune diseases is probably the most relevant risk of the treatment with alemtuzumab. Although it remains unclear why only a subset of patients develops autoimmune side effects, hyperrepopulation of B lymphocytes is likely to be a major driver [93]. Elevated levels of interleukin (IL) 21 have been suggested to be predictive of this secondary autoimmunity but this remains contentious [94, 95]. Secondary autoimmune disorders can occur up to 5 years after treatment with a frequency peak at 12–18 months [93]. The most commonly reported autoimmune adverse effect is thyroid dysfunction with either hyper- or hypothyroidism, reported in approximately 36% of patients in a 4-year follow-up of the CARE-MS I and CARE-MS II trials [96]. In the case of hypothyroidism, thyroid hormone replacement therapy should be considered, with patients monitored every 4–8 weeks to adjust thyroid hormone dosages. Hyperthyroidism following alemtuzumab treatment is most likely due to Graves’ disease and should be managed initially with anti-thyroid medication which has been associated with a high likelihood of remission. Thyroidectomy or radioactive iodine would only be indicated following failure of anti-thyroid medication. Where subacute painless thyroiditis is suspected, β-adrenergic blockers or corticosteroids in severe cases may be considered, but not anti-thyroid medications as thyroid hormone synthesis in those patients is already low [96, 97].
Immune thrombocytopenic purpura (ITP) is also one of the potential autoimmune conditions and has been detected in approximately 2% of patients. It is in most instances responsive to first-line therapy with corticosteroids, platelet replacement, and/or intravenous immunoglobulins [98, 99]. Apparently, the risk of this complication is not further increased in the subset of patients receiving additional alemtuzumab beyond the initial two courses [100]. Rarely, nephropathies such as Goodpasture disease with anti-glomerular basement membrane (anti-GBM) antibodies also occur [101].
In a recently published study, five patients received at least one infusion of low-dose rituximab following alemtuzumab treatment, with none of them developing secondary autoimmune disorders [98]. This speaks in favor of the imbalance in B- and T-cell regulatory networks during immune reconstitution as the driving force of autoimmune disorders following alemtuzumab treatment.
In the postmarketing surveillance phase, additional serious safety concerns of cardiovascular complications were identified [102, 103]. Among those, cardiac ischemia and myocardial infarction (2.0/10,000), ischemic and hemorrhagic stroke (3.6/10,000), arterial dissection (1.6/10,000), pulmonary hemorrhage and embolism (4.3/10,000), and vasculitis seem to be those of greatest concern [62, 92, 104]. The underlying pathophysiology remains to be elucidated. Cytokine-release syndrome caused by increased levels of serum tumor necrosis factor (TNF), IFN, and IL-6, leading to vasospasm or transient myocardial dysfunction have been pathomechanistically invoked [105, 106]. Another potential explanation could be direct cardiac myocytotoxicity causing myocyte dysfunction or electrical disturbances [107,108,109].
Beyond well-known adverse effects, rarer but still significant serious adverse events have been reported in patients during and following alemtuzumab treatment, e.g., exacerbated CNS inflammation with tumefactive demyelination, acute cholecystitis, vasculitis, sarcoidosis, listeria meningitis and meningoencephalitis, hemolytic anemia, hemophagocytic lymphohistiocytosis, opportunistic infections, and acute pneumonitis and pericarditis [110,111,112,113,114,115,116,117,118,119,120,121,122].
Several cases of malignancy have also been reported in patients receiving alemtuzumab, but causality is not established. It may represent a random finding because of effective monitoring bias [123]. Reported malignancies encompass papillary thyroid cancer, basalioma, non-EBV-associated Burkitt’s lymphoma, breast cancer, and cancer of the uterus.
Monitoring and Screening
On the basis of reported side effects recommendations have been formulated. Baseline routine screening of blood (thyroid panel, cell count inclusive of CD4/CD8 ratio, liver function tests, basic metabolic panel, HIV, HBV, HCV, VZV, and \(\beta\)-HCG), dermatologic examination and urinalysis within 30 days prior to the first infusion should be conducted. Thereafter, cell counts (inclusive of CD4/CD8 ratio), TSH, creatinine should be determined and urinalysis performed every month, and dermatologic examination performed yearly for 5 years after the last treatment cycle.
Prophylaxis with oral antiviral (acyclovir) is commenced one week prior to the first infusion and discontinued when CD4 count \(\ge\) 200, and listeria prophylaxis with listeria-free diet or co-trimoxazole is recommended. Patients are also pretreated with steroids, antihistamines and acetaminophen on infusion day.
Pregnancy and Breastfeeding
Alemtuzumab is classified as a pregnancy category C drug, as alemtuzumab was embryolethal in pregnant huCD52 transgenic mice when administered during organogenesis [62]. According to the Summary of Product Characteristics (SmPC), serum concentrations of alemtuzumab are low or undetectable within 30 days of each treatment course [62]. Therefore, women of childbearing potential should use effective contraception when receiving a course of alemtuzumab, and for 4 months following each course of treatment [124]. A study analyzing pregnancy outcomes in women treated with alemtuzumab, reported 66% healthy live births, 22% spontaneous abortions, 11% elective abortions, and 0.6% stillbirth (n = 167) [125]. Maternal age seemed to be associated with an elevated risk of spontaneous abortion (relative risk [RR] 2.46 in patients \(\ge\) 35 years) [126]. However, the risk of spontaneous abortion was not increased in patients becoming pregnant \(\le\) 4 months versus > 4 months since alemtuzumab exposure (19% vs. 23%, RR 1.08) [126]. The risk of autoimmune thyroid disease remains increased for 4 years after completing alemtuzumab treatment, therefore thyroid function should be tested regularly in newborns [124, 127].
Although it is unclear whether alemtuzumab is excreted in human breast milk, it falls into class C category as it has been detected in the milk of lactating mice. Hence, women should be advised to discontinue breastfeeding during each course of treatment, and for at least 4 months after each course [128].
As a cyclically administered treatment, alemtuzumab may be considered in women with very high disease activity and without acute plans to become pregnant.
Vaccination
According to EMA and FDA label, inactivated vaccines can be given to patients receiving alemtuzumab, whereas live and live-attenuated vaccines have not been studied in those patients and should, therefore, be avoided [128]. The SmPC suggests that vaccination before alemtuzumab should be considered in patients who have not completed standard required vaccinations, and for those without immunity to chickenpox [62]. Required vaccinations should be given at least 6 weeks before treatment [62].
The ability to mount effective immune responses to vaccines following alemtuzumab has not been studied extensively. Diphtheria, tetanus, poliomyelitis, and pneumococcus vaccines have been shown to evoke a normal T-cell response upon administration in patients treated with alemtuzumab despite the relatively prolonged T- and B-cell suppression [129, 130]. However, one patient was vaccinated within two months of alemtuzumab treatment and developed a poor response to several vaccines, suggesting immunization very early after alemtuzumab may not be effective [129]. Currently available data also indicates nearly normal humoral immune response to SARS-CoV2 vaccines in patients on alemtuzumab and healthy controls, depending on lymphocyte counts and time since last application of alemtuzumab [56, 58, 131].
B-Cell Depletion Therapy
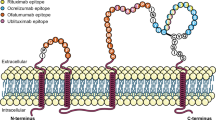
mAbs targeting CD20-expressing lymphocytes B represent an important treatment option for patients with MS. Spared from anti-CD20 lysis are stem cells (pro-B cells), many plasmablasts, and terminally differentiated antibody-producing plasma cells [132]. Anti-CD20 mAb further differ in their structure (chimeric, humanized, fully human), relative potency to drive ADCC and CDC, route of administration (intravenous or subcutaneous), pharmacokinetics, and required infusion times (Table 3) [133]. Three anti-CD20 mAbs are currently available with ocrelizumab and ofatumumab labeled for treatment of MS and rituximab frequently used off-label. Another one, ublituximab, is expected to be approved in 2022.
Rituximab
Rituximab is a second-generation chimeric mouse-human anti-CD20 mAb that was approved in 1997 for B-cell lymphoma but is being used off-label in several neurological diseases, including neuromyelitis optica spectrum disorder (NMOSD), myasthenia gravis, and MS. Several different protocols of rituximab dosage have been used, with patients being most commonly treated with 500 or 1000 mg rituximab intravenously every 6–12 months, in some cases after two initial application held 2 weeks apart [134,135,136].
Pharmacology and Pharmacodynamics
Rituximab works primarily through CDC of B cells but also has significant ADCC activity. Due to its intravenous route of application, its bioavailability is 100%. The replenishment of B cells is subject to individual variability, with a study with 26 RRMS patients showing a reconstitution to a mean of 35% of baseline counts by week 72, with the vast majority being naïve B cells rather than memory B cells [137]. The elimination half-life for intravenous rituximab 2 times 1000 mg administered 2 weeks apart is around 20 days but depends on sex, body weight, and renal clearance [138].
Clinical Trials
In spite of the overall positive efficacy with only rare serious adverse events, rituximab was never tested in phase 3 trials for efficacy in RMS. However, there is growing evidence from real-world evidence studies strengthening the case for rituximab as a potent treatment option for RMS [135, 139, 140].
IRRs are relatively common with use of rituximab in MS, appearing in 67.1–78.3% of treated patients after first infusion compared to 23.1–40.0% placebo-treated patients [141, 142]. However, they decrease to placebo levels with successive infusions, are only mild to moderate in severity, and include fever, rush, chills, throat irritation, nausea, headache, cough, tiredness, headache, hypotension, bronchospasm, or angioedema [141, 143].
Besides, treatment with rituximab is associated with an increased risk of infections [144]. Serious infections occur in 4.5% of treated patients compared to < 1.0% in placebo-treated patients with no clear association to the number of infusions [141]. Patients treated with rituximab should be screened for hypogammaglobulinemia and neutropenia, as these may present independent risk factors for developing infections [145, 146].
There is an increased PML risk with rituximab treatment (adjusted odds ratio = 3.22), but lower when compared to that of natalizumab [147]. Beside PML, reactivation of other latent infections such as tuberculosis, hepatitis, or HIV upon rituximab treatment has been reported [148, 149]. Therefore, patients should be thoroughly screened for such infections prior to rituximab treatment.
A low frequency of all types of malignancies was reported for rituximab in MS patients, which did not differ significantly from the general population (26.6 vs. 28.9 per 10,000 patient years, respectively) [150].
Monitoring and Screening
Cases of hepatitis B reactivation have been reported in subjects receiving anti-CD20 mAb [151]; therefore, HBV screening should be performed in all patients before initiation of treatment (HBsAg, HBcAg) [152]. Patients with active hepatitis B disease should not be treated with rituximab [153].
Apart from routine laboratory tests, baseline immunoglobulin levels should be determined as a reduced baseline level of IgG has been associated with higher risk for severe infections with rituximab in patients with rheumatoid arthritis [144]. Currently, there is no evidence to suggest monitoring anti-JCV antibodies in patients on rituximab.
Pregnancy and Breastfeeding
Rituximab is classified as a pregnancy category C drug as there are no adequate and well-controlled studies of rituximab in pregnant women [153]. Although at least a period of 6–12 months (FDA/EMA) after the last injection of rituximab is recommended before conceiving, a study analyzing 90 live birth outcomes of women inadvertently conceiving during or less than 12 months after the treatment of rituximab reported 22 premature births, one neonatal death after 6 weeks, 11 newborns with hematological changes (B-cell deficiency, neutropenia, thrombocytopenia, anemia, and lymphopenia), and two inborn malformations [154]. Besides, a recent systematic review and case series of MS and NMOSD patients assessed the safety of rituximab before and during pregnancy, with no major safety signal being found with rituximab use withing 6 months of conception [155]. However, as anti-CD20 mAbs can be actively transported across placental barrier and subsequently deplete fetal B cells, women are advised to use effective contraception for at least 3–4 months after the last rituximab infusion [47, 155, 156].
A case report from a breastfeeding patient found 0.42% of rituximab serum concentration in the milk, and similar concentrations were found in monkeys as well (0.19–0.26%) [157, 158]. As IgG is degraded in the gut of newborns, administration of rituximab is highly unlikely to pose clinically relevant risk for the infant, but any recommendation regarding its use in breastfeeding women should await more safety data [157]. To avoid potential harm to the newborn, women are still advised not to breastfeed during and up to 6 months after discontinuing the treatment.
Vaccination
EMA and FDA labels allow inactivated vaccines to be given to patients receiving rituximab, whereas live and live-attenuated vaccines have not been studied in those patients and should, therefore, be avoided [153].
Response to vaccination in patients receiving rituximab was only studied in non-MS populations. Patients with rheumatoid arthritis showed a reduced response to pneumococcal vaccine when treated with both rituximab and methotrexate compared to methotrexate alone (57% vs. 82%, respectively) [159].
Available data indicates significantly reduced humoral immune response to SARS-CoV2 vaccines (15–60% developing antibodies) in patients on rituximab compared to healthy controls, depending on B cell counts and time since last application [56, 58, 160]. Moreover, the development of a humoral immune response remains rare in seronegative patients with MS on anti-CD20 mAb even after a third dose of SARS-CoV-2 vaccine unless patients have measurable B-cell counts [161]. However, there is growing evidence that T cell responses may be preserved or even augmented under anti-CD20 mAbs, potentially mitigating the loss of antibody-mediated vaccine efficacy [162, 163].
Therefore, every patient considered for rituximab therapy should receive all indicated vaccines (hepatitis B for at-risk population, pneumococcus, tetanus toxoid every 10 years, influenza annually) before treatment. Ideally, vaccination should be undertaken at least 4 weeks before treatment initiation [164].
Ocrelizumab
Ocrelizumab is a second-generation recombinant humanized mAb targeting CD20-expressing B cells that is approved for RMS and primary progressive MS [165]. According to the label, it is administered with two starting doses of 300 mg 2 weeks apart, and after that 600 mg every 6 months. Patients should be premedicated at least 30–60 min prior ocrelizumab infusion with 100 mg methylprednisolone and an antihistamine in order to avoid IRRs. Patients need to be observed for at least 60 min following ocrelizumab infusion [166].
Pharmacology and Pharmacokinetics
Ocrelizumab binds to the extracellular loop of CD20 causing ADCC and CDC of circulating B cells [143, 166]. Interestingly, recent studies have shown that ocrelizumab may also target CD20 + T cells, which are present in low frequencies in MS patients, suggesting an alternative contributing mechanism of action [167]. As it is administered intravenously, its bioavailability is 100%. The median time to B-cell repletion was 72 weeks, with 90% of patients reaching pre-treatment levels by approximately 2.5 years after the last infusion [168]. Ocrelizumab has a half-life of 26 days [165]. It is expected to enter the metabolic pathway of endogenous antibodies; in that way, no studies concerning its metabolism and elimination were performed [168].
Clinical Trials
The efficacy and safety of ocrelizumab versus IFN \(\beta\)-1a for the treatment of RMS was reported in two phase 3 clinical trials named OPERA I and OPERA II [169]. In both trials, treatment with ocrelizumab lowered ARR (0.16 vs. 0.29; p < 0.001), and led to lower percentage of patients with CDP at 12 weeks (9.1% vs. 13.6%, p < 0.001) and lower number of Gd-enhancing lesions. 47.9% and 47.5% of ocrelizumab-treated patients (OPERA I and II, respectively) had no evidence of disease activity at 96 weeks compared to 29.2% and 25.1% on IFN \(\beta\)-1a, respectively [170].
Safety and Adverse Effects
IRRs occurred in 34.3% of the treated patients with ocrelizumab (vs. 9.7% on IFN \(\beta\)-1a); a shorter infusion period (2 h instead of 3.5 h) did not increase the risk of IRRs in one recently published study [169, 171]. Although CDC activity was believed to play an important role in triggering infusion-related reactions, IRRs seem to be mainly associated with cytokine release by immune cells (lymphocytes B and natural killers) [172, 173]. Current recommendations to reduce the risk of an IRR include premedication with methylprednisolone and an antihistamine [171].
The most common adverse events are infections with the overall rate of 84.5% in the period up to 8 years [174,175,176]. Most common infections were upper respiratory tract infections (predominantly nasopharyngitis) and urinary tract infections. Serious infections occurred in 1.3% of patients treated with ocrelizumab (vs. 2.9% on IFN \(\beta\)-1a) [169]. Approximately 30% of patients show hypogammaglobulinemia, which significantly increases infection risk.
Neoplasms occurred in 1.1% of patients treated with ocrelizumab and 0.4% patients treated with IFN \(\beta\)-1a [169, 174]. From those, 6 were breast cancer cases, while no such cases were observed in the placebo or IFN \(\beta\)-1a group. The total number of patients with breast or other cancers in the ocrelizumab-treated populations was, however, not higher than expected as background from epidemiological studies of the general population. Also, the incidence of cancer has fallen during the subsequent open-label extension studies [175, 177].
Currently, 8 cases of PML have been identified in patients treated with ocrelizumab, which were judged related to previous treatment with natalizumab or fingolimod, while one PML case was considered to be directly associated with ocrelizumab treatment as the patient had no prior DMT exposure (progressive MS) [175, 178].
Monitoring and Screening
Cases of hepatitis B reactivation have been reported in subjects receiving anti-CD20 mAb; therefore, HBV screening should be performed in all patients before initiation of treatment (HBsAg, HBcAg) [151]. Patients with active hepatitis B disease should not be treated with ocrelizumab [168]. Cases of late-onset neutropenia have been reported, with the majority being reported at least 4 weeks after last ocrelizumab infusion (grade 1 or 2). In patients with signs and symptoms of infection, measurement of blood neutrophils is recommended.
Given the observation of malignancies in the pivotal trials, patients with a known active malignancy should not be treated with ocrelizumab, and every patient should follow standard breast cancer screening per local guidelines [168]. There is no evidence to support monitoring anti-JCV antibodies in patients treated with ocrelizumab.
Pregnancy and Breastfeeding
Ocrelizumab is classified as a pregnancy category C drug as there are no adequate data on the developmental risk associated with the use of ocrelizumab in pregnant women [168]. According to EMA, animal studies do not indicate teratogenic effects of ocrelizumab, but B cell depletion was detected in utero and reproductive toxicity was observed in pre- and post-natal development studies [168]. Besides, ocrelizumab is a humanized mAb of an immunoglobulin G1 subtype that is known to cross the placental barrier. Therefore, women of childbearing potential are advised to use contraception while receiving ocrelizumab and for 12 months after the last infusion [168].
As this label appears very conservative given the available pharmacological data, a growing number of experts and guidelines recommends women to use effective contraception for at least 3–4 months after the last ocrelizumab infusion [47, 155].
Recently, a study of the German MS and Pregnancy Registry was published claiming B cells to be normal in infants breastfed by mothers receiving anti-CD20 mAb [179]. However, women are still advised to discontinue breastfeeding during ocrelizumab therapy [168].
Vaccination
Current EMA and FDA labelling allows application of inactivated vaccines to patients receiving ocrelizumab. Live or live-attenuated vaccines have not been studied in those patients and should, therefore, be avoided during treatment and until B-cell repletion [168]. The VELOCE study, which evaluated the effects of ocrelizumab on immune response to various vaccines in patients with RMS, confirmed that patients treated with OCR can mount humoral responses, albeit attenuated, to the inactivated vaccine studied (tetanus, pneumococcal and influenza vaccine) [180]. Available data indicates significantly reduced humoral immune response to SARS-CoV2 vaccines in patients on ocrelizumab compared to healthy controls, depending on B cell counts and time since last application [56, 58, 160, 164, 181, 182]. Therefore, some authors suggest extending dosage intervals in order to improve chances of building up a sufficient immune response [183]. However, it has to be kept in mind that many patients (37–53%) still develop humoral response under ocrelizumab and that T cell response seems to be unaffected under B cell depletion [58, 184,185,186].
Patients should be reviewed for their immunization status before embarking on treatment with ocrelizumab. Patients who require vaccination should complete it at least 6 weeks prior to treatment initiation. It is recommended to vaccinate patients with ocrelizumab with seasonal influenza vaccines that are inactivated [168].
Ofatumumab
Ofatumumab is a fully human mAb targeting CD20-positive B cell lineage cells but recognizing a different epitope than either rituximab or ocrelizumab. It was originally approved by the FDA in 2009 for use in chronic lymphocytic leukemia but has been also approved for use in MS in 2020.
Dosage and Administration
Its notable strength is its subcutaneous application with an auto-injector pen which is administered at four-week intervals with the first three doses delivered on days 1, 8, and 15. Despite its differing route of application, it does not seem to be inferior to other mAbs used in treatment of MS [187].
Pharmacology and Pharmacokinetics
Ofatumumab binds to an epitope encompassing both small and large loops of the extracellular domain of the CD20 protein, causing ADCC and CDC of circulating B cells [188]. The mechanisms are similar to ocrelizumab, although ofatumumab causes more CDC than ADCC, and in this regard, resembles rituximab [189]. Its bioavailability is 85% and 40% on day 1 and day 15, respectively [190]. After several subcutaneous applications of ofatumumab, its half-life is 16 days.
Low-dose subcutaneous ofatumumab treatment provides effective B cell depletion within lymphoid tissues, comparable to high-dose intravenous rituximab. However, subcutaneous administration may facilitate ofatumumab entry into lymphatic drainage and lymph nodes [191]. Before reaching the maintenance dose by week 4, 94% of patients had levels of B lymphocytes < 10 cells/µl. Pre-depletion levels of B cells are reached in 24.6 weeks after treatment discontinuation [190]. Modes of metabolism and excretion are anticipated to be similar to endogenous antibodies, but no studies were performed specifically with ofatumumab.
Clinical Trials
The efficacy and safety of ofatumumab was investigated in two double-blind, double-dummy phase 3 clinical trials called ASCLEPIOS I and ASCLEPIOS II with teriflunomide as an active comparator [192]. ARR was lower with ofatumumab in both studies (0.11 vs. 0.22 ASCLEPIOS I; 0.10 vs. 0.25 in ASCLEPIOS II). The decrease in the number of Gd-enhancing lesions was greater with ofatumumab (0.01 vs. 0.45 in ASCLEPIOS I; 0.03 vs. 0.51 in ASCLEPIOS II) and the numbers of new or enlarging lesions per year (0.72 vs. 4.00 in ASCLEPIOS I; 0.64 vs. 4.15 in ASCLEPIOS II) were lower than with teriflunomide. In the pooled trials, the percentage of patients with CDP at 3 and 6 months was 10.9% and 8.1% with ofatumumab and 15.0% and 12.0% with teriflunomide, respectively (hazard ratio 0.66 and 0.68, respectively). The rate of brain atrophy did not differ significantly between the ofatumumab group and the teriflunomide group (− 0.28% and − 0.29% with ofatumumab and -0.35% with teriflunomide in ASCLEPIOS I and ASCLEPIOS II, respectively) [192].
Safety and Adverse Effects
The most common adverse effect was the injection-related reaction which occurred in 20.6% in the ofatumumab group but also in 15.0% in the teriflunomide group [192]. The injection-related reaction is most marked after the first application (14.4%) and seems to diminish subsequently (4.4% after the second, < 3% after the third application) [192]. The most commonly reported symptoms were fever, headache, myalgia, and fatigue.
Serious infections occurred in 2.5% and 1.8% of the patients in the respective groups [192]. Most common were upper-respiratory (39.4%) and urinary tract infections (11.9%) which were mostly mild to moderate. Like other B-cell depleting therapies, ofatumumab causes hypogammaglobulinemia, although there is currently no evidence indicating an elevated risk for infections in patients these patients.
Five neoplasms (0.5%) occurred in the ofatumumab group (two cases of basal-cell carcinoma and one case of malignant melanoma, recurrent non-Hodgkin’s lymphoma, and invasive breast carcinoma, each) and four (0.4%) in the teriflunomide group.
Monitoring and Screening
Since hepatitis B reactivation can occur in patients treated with anti-CD20 mAb; patients with active hepatitis B disease should not receive ofatumumab, and HBV screening should be performed in all patients before initiation of treatment (HBsAg and HBcAb) [151, 190].
Pregnancy and Breastfeeding
Ofatumumab is classified as a pregnancy category C drug as there are no adequate or well-controlled studies of ofatumumab in pregnant women [190]. Recently, a study on cynomolgus monkeys proved that intravenous application of ofatumumab from gestation day 20 until parturition does not affect pre- or postnatal development [193]. As of 31 August 2021, 32 pregnancies were reported in women with MS exposed to ofatumumab; no birth defects or congenital anomalies were reported in 23 pregnant women with known outcomes [194]. However, as ofatumumab crosses the placental barrier and fetuses exhibit depletion of peripheral B cells and decreased spleen and placental weights, treatment with ofatumumab should be avoided during pregnancy unless the potential benefit to the mother outweighs the potential risk to the fetus [190, 194].
No information is available on the clinical use of ofatumumab during breastfeeding. However, as ofatumumab is a large protein molecule (146 kDa), its amount in milk is likely to be very low, confirmed by some studies evaluating transfer of other mAb into breastmilk with comparable molecular weight [195, 196]. Furthermore, it is also partially destroyed in the infant’s gastrointestinal tract and absorption by the infant will be minimal. Therefore, if clinically needed, ofatumumab can be used during breast-feeding [190].
Vaccination
According to EMA and FDA, inactivated vaccines can be administered to patients receiving ofatumumab, whereas live or live-attenuated vaccines have not been studied in these patients and should, therefore, be avoided during treatment and after discontinuation until B cell repletion [190]. Immunization with live or live-attenuated vaccines should be performed at least 4 weeks prior to initiation of ofatumumab whereas at least 2 weeks should elapse before immunization with inactivated vaccines [190].
The safety of and ability to generate an antibody response to vaccination during treatment with ofatumumab has not been studied yet. The response to vaccination could be, however, impaired when lymphocytes B are depleted, which of course also applies to SARS-CoV2 vaccination [58, 160, 190].
Ublituximab
Ublituximab is a novel chimeric mAb against CD20-positive lymphocytes B that targets an epitope on CD20 not targeted by other anti-CD20 mAb, allowing lower doses and shorter infusion times in comparison to other anti-CD20 mAb [189, 197]. It has been glycoengineered to exhibit a low-fucose fragment crystallizable (Fc) region, demonstrating 100 times greater ADCC in vitro than rituximab in cells from patients with chronic lymphocytic leukemia [197, 198]. This activity is evident regardless of CD20 surface expression level on target cells as opposed to ofatumumab which demonstrates superiority to CDC-mediated killing of target cells expressing high levels of CD20 only [199]. All patients reached \(\ge\) 95% B cell depletion from baseline within 2 weeks after the second ublituximab infusion, with depletion occurring already within 24 h of the initial dose in most patients. B cell depletion was sustained pre-dose at weeks 24 and 48 [200].
The efficacy and safety of ublituximab was investigated in ULTIMATE I and ULTIMATE II clinical trials using teriflunomide as an active comparator, with patients being randomized to receive ublituximab 150 mg on day 1, and 450 mg on day 15, and weeks 24, 48, and 72. Their primary endpoint, ARR after 96 weeks of treatment, was reduced in both studies (0.08 vs. 0.19 [59.6%] and 0.09 vs. 0.18 [48.9%], respectively). A pooled analysis of CDP from both ULTIMATE studies at 12 and 24 weeks showed a 15.7% and 34.3% reduction for ublituximab compared to teriflunomide, although this was not statistically significant. There was a strong reduction of the total number of Gd-enhancing lesions (lesions per scan per participant: 0.02 vs. 0.49 [96.7%] and 0.01 vs. 0.25 [96.4%], respectively), and the number of new or enlarging T2L (0.21 vs. 2.79 [92.4%] and 0.28 vs. 2.83 [90.0%], respectively), while post hoc analysis of brain volume change between week 24 and 96 showed no difference between treatment arms. NEDA was reached in 43.0–44.6% of patients on ublituximab, and in 11.4–15.0% on teriflunomide (p < 0.0001) [201]. Ublituximab also demonstrated significant improvement in the overall MSFC scores in both ULTIMATE I and II (p = 0.0484 and p = 0.0171, respectively), with 9HPT being statistically significant in both groups and T25FW in ULTIMATE II but not in ULTIMATE I [202].
A single-arm extension study of those studies was initiated in November 2019 to study the long-term efficiency and safety profile of ublituximab; results from the open-label extension are expected in 2023.
Ublituximab was generally well tolerated, and the most common adverse effect was an IRR, occurring in 43.4% of patients (most commonly grade 1 or 2) [200, 201]. These were most frequent at the first dose, and decreased in frequency with subsequent dosing [201]. Respiratory tract infections occurred in 15.0–17.2% of patients [200, 201]. Proportion of patients with IgM levels under the lower limit of normal after week 96 was 20.9% in the ublituximab and 4.9% in the teriflunomide group [201].
Serious adverse events were reported in 52 (9.5%) patients, the most common being infections (4.0%) and nervous system disorders (0.9%). In total, two malignancies were reported (endometrial and uterine cancer). Three deaths occurred in patients treated with ublituximab due to encephalitis (post-measles), salpingitis and pneumonia, the latter being possibly related to treatment [201]. No case of PML was reported.
Currently, no data are available for the use of ublituximab during pregnancy and breast feeding nor is there any published data available on vaccination. As a member of the anti-CD20 mAb class, recommendations are based on other anti-CD20 mAb, including SARS-CoV2 vaccination.
Conclusion
mAb have become a mainstay of treatment in patients with MS who are in need of HET. The arsenal will most likely be further broadened by the approval of ublituximab in 2022. Further investigations will analyze safety and efficacy of different administration regimes. While all mAb in use have shown high efficacy, serious adverse events may occur with different frequency and require appropriate monitoring and risk management.
References
Thompson AJ, Baranzini SE, Geurts J, et al. Multiple sclerosis. Lancet. 2018;391(10130):1622–36.
Katz SI. Classification, diagnosis, and differential diagnosis of multiple sclerosis. Curr Opin Neurol. 2015;28(3):193–205.
Hegen H, Bsteh G, Berger T. No evidence of disease activity - is it an appropriate surrogate in multiple sclerosis? Eur J Neurol. 2018;25(9):1107-e101.
Diaz C, Zarco LA, Rivera DM. Highly active multiple sclerosis: An update. Mult Scler Relat Disord. 2019;30:215–24.
Smith SL. Ten years of Orthoclone OKT3 (muromonab-CD3): a review. J Transpl Coord. 1996;6(3):109–19; quiz 120–1.
Ober RJ, Radu CG, Ghetie V, et al. Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol. 2001;13(12):1551–9.
Orthmann-Murphy JL, Calabresi PA. Therapeutic application of monoclonal antibodies in multiple sclerosis. Clin Pharmacol Ther. 2017;101(1):52–64.
Bruno V, Battaglia G, Nicoletti F. The advent of monoclonal antibodies in the treatment of chronic autoimmune diseases. Neurol Sci. 2011;31(Suppl 3):283–8.
Saunders KO. Conceptual approaches to modulating antibody effector functions and circulation half-life. Front Immunol. 2019;10:1296.
Goulet DR, Atkins WM. Considerations for the design of antibody-based therapeutics. J Pharm Sci. 2020;109(1):74–103.
Yednock TA, Cannon C, Fritz LC, et al. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature. 1992;356(6364):63–6.
Frisullo G, Iorio R, Plantone D, et al. CD4+T-bet+, CD4+pSTAT3+ and CD8+T-bet+ T cells accumulate in peripheral blood during NZB treatment. Mult Scler. 2011;17(5):556–66.
Khatri BO, Man S, Giovannoni G, et al. Effect of plasma exchange in accelerating natalizumab clearance and restoring leukocyte function. Neurology. 2009;72(5):402–9.
Muralidharan KK, Kuesters G, Plavina T, et al. Population pharmacokinetics and target engagement of natalizumab in patients with multiple sclerosis. J Clin Pharmacol. 2017;57(8):1017–30.
Foley JF, Goelz S, Hoyt T, et al. Evaluation of natalizumab pharmacokinetics and pharmacodynamics with standard and extended interval dosing. Mult Scler Relat Disord. 2019;31:65–71.
Zhovtis Ryerson L, Li X, Goldberg JD, et al. Pharmacodynamics of natalizumab extended interval dosing in MS. Neurol Neuroimmunol Neuroinflamm. 2020;7(2).
Serra Lopez-Matencio JM, Perez Garcia Y, Meca-Lallana V, et al. Evaluation of Natalizumab Pharmacokinetics and Pharmacodynamics: Toward Individualized Doses. Front Neurol. 2021;12:716548.
Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899–910.
Rudick RA, Stuart WH, Calabresi PA, et al. Natalizumab plus interferon beta-1a for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):911–23.
Miller DH, Soon D, Fernando KT, et al. MRI outcomes in a placebo-controlled trial of natalizumab in relapsing MS. Neurology. 2007;68(17):1390–401.
Phillips JT, Giovannoni G, Lublin FD, et al. Sustained improvement in Expanded Disability Status Scale as a new efficacy measure of neurological change in multiple sclerosis: treatment effects with natalizumab in patients with relapsing multiple sclerosis. Mult Scler. 2011;17(8):970–9.
Balcer LJ, Galetta SL, Calabresi PA, et al. Natalizumab reduces visual loss in patients with relapsing multiple sclerosis. Neurology. 2007;68(16):1299–304.
Rudick RA, Miller D, Hass S, et al. Health-related quality of life in multiple sclerosis: effects of natalizumab. Ann Neurol. 2007;62(4):335–46.
Guger M, Enzinger C, Leutmezer F, et al. Long-term outcome and predictors of long-term disease activity in natalizumab-treated patients with multiple sclerosis: real life data from the Austrian MS Treatment Registry. J Neurol. 2021;268(11):4303–10.
Bloomgren G, Richman S, Hotermans C, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. 2012;366(20):1870–80.
Ho PR, Koendgen H, Campbell N, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: a retrospective analysis of data from four clinical studies. Lancet Neurol. 2017;16(11):925–33.
European Medical Agency. Tysabri summary of product characteristics. 2009.
Zhovtis Ryerson L, Frohman TC, Foley J, et al. Extended interval dosing of natalizumab in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2016;87(8):885–9.
Yamout BI, Sahraian MA, Ayoubi NE, et al. Efficacy and safety of natalizumab extended interval dosing. Mult Scler Relat Disord. 2018;24:113–6.
Foley J, Defer G, Zhovtis Ryerson L, et al. Primary results of NOVA: a randomised controlled study of the efficacy of 6-week dosing of natalizumab versus continued 4-week treatment for multiple sclerosis, in ECTRIMS. 2021: Vienna.
Chang I, Muralidharan KK, Campbell N, et al. Modeling the efficacy of natalizumab in multiple sclerosis patients who switch from every-4-week dosing to extended-interval dosing. J Clin Pharmacol. 2021;61(3):339–48.
Clerico M, De Mercanti SF, Signori A, et al. Extending the interval of natalizumab dosing: is efficacy preserved? Neurotherapeutics. 2020;17(1):200–7.
Riancho J, Setien S, Sanchez de la Torre JR, et al. Does extended interval dosing natalizumab preserve effectiveness in multiple sclerosis? A 7 year-retrospective observational study. Front Immunol. 2021;12:614715.
Tan IL, McArthur JC, Clifford DB, et al. Immune reconstitution inflammatory syndrome in natalizumab-associated PML. Neurology. 2011;77(11):1061–7.
Wehner NG, Shopp G, Oneda S, et al. Embryo/fetal development in cynomolgus monkeys exposed to natalizumab, an alpha4 integrin inhibitor. Birth Defects Res B Dev Reprod Toxicol. 2009;86(2):117–30.
Wehner NG, Skov M, Shopp G, et al. Effects of natalizumab, an alpha4 integrin inhibitor, on fertility in male and female guinea pigs. Birth Defects Res B Dev Reprod Toxicol. 2009;86(2):108–16.
Wehner NG, Shopp G, Osterburg I, et al. Postnatal development in cynomolgus monkeys following prenatal exposure to natalizumab, an alpha4 integrin inhibitor. Birth Defects Res B Dev Reprod Toxicol. 2009;86(2):144–56.
Rasenack M, Derfuss T. Disease activity return after natalizumab cessation in multiple sclerosis. Expert Rev Neurother. 2016;16(5):587–94.
Confavreux C, Hutchinson M, Hours MM, et al. Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in Multiple Sclerosis Group. N Engl J Med. 1998;339(5):285–91.
Portaccio E, Ghezzi A, Hakiki B, et al. Postpartum relapses increase the risk of disability progression in multiple sclerosis: the role of disease modifying drugs. J Neurol Neurosurg Psychiatry. 2014;85(8):845–50.
Vukusic S, Hutchinson M, Hours M, et al. Pregnancy and multiple sclerosis (the PRIMS study): clinical predictors of post-partum relapse. Brain. 2004;127(Pt 6):1353–60.
Bsteh G, Algrang L, Hegen H, et al. Pregnancy and multiple sclerosis in the DMT era: A cohort study in Western Austria. Mult Scler. 2020;26(1):69–78.
Schneider H, Miller RK. Receptor-mediated uptake and transport of macromolecules in the human placenta. Int J Dev Biol. 2010;54(2–3):367–75.
Morell A, Skvaril F, van Loghem E, et al. Human IgG subclasses in maternal and fetal serum. Vox Sang. 1971;21(6):481–92.
Gusdon JP Jr. Fetal and maternal immunoglobulin levels during pregnancy. Am J Obstet Gynecol. 1969;103(7):895–900.
Yeh WZ, Widyastuti PA, Van der Walt A, et al. Natalizumab, Fingolimod and Dimethyl Fumarate Use and Pregnancy-Related Relapse and Disability in Women With Multiple Sclerosis. Neurology. 2021.
Wiendl H, Gold R, Berger T, et al. Multiple Sclerosis Therapy Consensus Group (MSTCG): position statement on disease-modifying therapies for multiple sclerosis (white paper). Ther Adv Neurol Disord. 2021;14:17562864211039648.
Friend S, Richman S, Bloomgren G, et al. Evaluation of pregnancy outcomes from the Tysabri(R) (natalizumab) pregnancy exposure registry: a global, observational, follow-up study. BMC Neurol. 2016;16(1):150.
Guger M, Traxler G, Drabauer M, et al. Pregnancy Outcomes in Patients With Multiple Sclerosis Exposed to Natalizumab-A Retrospective Analysis From the Austrian Multiple Sclerosis Treatment Registry. Front Neurol. 2020;11:676.
Haghikia A, Langer-Gould A, Rellensmann G, et al. Natalizumab use during the third trimester of pregnancy. JAMA Neurol. 2014;71(7):891–5.
Alroughani R, Altintas A, Al Jumah M, et al. Pregnancy and the use of disease-modifying therapies in patients with multiple sclerosis: benefits versus risks. Mult Scler Int. 2016;2016:1034912.
Vagberg M, Kumlin U, Svenningsson A. Humoral immune response to influenza vaccine in natalizumab-treated MS patients. Neurol Res. 2012;34(7):730–3.
Olberg HK, Eide GE, Cox RJ, et al. Antibody response to seasonal influenza vaccination in patients with multiple sclerosis receiving immunomodulatory therapy. Eur J Neurol. 2018;25(3):527–34.
Metze C, Winkelmann A, Loebermann M, et al. Immunogenicity and predictors of response to a single dose trivalent seasonal influenza vaccine in multiple sclerosis patients receiving disease-modifying therapies. CNS Neurosci Ther. 2019;25(2):245–54.
Kaufman M, Pardo G, Rossman H, et al. Natalizumab treatment shows no clinically meaningful effects on immunization responses in patients with relapsing-remitting multiple sclerosis. J Neurol Sci. 2014;341(1–2):22–7.
Sormani MP, Inglese M, Schiavetti I, et al. Effect of SARS-CoV-2 mRNA vaccination in MS patients treated with disease modifying therapies. EBioMedicine. 2021;72:103581.
Al Jumah M, Abulaban A, Aggad H, et al. Managing multiple sclerosis in the Covid19 era: a review of the literature and consensus report from a panel of experts in Saudi Arabia. Mult Scler Relat Disord. 2021;51:102925.
Bsteh G, Hegen H, Traxler G, et al. Comparing humoral immune response to SARS-CoV2 vaccines in people with multiple sclerosis and healthy controls: An Austrian prospective multicenter cohort study. Eur J Neurol. 2022.
Buggins AG, Mufti GJ, Salisbury J, et al. Peripheral blood but not tissue dendritic cells express CD52 and are depleted by treatment with alemtuzumab. Blood. 2002;100(5):1715–20.
Hu Y, Turner MJ, Shields J, et al. Investigation of the mechanism of action of alemtuzumab in a human CD52 transgenic mouse model. Immunology. 2009;128(2):260–70.
Watanabe T, Masuyama J, Sohma Y, et al. CD52 is a novel costimulatory molecule for induction of CD4+ regulatory T cells. Clin Immunol. 2006;120(3):247–59.
Lemtrada. Summary of product characteristics: Sanofi Belgium. 2020: Diegem, Belgium.
Bindon CI, Hale G, Waldmann H. Importance of antigen specificity for complement-mediated lysis by monoclonal antibodies. Eur J Immunol. 1988;18(10):1507–14.
Hale C, Bartholomew M, Taylor V, et al. Recognition of CD52 allelic gene products by CAMPATH-1H antibodies. Immunology. 1996;88(2):183–90.
Hale G, Rebello P, Brettman LR, et al. Blood concentrations of alemtuzumab and antiglobulin responses in patients with chronic lymphocytic leukemia following intravenous or subcutaneous routes of administration. Blood. 2004;104(4):948–55.
Hill-Cawthorne GA, Button T, Tuohy O, et al. Long term lymphocyte reconstitution after alemtuzumab treatment of multiple sclerosis. J Neurol Neurosurg Psychiatry. 2012;83(3):298–304.
Baker D, Herrod SS, Alvarez-Gonzalez C, et al. Interpreting lymphocyte reconstitution data from the pivotal phase 3 trials of alemtuzumab. JAMA Neurol. 2017;74(8):961–9.
Kousin-Ezewu O, Azzopardi L, Parker RA, et al. Accelerated lymphocyte recovery after alemtuzumab does not predict multiple sclerosis activity. Neurology. 2014;82(24):2158–64.
Jones JL, Thompson SA, Loh P, et al. Human autoimmunity after lymphocyte depletion is caused by homeostatic T-cell proliferation. Proc Natl Acad Sci U S A. 2013;110(50):20200–5.
Zhang X, Tao Y, Chopra M, et al. Differential reconstitution of T cell subsets following immunodepleting treatment with alemtuzumab (anti-CD52 monoclonal antibody) in patients with relapsing-remitting multiple sclerosis. J Immunol. 2013;191(12):5867–74.
Gross CC, Ahmetspahic D, Ruck T, et al. Alemtuzumab treatment alters circulating innate immune cells in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2016;3(6):e289.
Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1819–28.
Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380(9856):1829–39.
Arnold DL, Fisher E, Brinar VV, et al. Superior MRI outcomes with alemtuzumab compared with subcutaneous interferon beta-1a in MS. Neurology. 2016;87(14):1464–72.
Coles AJ, Arnold DL, Bass AD, et al. Efficacy and safety of alemtuzumab over 6 years: final results of the 4-year CARE-MS extension trial. Ther Adv Neurol Disord. 2021;14:1756286420982134.
Havrdova E, Arnold DL, Cohen JA, et al. Alemtuzumab CARE-MS I 5-year follow-up: Durable efficacy in the absence of continuous MS therapy. Neurology. 2017;89(11):1107–16.
Coles AJ, Cohen JA, Fox EJ, et al. Alemtuzumab CARE-MS II 5-year follow-up: efficacy and safety findings. Neurology. 2017;89(11):1117–26.
Ziemssen T, Bass AD, Berkovich R, et al. Efficacy and safety of alemtuzumab through 9 years of follow-up in patients with highly active disease: post hoc analysis of CARE-MS I and II patients in the TOPAZ Extension Study. CNS Drugs. 2020;34(9):973–88.
Chan JK, Hernandez Martinez de Lapiscina E, Taylor C, et al. Long-term stability of neuroaxonal structure in alemtuzumab-treated relapsing-remitting multiple sclerosis patients. J Neuroophthalmol. 2020;40(1):37–43.
Vavasour IM, Tam R, Li DK, et al. A 24-month advanced magnetic resonance imaging study of multiple sclerosis patients treated with alemtuzumab. Mult Scler. 2019;25(6):811–8.
Button T, Altmann D, Tozer D, et al. Magnetization transfer imaging in multiple sclerosis treated with alemtuzumab. Mult Scler. 2013;19(2):241–4.
Brown JWL, Prados Carrasco F, Eshaghi A, et al. Periventricular magnetisation transfer ratio abnormalities in multiple sclerosis improve after alemtuzumab. Mult Scler. 2020;26(9):1093–101.
Guarnera C, Bramanti P, Mazzon E. Alemtuzumab: a review of efficacy and risks in the treatment of relapsing remitting multiple sclerosis. Ther Clin Risk Manag. 2017;13:871–9.
Evan JR, Bozkurt SB, Thomas NC, et al. Alemtuzumab for the treatment of multiple sclerosis. Expert Opin Biol Ther. 2018;18(3):323–34.
Sega-Jazbec S, Barun B, Horvat Ledinek A, et al. Management of infusion related reactions associated with alemtuzumab in patients with multiple sclerosis. Mult Scler Relat Disord. 2017;17:151–3.
Willis MD, Harding KE, Pickersgill TP, et al. Alemtuzumab for multiple sclerosis: Long term follow-up in a multi-centre cohort. Mult Scler. 2016;22(9):1215–23.
Havrdova E, Cohen JA, Horakova D, et al. Understanding the positive benefit:risk profile of alemtuzumab in relapsing multiple sclerosis: perspectives from the Alemtuzumab Clinical Development Program. Ther Clin Risk Manag. 2017;13:1423–37.
Wray S, Havrdova E, Snydman DR, et al. Infection risk with alemtuzumab decreases over time: pooled analysis of 6-year data from the CAMMS223, CARE-MS I, and CARE-MS II studies and the CAMMS03409 extension study. Mult Scler. 2019;25(12):1605–17.
Buonomo AR, Zappulo E, Viceconte G, et al. Risk of opportunistic infections in patients treated with alemtuzumab for multiple sclerosis. Expert Opin Drug Saf. 2018;17(7):709–17.
de Noordhout CM, Devleesschauwer B, Angulo FJ, et al. The global burden of listeriosis: a systematic review and meta-analysis. Lancet Infect Dis. 2014;14(11):1073–82.
Holmoy T, von der Lippe H, Leegaard TM. Listeria monocytogenes infection associated with alemtuzumab - - a case for better preventive strategies. BMC Neurol. 2017;17(1):65.
Hartung HP, Mares J, Barnett MH. Alemtuzumab: Rare serious adverse events of a high-efficacy drug. Mult Scler. 2020;26(6):737–40.
Cossburn M, Pace AA, Jones J, et al. Autoimmune disease after alemtuzumab treatment for multiple sclerosis in a multicenter cohort. Neurology. 2011;77(6):573–9.
Jones JL, Phuah CL, Cox AL, et al. IL-21 drives secondary autoimmunity in patients with multiple sclerosis, following therapeutic lymphocyte depletion with alemtuzumab (Campath-1H). J Clin Invest. 2009;119(7):2052–61.
Rotondi M, Molteni M, Leporati P, et al. Autoimmune thyroid diseases in patients treated with alemtuzumab for multiple sclerosis: an example of selective anti-TSH-receptor immune response. Front Endocrinol (Lausanne). 2017;8:254.
Mahzari M, Arnaout A, Freedman MS. Alemtuzumab induced thyroid disease in multiple sclerosis: a review and approach to management. Can J Neurol Sci. 2015;42(5):284–91.
Berger T, Elovaara I, Fredrikson S, et al. Alemtuzumab use in clinical practice: recommendations from European multiple sclerosis experts. CNS Drugs. 2017;31(1):33–50.
Meltzer E, Campbell S, Ehrenfeld B, et al. Mitigating alemtuzumab-associated autoimmunity in MS: A whack-a-mole B-cell depletion strategy. Neurol Neuroimmunol Neuroinflamm. 2020;7(6).
Cuker A, Coles AJ, Sullivan H, et al. A distinctive form of immune thrombocytopenia in a phase 2 study of alemtuzumab for the treatment of relapsing-remitting multiple sclerosis. Blood. 2011;118(24):6299–305.
Cuker A, Bass AD, Nadj C, et al. Immune thrombocytopenia in alemtuzumab-treated MS patients: Incidence, detection, and management. Mult Scler. 2020;26(1):48–56.
Hartung HP, Aktas O, Boyko AN. Alemtuzumab: a new therapy for active relapsing-remitting multiple sclerosis. Mult Scler. 2015;21(1):22–34.
Coles AJ, Jones JL, Vermersch P, et al. Autoimmunity and long-term safety and efficacy of alemtuzumab for multiple sclerosis: Benefit/risk following review of trial and post-marketing data. Mult Scler. 2021:13524585211061335.
Muraro PA, Scolding NJ, Fox RJ. Rare side effects of alemtuzumab remind us of the need for postmarketing surveillance. Neurology. 2018;90(18):819–20.
Killestein J, van Oosten B. Emerging safety issues in alemtuzumab-treated MS patients. Mult Scler. 2019;25(9):1206–8.
Lenihan DJ, Alencar AJ, Yang D, et al. Cardiac toxicity of alemtuzumab in patients with mycosis fungoides/Sezary syndrome. Blood. 2004;104(3):655–8.
Basquiera AL, Berretta AR, Garcia JJ, et al. Coronary ischemia related to alemtuzumab therapy. Ann Oncol. 2004;15(3):539–40.
Coles AJ, Wing MG, Molyneux P, et al. Monoclonal antibody treatment exposes three mechanisms underlying the clinical course of multiple sclerosis. Ann Neurol. 1999;46(3):296–304.
Moreau T, Coles A, Wing M, et al. Transient increase in symptoms associated with cytokine release in patients with multiple sclerosis. Brain. 1996;119(Pt 1):225–37.
Ottaviani G, Matturri L, Rossi L, et al. Sudden death due to lymphomatous infiltration of the cardiac conduction system. Cardiovasc Pathol. 2003;12(2):77–81.
Haghikia A, Dendrou CA, Schneider R, et al. Severe B-cell-mediated CNS disease secondary to alemtuzumab therapy. Lancet Neurol. 2017;16(2):104–6.
Wehrum T, Beume LA, Stich O, et al. Activation of disease during therapy with alemtuzumab in 3 patients with multiple sclerosis. Neurology. 2018;90(7):e601–5.
Barton J, Hardy TA, Riminton S, et al. Tumefactive demyelination following treatment for relapsing multiple sclerosis with alemtuzumab. Neurology. 2017;88(10):1004–6.
Blasco MR, Ramos A, Malo CG, et al. Acute pneumonitis and pericarditis related to alemtuzumab therapy in relapsing-remitting multiple sclerosis. J Neurol. 2017;264(1):168–9.
Pfeuffer S, Beuker C, Ruck T, et al. Acute cholecystitis during treatment with alemtuzumab in 3 patients with RRMS. Neurology. 2016;87(22):2380–1.
Sauer EM, Schliep S, Manger B, et al. Microscopic polyangiitis after alemtuzumab treatment in relapsing-remitting MS. Neurol Neuroimmunol Neuroinflamm. 2018;5(5):e488.
Graf J, Ringelstein M, Lepka K, et al. Acute sarcoidosis in a multiple sclerosis patient after alemtuzumab treatment. Mult Scler. 2018;24(13):1776–8.
Rau D, Lang M, Harth A, et al. Listeria meningitis complicating alemtuzumab treatment in multiple sclerosis–report of two cases. Int J Mol Sci. 2015;16(7):14669–76.
Canham LJW, Manara A, Fawcett J, et al. Mortality from Listeria monocytogenes meningoencephalitis following escalation to alemtuzumab therapy for relapsing-remitting Multiple Sclerosis. Mult Scler Relat Disord. 2018;24:38–41.
Meunier B, Rico A, Seguier J, et al. Life-threatening autoimmune warm hemolytic anemia following treatment for multiple sclerosis with alemtuzumab. Mult Scler. 2018;24(6):811–3.
Saarela M, Senthil K, Jones J, et al. Hemophagocytic lymphohistiocytosis in 2 patients with multiple sclerosis treated with alemtuzumab. Neurology. 2018;90(18):849–51.
Brownlee WJ, Chataway J. Opportunistic infections after alemtuzumab: New cases of norcardial infection and cytomegalovirus syndrome. Mult Scler. 2017;23(6):876–7.
Clerico M, De Mercanti S, Artusi CA, et al. Active CMV infection in two patients with multiple sclerosis treated with alemtuzumab. Mult Scler. 2017;23(6):874–6.
Berker D, Isik S, Ozuguz U, et al. Prevalence of incidental thyroid cancer and its ultrasonographic features in subcentimeter thyroid nodules of patients with hyperthyroidism. Endocrine. 2011;39(1):13–20.
Kaplan TB. Management of demyelinating disorders in pregnancy. Neurol Clin. 2019;37(1):17–30.
Oh J, Achiron A, Chambers S, et al. Pregnancy outcomes in patients with RRMS who received alemtuzumab in the clinical development program. Neurology. 2016;86:S24.008.
Oh J, Achiron A, Celius EG, et al. Pregnancy outcomes and postpartum relapse rates in women with RRMS treated with alemtuzumab in the phase 2 and 3 clinical development program over 16 years. Mult Scler Relat Disord. 2020;43:102146.
Dobson R, Dassan P, Roberts M, et al. UK consensus on pregnancy in multiple sclerosis: Association of British Neurologists guidelines. Pract Neurol. 2019;19(2):106–14.
LEMTRADA (Alemtuzumab) Injection, for Intravenous Use. Full Prescribing Information. [cited 2021 October 25]; Available from: https://products.sanofi.us/Lemtrada/Lemtrada.pdf.
McCarthy CL, Tuohy O, Compston DA, et al. Immune competence after alemtuzumab treatment of multiple sclerosis. Neurology. 2013;81(10):872–6.
Thompson SA, Jones JL, Cox AL, et al. B-cell reconstitution and BAFF after alemtuzumab (Campath-1H) treatment of multiple sclerosis. J Clin Immunol. 2010;30(1):99–105.
Achiron A, Mandel M, Dreyer-Alster S, et al. Humoral immune response in multiple sclerosis patients following PfizerBNT162b2 COVID19 vaccination: up to 6 months cross-sectional study. J Neuroimmunol. 2021;361:577746.
Greenfield AL, Hauser SL. B-cell therapy for multiple sclerosis: entering an era. Ann Neurol. 2018;83(1):13–26.
Franks SE, Getahun A, Hogarth PM, et al. Targeting B cells in treatment of autoimmunity. Curr Opin Immunol. 2016;43:39–45.
Etemadifar M, Salari M, Mirmosayyeb O, et al. Efficacy and safety of rituximab in neuromyelitis optica: Review of evidence. J Res Med Sci. 2017;22:18.
Salzer J, Svenningsson R, Alping P, et al. Rituximab in multiple sclerosis: a retrospective observational study on safety and efficacy. Neurology. 2016;87(20):2074–81.
Tandan R, Hehir MK 2nd, Waheed W, et al. Rituximab treatment of myasthenia gravis: A systematic review. Muscle Nerve. 2017;56(2):185–96.
Bar-Or A, Calabresi PA, Arnold D, et al. Rituximab in relapsing-remitting multiple sclerosis: a 72-week, open-label, phase I trial. Ann Neurol. 2008;63(3):395–400.
Ng CM, Bruno R, Combs D, et al. Population pharmacokinetics of rituximab (anti-CD20 monoclonal antibody) in rheumatoid arthritis patients during a phase II clinical trial. J Clin Pharmacol. 2005;45(7):792–801.
Tobias Z, Esther D, Niklas S, et al. Rituximab versus mitoxantrone: comparing effectiveness and safety in advanced relapsing multiple sclerosis. Ther Adv Chronic Dis. 2021;12:20406223211024370.
He A, Merkel B, Brown JWL, et al. Timing of high-efficacy therapy for multiple sclerosis: a retrospective observational cohort study. Lancet Neurol. 2020;19(4):307–16.
Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol. 2009;66(4):460–71.
Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358(7):676–88.
Gelfand JM, Cree BAC, Hauser SL. Ocrelizumab and Other CD20(+) B-Cell-Depleting Therapies in Multiple Sclerosis. Neurotherapeutics. 2017;14(4):835–41.
Gottenberg JE, Ravaud P, Bardin T, et al. Risk factors for severe infections in patients with rheumatoid arthritis treated with rituximab in the autoimmunity and rituximab registry. Arthritis Rheum. 2010;62(9):2625–32.
Tallantyre EC, Whittam DH, Jolles S, et al. Secondary antibody deficiency: a complication of anti-CD20 therapy for neuroinflammation. J Neurol. 2018;265(5):1115–22.
Rissanen E, Remes K, Airas L. Severe neutropenia after rituximab-treatment of multiple sclerosis. Mult Scler Relat Disord. 2018;20:3–5.
Oshima Y, Tanimoto T, Yuji K, et al. Drug-associated progressive multifocal leukoencephalopathy in multiple sclerosis patients. Mult Scler. 2019;25(8):1141–9.
Mok CC. Rituximab for the treatment of rheumatoid arthritis: an update. Drug Des Devel Ther. 2013;8:87–100.
Mitka M. FDA: Increased HBV reactivation risk with ofatumumab or rituximab. JAMA. 2013;310(16):1664.
Alping P, Askling J, Burman J, et al. Cancer risk for fingolimod, natalizumab, and rituximab in multiple sclerosis patients. Ann Neurol. 2020;87(5):688–99.
Smalls DJ, Kiger RE, Norris LB, et al. Hepatitis B Virus Reactivation: Risk Factors and Current Management Strategies. Pharmacotherapy. 2019;39(12):1190–203.
Villadolid J, Laplant KD, Markham MJ, et al. Hepatitis B reactivation and rituximab in the oncology practice. Oncologist. 2010;15(10):1113–21.
European Medical Agency. Mabthera summary of product characteristics. 2009.
Chakravarty EF, Murray ER, Kelman A, et al. Pregnancy outcomes after maternal exposure to rituximab. Blood. 2011;117(5):1499–506.
Das G, Damotte V, Gelfand JM, et al. Rituximab before and during pregnancy: a systematic review, and a case series in MS and NMOSD. Neurol Neuroimmunol Neuroinflamm. 2018;5(3):e453.
Klink DT, van Elburg RM, Schreurs MW, et al. Rituximab administration in third trimester of pregnancy suppresses neonatal B-cell development. Clin Dev Immunol. 2008;2008:271363.
Bragnes Y, Boshuizen R, de Vries A, et al. Low level of Rituximab in human breast milk in a patient treated during lactation. Rheumatology (Oxford). 2017;56(6):1047–8.
Vaidyanathan A, McKeever K, Anand B, et al. Developmental immunotoxicology assessment of rituximab in cynomolgus monkeys. Toxicol Sci. 2011;119(1):116–25.
Bingham CO 3rd, Looney RJ, Deodhar A, et al. Immunization responses in rheumatoid arthritis patients treated with rituximab: results from a controlled clinical trial. Arthritis Rheum. 2010;62(1):64–74.
Kornek B, Leutmezer F, Rommer PS, et al. B Cell Depletion and SARS-CoV-2 Vaccine Responses in Neuroimmunologic Patients. Ann Neurol. 2022.
Achtnichts L, Jakopp B, Oberle M, et al. Humoral immune response after the third SARS-CoV-2 mRNA vaccination in CD20 depleted people with multiple sclerosis. Vaccines (Basel). 2021;9(12).
Apostolidis SA, Kakara M, Painter MM, et al. Cellular and humoral immune responses following SARS-CoV-2 mRNA vaccination in patients with multiple sclerosis on anti-CD20 therapy. Nat Med. 2021.
Brill L, Rechtman A, Zveik O, et al. Humoral and T-cell response to SARS-CoV-2 vaccination in patients with multiple sclerosis treated with ocrelizumab. JAMA Neurol. 2021.
Moor MB, Suter-Riniker F, Horn MP, et al. Humoral and cellular responses to mRNA vaccines against SARS-CoV-2 in patients with a history of CD20 B-cell-depleting therapy (RituxiVac): an investigator-initiated, single-centre, open-label study. Lancet Rheumatol. 2021;3(11):e789–97.
Ocrelizumab (Ocrevus) for MS. Med Lett Drugs Ther. 2017;59(1523):98–101.
Klein C, Lammens A, Schafer W, et al. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. MAbs. 2013;5(1):22–33.
Gingele S, Jacobus TL, Konen FF, et al. Ocrelizumab depletes CD20(+) T cells in multiple sclerosis patients. Cells. 2018;8(1).
European Medical Agency. Ocrevus summary of product characteristics. 2018
Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med. 2017;376(3):221–34.
Havrdova E, Arnold DL, Bar-Or A, et al. No evidence of disease activity (NEDA) analysis by epochs in patients with relapsing multiple sclerosis treated with ocrelizumab vs interferon beta-1a. Mult Scler J Exp Transl Clin. 2018;4(1):2055217318760642.
Hartung HP; ENSEMBLE Steering Committee members and study investigators. Ocrelizumab shorter infusion: Primary results from the ENSEMBLE PLUS substudy in patients with MS. Neurol Neuroimmunol Neuroinflamm. 2020;7(5):e807.
van der Kolk LE, Grillo-Lopez AJ, Baars JW, et al. Complement activation plays a key role in the side-effects of rituximab treatment. Br J Haematol. 2001;115(4):807–11.
Paul F, Cartron G. Infusion-related reactions to rituximab: frequency, mechanisms and predictors. Expert Rev Clin Immunol. 2019;15(4):383–9.
Ng HS, Rosenbult CL, Tremlett H. Safety profile of ocrelizumab for the treatment of multiple sclerosis: a systematic review. Expert Opin Drug Saf. 2020;19(9):1069–94.
Hauser SL, Kappos L, Montalban X, et al. Safety of ocrelizumab in patients with relapsing and primary progressive multiple sclerosis. Neurology. 2021;97(16):e1546–59.
Hauser S, Kappos L, Montalban X, et al. Safety of ocrelizumab in multiple sclerosis: updated analysis in patients with relapsing and primary progressive multiple sclerosis, in ECTRIMS. 2021:Vienna.
Hauser SL, Kappos L, Arnold DL, et al. Five years of ocrelizumab in relapsing multiple sclerosis: OPERA studies open-label extension. Neurology. 2020;95(13):e1854–67.
Patel A, Sul J, Gordon ML, et al. Progressive Multifocal Leukoencephalopathy in a Patient With Progressive Multiple Sclerosis Treated With Ocrelizumab Monotherapy. JAMA Neurol. 2021;78(6):736–40.
Ciplea AI, Langer-Gould A, de Vries A, et al. Monoclonal antibody treatment during pregnancy and/or lactation in women with MS or neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm. 2020;7(4).
Bar-Or A, Calkwood JC, Chognot C, et al. Effect of ocrelizumab on vaccine responses in patients with multiple sclerosis: The VELOCE study. Neurology. 2020;95(14):e1999–2008.
Guerrieri S, Lazzarin S, Zanetta C, et al. Serological response to SARS-CoV-2 vaccination in multiple sclerosis patients treated with fingolimod or ocrelizumab: an initial real-life experience. J Neurol. 2022;269(1):39-43.
Disanto G, Sacco R, Bernasconi E, et al. Association of disease-modifying treatment and anti-CD20 infusion timing with humoral response to 2 SARS-CoV-2 vaccines in patients with multiple sclerosis. JAMA Neurol. 2021.
Rolfes L, Pawlitzki M, Pfeuffer S, et al. Ocrelizumab extended interval dosing in multiple sclerosis in times of COVID-19. Neurol Neuroimmunol Neuroinflamm. 2021;8(5).
Pompsch M, Fisenkci N, Horn PA, et al. Evidence of extensive cellular immune response after SARS-CoV-2 vaccination in ocrelizumab-treated patients with multiple sclerosis. Neurol Res Pract. 2021;3(1):60.
Iannetta M, Landi D, Cola G, et al. B- and T-cell responses after SARS-CoV-2 vaccination in patients with multiple sclerosis receiving disease modifying therapies: immunological patterns and clinical implications. Front Immunol. 2021;12:796482.
Novak F, Nilsson AC, Nielsen C, et al. Humoral immune response following SARS-CoV-2 mRNA vaccination concomitant to anti-CD20 therapy in multiple sclerosis. Mult Scler Relat Disord. 2021;56:103251.
Samjoo IA, Worthington E, Drudge C, et al. Comparison of ofatumumab and other disease-modifying therapies for relapsing multiple sclerosis: a network meta-analysis. J Comp Eff Res. 2020;9(18):1255–74.
Masoud S, McAdoo SP, Bedi R, et al. Ofatumumab for B cell depletion in patients with systemic lupus erythematosus who are allergic to rituximab. Rheumatology (Oxford). 2018;57(7):1156–61.
Babiker HM, Glode AE, Cooke LS, et al. Ublituximab for the treatment of CD20 positive B-cell malignancies. Expert Opin Investig Drugs. 2018;27(4):407–12.
European Medical Agency. Kesimpta summary of product characteristics. 2021.
Smith P, Huck C, Schmid C, et al. Ofatumumab differs from rituximab by effectively targeting lymph node B cells and achieving faster post-treatment repletion (S24.003). Neurology. 2017;88(16 Supplement):S24.003.
Hauser SL, Bar-Or A, Cohen JA, et al. Ofatumumab versus teriflunomide in multiple sclerosis. N Engl J Med. 2020;383(6):546–57.
Bellot M, Bagger M, Horvat C, et al. Effect of ofatumumab on pregnancy, parturition and lactation in cynomolgus monkeys (2265). Neurology. 2021;96.
Pregnancy Outcomes. 2021 [cited 2021 27 October]; Available from: https://www.ofatumumabinfo.com/en/pregnancy.
LaHue SC, Anderson A, Krysko KM, et al. Transfer of monoclonal antibodies into breastmilk in neurologic and non-neurologic diseases. Neurol Neuroimmunol Neuroinflamm. 2020;7(4).
Krysko KM, LaHue SC, Anderson A, et al. Minimal breast milk transfer of rituximab, a monoclonal antibody used in neurological conditions. Neurol Neuroimmunol Neuroinflamm. 2020;7(1).
Le Garff-Tavernier M, Herbi L, de Romeuf C, et al. Antibody-dependent cellular cytotoxicity of the optimized anti-CD20 monoclonal antibody ublituximab on chronic lymphocytic leukemia cells with the 17p deletion. Leukemia. 2014;28(1):230–3.
Sharman JP, Farber CM, Mahadevan D, et al. Ublituximab (TG-1101), a novel glycoengineered anti-CD20 antibody, in combination with ibrutinib is safe and highly active in patients with relapsed and/or refractory chronic lymphocytic leukaemia: results of a phase 2 trial. Br J Haematol. 2017;176(3):412–20.
Sawas A, Farber CM, Schreeder MT, et al. A phase 1/2 trial of ublituximab, a novel anti-CD20 monoclonal antibody, in patients with B-cell non-Hodgkin lymphoma or chronic lymphocytic leukaemia previously exposed to rituximab. Br J Haematol. 2017;177(2):243–53.
Fox E, Lovett-Racke AE, Gormley M, et al. A phase 2 multicenter study of ublituximab, a novel glycoengineered anti-CD20 monoclonal antibody, in patients with relapsing forms of multiple sclerosis. Mult Scler. 2021;27(3):420–9.
Steinman L, Fox E, Hartung HP, et al. Phase 3 results of the ULTIMATE I & II global studies: ublituximab versus teriflunomide in relapsing multiple sclerosis, in ECTRIMS. 2021: Vienna.
Steinman L, Alvarez E, Fox E, et al. Ublituximab is associated with significant improvement in the multiple sclerosis functional composite (MSFC): results from the Phase 3 ULTIMATE I & II studies, in ECTRIMS. 2021: Vienna.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Nik Krajnc has participated in meetings sponsored by, received speaker honoraria or travel funding from Roche, Novartis, and Merck and holds a grant for a Multiple Sclerosis Clinical Training Fellowship Programme from the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS). Gabriel Bsteh has participated in meetings sponsored by, received speaker honoraria or travel funding from Biogen, Celgene, Lilly, Merck, Novartis, Sanofi-Genzyme, and Teva and received honoraria for consulting Biogen, Celgene, Novartis, Sanofi-Genzyme, Roche, and Teva. Thomas Berger has participated in meetings sponsored by and received honoraria (lectures, advisory boards, consultations) from pharmaceutical companies marketing treatments for MS: Allergan, Bayer, Biogen, Bionorica, Celgene, MedDay, Merck, Novartis, Octapharma, Roche, Sanofi-Genzyme, Teva. His institution has received financial support in the past 12 months by unrestricted research grants (Biogen, Bayer, Merck, Novartis, Sanofi Aventis, Teva and for participation in clinical trials in multiple sclerosis sponsored by Alexion, Bayer, Biogen, Merck, Novartis, Octapharma, Roche, Sanofi-Genzyme, Teva. Jan Mares declares no conflict of interest relevant to this manuscript. Hans-Peter Hartung has participated in meetings sponsored by and received honoraria (lectures, advisory boards, consultations) from pharmaceutical companies marketing treatments for MS: Alexion, Bayer, Biogen, Bristol Myers Squibb, GeNeuro, Horizon Therapeutics, Merck, Novartis, Octapharma, Roche, TG Therapeutics, UCB.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Krajnc, N., Bsteh, G., Berger, T. et al. Monoclonal Antibodies in the Treatment of Relapsing Multiple Sclerosis: an Overview with Emphasis on Pregnancy, Vaccination, and Risk Management. Neurotherapeutics 19, 753–773 (2022). https://doi.org/10.1007/s13311-022-01224-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-022-01224-9