
Abstract
Selective depletion of CD20+ B cells by anti-CD20 monoclonal antibodies as monotherapy in multiple sclerosis (MS) profoundly suppresses acute inflammatory disease activity and signifies an important advance in the treatment of relapsing-remitting MS. Ocrelizumab, a humanized anti-CD20 monoclonal antibody, is also the first proven therapy to lessen disability progression in primary progressive MS—a breakthrough for patients with a disease that had no proven therapy. Ocrelizumab is generally well tolerated, with the most common adverse events experienced being infusion reactions and infections. In ocrelizumab trials in MS a numerical imbalance in the risk of malignancies was observed. In this article, we review advances in anti-CD20 B-cell-depleting biological therapies for MS, including ocrelizumab, rituximab, and ofatumumab.
Similar content being viewed by others


Introduction
Although multiple sclerosis (MS) was traditionally considered to be a T-cell-mediated autoimmune disease, the success of several clinical trials with B-cell-depleting therapies [anti-CD20 monoclonal antibodies (mAbs)] has highlighted an essential role for B cells in MS pathogenesis. Histopathologically, MS is characterized by inflammation, demyelination, and neurodegeneration, and the inflammatory infiltrate observed in active lesions consists of both T and B lymphocytes [1]. Most proven therapies for relapsing forms of MS target both cell types, by inhibiting replication, altering trafficking, modulating function, or depleting subpopulations of these cells [2]. It now seems likely that B-cell effects are integral to the mechanism of action of many proven MS therapies [3], whereas treatment strategies that purely target T cells, such as selective CD4+ T-cell depletion [4], have thus far not been fully effective.
B cells secrete cytokines that modulate a variety of immune responses via proinflammatory and regulatory effects. B cells also serve as highly effective antigen presenting cells that present selected antigens recognized by the surface immunoglobulin molecule that are then internalized and presented to T cells in the context of histocompatibility proteins [5]. B cells and their plasma cell derivatives also produce antibodies, including clonally expanded IgG oligoclonal bands detectable in the cerebrospinal fluid of about 90% of patients with MS [6]. Although still controversial, the presence of B-cell-rich ectopic meningeal lymphoid aggregates—a central nervous system (CNS) manifestation of tertiary lymphoid organ formation recognized to occur in other organ systems in other autoimmune diseases—has recently been recognized in MS, particularly in the later stages of disease. These meningeal lymphoid follicles are sometimes associated with adjacent cortical demyelination and nerve and axon loss [7, 8]. In patients with MS, B cells also appear to be polarized to augment proinflammatory immune functions [9], display deficits in peripheral but not central tolerance [10], and to harbor numerous genetic variants that influence MS risk [11]. Selective depletion of CD20+ B cells has proven to be an extremely robust treatment strategy for suppressing inflammatory disease activity in relapsing-remitting MS (RRMS) and is also the first proven strategy to reduce disability progression in primary progressive MS (PPMS). In this article, we review advances in anti-CD20 B-cell-depleting biological therapies.
CD20 in the B-Cell Lineage
CD20 is a cell-surface antigen expressed on most B cells [12]. Lymphoid stem cells, most plasmablasts, and nearly all plasma cells do not express CD20 [13]. The practical implication of differential CD20 expression is that anti-CD20 mAbs tend not to substantially reduce IgG antibody levels despite profound depletion of CD20+ B cells, because plasma cells that produce most IgG are not depleted by anti-CD20 mAbs. B cells reconstitute after discontinuation of CD20+-depleting therapy, particularly by CD20-negative early B-cell precursors that are not affected by anti-CD20 therapies. Another noteworthy feature of therapy with anti-CD20 antibodies is that removal of B cells occurs more efficiently in peripheral blood than in lymphoid organs, including meningeal lymphoid follicles discussed above; as only 2% of the body’s total B-cell pool is in the peripheral blood, the resistance of tissue-residing B cells might account for the relative safety of these therapies. Following B-cell therapy, repopulating B cells consist of larger numbers of naïve B cells and fewer antigen-educated memory B cells and plasmablasts [14], possibly explaining the continuing suppression of MS disease activity noted even after B-cell reconstitution has occurred [14,15,16].
There is also a rare, heterogeneous population of CD3+ T cells that express CD20 and that are also depleted with anti-CD20 therapy, but the relevance, if any, of CD3+CD20+ T cells to MS pathogenesis or treatment response is unknown [17].
CD19 is another pan B-cell marker, with the additional spectrum of CD19+ expression on some plasmablasts that are CD20−. Because anti-CD20 therapies interfere with detection of the CD20 antigen, in clinical practice CD19+ B-cell levels are widely used as a proxy to measure the extent of B-cell depletion accompanying anti-CD20 therapy.
Anti-CD20 B-Cell Depleting Therapies in RRMS
Rituximab
Rituximab (RTX), an IgG1 mouse–human mAb against CD20, was the first monoclonal anti-CD20 antibody to be licensed for use in human disease. RTX was initially approved for CD20+ non-Hodgkin lymphoma and subsequently for CD20+ chronic lymphocytic leukemia, rheumatoid arthritis (with methotrexate, after failure of a tumor necrosis factor-α antagonist), and granulomatosis with polyangiitis and microscopic polyangiitis. RTX was also the first anti-CD20 agent used in neuromyelitis optica [18] and MS [14, 19], and is widely used as an “off label” therapy for both conditions [20]. CSF concentrations of intravenous RTX were measured at about 0.1% in a series of patients with CNS lymphoma also treated with intrathecal methotrexate or Ara-C [21]—but, nevertheless, there still appears to be a profound depletion of intrathecal B cells with standard intravenous dosing [22].
In a phase II, double-blind, placebo-controlled manufacturer-sponsored 48-week trial in 104 patients with RRMS [14], intravenous RTX at a dose of 1000 mg intravenously on days 1 and 15 resulted in a marked reduction of total gadolinium-enhancing magnetic resonance imaging (MRI) lesions and total new gadolinium-enhancing lesions at weeks 12, 16, 20, and 24 (with a relative reduction of 91%) that was sustained to 48 weeks. Relapse rates were also lower in the RTX group (40.0% vs 20.3% at week 48). At baseline, there was an imbalance in that the patients randomized to the placebo group had fewer gadolinium-enhancing MRI lesions; however, if this imbalance were to have an effect it would be expected to bias against RTX and, furthermore, the strong observed effect of RTX on the primary endpoint survived extensive sensitivity analysis testing. By week 48 after initial dosing, CD19+ B cells had returned to 30.7% of baseline values. While median levels of serum IgM, IgG, and IgA were normal in both the RTX and placebo groups for the study population as a whole, 22.4% of RTX-treated patients versus 8.6% of placebo-treated patients had serum IgM levels below the lower limit of normal. Of the RTX-treated patients, 7.4% reported grade 3 events with the infusion (none grade 4), whereas 92.6% reported grade 1 or 2 events (vs 40% in the placebo group). It should be noted, however, that in this study no glucocorticoid premedication was administered before RTX infusions. There was no difference in rates of infection between the RTX and placebo groups, with 2 serious infections (gastroenteritis and bronchitis) in the RTX group.
A second open-label manufacturer-sponsored 72-week trial of RTX in 26 patients with RRMS reported no serious adverse events but there were mild-to-moderate infusion-associated events [19].
In another investigator-initiated trial of intravenous RTX at a dose of 375 mg/m2 weekly × 4 doses in 30 patients with RRMS with breakthrough disease on injectable therapy with glatiramer acetate or an interferon (IFN)-β formulation, there was a 74% reduction in total gadolinium-enhancing lesions that was evident as early as 12 weeks postdose, the first surveillance MRI time point [23].
Off-label intravenous RTX (500–1000 mg every 6–12 months) in 557 patients with RRMS, 198 with secondary progressive, and 67 with PPMS was associated with markedly low relapse rates (including an annualized relapse rate of 0.044 for RRMS) in a retrospective, uncontrolled, observational, multicenter report from 3 university medical centers in Sweden [20]. Only 4.6% of RTX-treated patients exhibited any gadolinium-enhancing MRI lesions [20]. In a group of 72 patients from these same Swedish centers who switched from natalizumab to either RTX or fingolimod due to JC virus seropositivity, 1.8% of RTX-treated patients versus 17.6% of fingolimod-treated patients experienced a clinical relapse [hazard ratio (HR) for RTX of 0.1, 95% confidence interval 0.02–0.43] and the odds ratio of any gadolinium-enhancing lesions was 0.05 for RTX (with 1.4% of RTX-treated patients vs 24.2% with fingolimod exhibiting gadolinium-enhancing lesions) [24].
There are limited data about the safety and efficacy of RTX in children with MS and other CNS inflammatory conditions, with benefit observed in uncontrolled studies in most treated children; however, infusion reactions (including rare anaphylaxis) and infections (including rare severe and fatal infections) have also been reported [25, 26].
Ocrelizumab
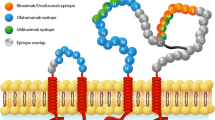
Ocrelizumab (OCR) is a humanized anti-CD20 IgG1 mAb that targets the large extracellular loop of CD20 [27], a different binding epitope than that of RTX [28]. After binding to CD20, OCR depletes B cells via apoptosis, antibody-dependent cellular cytotoxicity (ADCC), antibody-dependent cell-mediated phagocytosis, and complement-dependent cytotoxicity (CDC), mechanisms thought to be similar with other type 1 CD20 antibodies, including RTX and ofatumumab [28]. OCR, compared with RTX, has relatively stronger ADCC and relatively weaker CDC activities.
In a phase II randomized, parallel, double-blind, placebo-controlled manufacturer-sponsored trial in 220 patients testing intravenous OCR 600 mg (lower dose) and 2000 mg (high dose) versus subcutaneous IFN-β1a (30 μg), the number of gadolinium-enhancing lesions at week 24 was 89% lower in the 600 mg group and 96% lower in the 2000 mg group [16]. Infusion-related adverse events were recorded in 35% of patients treated with 600 mg, 44% with 2000 mg, and 9% with IFN-β1a. There was no difference in serum IgG over time in any group. B-cell counts were reduced by > 99% in both groups, an effect that persisted through week 24. There was no imbalance in adverse events between groups [16]. Based upon these data, a decision was made to move forward with the 600 mg dose for phase III trials.
Two identical phase III, double-blind, placebo-controlled, manufacturer-sponsored trials were conducted to test intravenous OCR 600 mg every 24 weeks (with the first dose split into two 300 mg infusions given 2 weeks apart followed by subsequent infusions of 600 mg at 24-week intervals) or subcutaneous IFN-β1a 44 μg 3× weekly [27]. Both studies were 96 weeks in duration and recruited 821 and 835 patients with relapsing MS, respectively, aged 18 to 55 years, Expanded Disability Status Scale (EDSS) 0 to 5.5 with recent disease activity (≥2 documented clinical relapses within the last 2 years or 1 clinical relapse within the past year). The annualized relapse rate was 46% lower with OCR (0.16) than IFN-β1a (0.29). The hazard of disability progression confirmed at 12 weeks was 40% lower in the OCR group (9.1% vs 13.6%), and at 24 weeks the risk was 40% lower with OCR. The mean number of gadolinium-enhancing lesions in the OCR arm was reduced by 94% and 95% in trials 1 and 2, respectively (0.02 vs 0.29 gadolinium-enhancing lesions in trial 1 and 0.02 vs 0.42 in trial 2). The mean number of new or newly enlarging T2 MRI lesions in the OCR arm was reduced by 77% and 83% in trials 1 and 2 versus IFN-β1a. As treatments can take time to achieve a maximal disease modifying effect, it can also be informative to analyze efficacy at later time points. From week 24 to 48 on treatment, the mean number of new or newly enlarging T2 MRI lesions was reduced by 94% and 96% in trials 1 and 2, respectively, and from week 48 to week 96 the mean number of lesions was reduced by 98% and 97%, respectively. No evidence of disease activity (NEDA) [29, 30], also sometimes referred to as disease activity-free status, was defined in the protocol as no relapse, no 12- or 24-week-confirmed disability progression, no new or newly enlarging T2 MRI lesions, and no new gadolinium-enhancing lesions by week 96. In trial 1, 47.9% of patients in the OCR group versus 29.2% in the IFN-β1a group met NEDA criteria; in trial 2, 47.5% in the OCR group versus 25.1% in the IFN-β1a group met NEDA criteria. Infusion-related reactions were reported in 34.3% of OCR treated patients (vs 9.7% treated with subcutaneous IFN-β1a and placebo infusions), with 1 patient in the OCR group experiencing a life-threatening bronchospasm event with recovery. No significant differences were observed in serious adverse events. Neoplasm was detected in 4 patients in the OCR group versus 2 in the IFN group, with 5 additional cases reported in the open-label phase [27]. The US Food and Drug Administration licensed OCR with an indication for relapsing MS in March 2017.
Ofatumumab
Ofatumumab (OFA) is a human IgG1 monoclonal antibody that binds to CD20. OFA binds to a membrane-proximal epitope different than RTX and OCR [28] that may lead to greater complement-dependent cytotoxicity [31]. In a phase II, randomized, double-blind, placebo-controlled, manufacturer-sponsored study, 38 patients with RRMS were randomized to intravenous OFA at doses of 100 mg, 300 mg, or 700 mg or placebo 2 weeks apart [31]. There was a 99% reduction in new brain MRI lesion activity in the first 24 weeks after OFA treatment. The most common adverse event was an infusion reaction with the first dose. There was no reduction in serum IgG. A second phase II manufacturer-sponsored study of 4 doses of subcutaneous OFA versus placebo in 238 patients with RRMS has only, to date, been presented in abstract form [32]. In that study, there was a 65% reduction in new gadolinium-enhancing lesions for each OFA dose with > 90% reductions for weeks 4 to 12. Two identical, manufacturer sponsored, head-to-head, double-blind, phase III trials testing OFA 20 mg subcutaneous versus teriflunomide are enrolling [33].
Anti-CD20 B-Cell-Depleting Therapies in Progressive MS
Ten to 15% of patients with MS do not experience clinical relapses [34,35,36], presenting instead with insidious worsening of function and accumulation of disability— [37] PPMS. PPMS is pathologically and genetically similar to relapse-onset MS, but differs in that males and females are approximately equally affected. With the notable exception of OCR, none of the other anti-inflammatory therapies for RRMS is of proven benefit in reducing disability accumulation in PPMS [38].
A phase II randomized, double-blind, placebo-controlled manufacturer-sponsored trial of RTX at a dose of 1000 mg intravenous × 2 doses every 24 weeks in 439 patients with PPMS randomized 2:1 to active therapy failed to hit its primary endpoint: time to confirmed disease progression sustained for 12 weeks (CDP12) at week 96 was not statistically different between groups (38.5% in the placebo group vs 30.2% in the RTX group; p = 0.14) [39]. However, there was a clear trend favoring RTX, and in a subgroup analysis of patients aged 50 years or younger, patients with gadolinium-enhancing lesion on brain MRI, or both, the time to CDP12 was delayed in the RTX group [39].
A phase III randomized, placebo-controlled, manufacturer-sponsored study of OCR at a dose of 600 mg, given as two 300 mg intravenous infusions administered 2 weeks apart every 24 weeks, was undertaken in 732 patients with PPMS aged < 56 years. The proportion of patients with CDP12 (the primary outcome) was 32.9% in the OCR group versus 39.3% with placebo (HR 0.75, 95% confidence interval 0.59–0.98; p = 0.03). The HR for confirmed disability progression at 24 weeks (CDP24) was similar [HR 0.75 (p = 0.04), 29.6% in the OCR group vs 35.7% in the placebo arm). Worsening on the 25-foot timed walk test was 29.3% less in the OCR group. OCR was also associated with less brain volume loss, as measured by the adjusted mean percent change in brain volume from week 24 to week 96. Infusion-related reactions were reported in 39.9% of OCR-treated patients versus 25.5% in the placebo group, including 2 withdrawals from the study from the OCR group due to infusion reactions. Infusion reactions were most severe and more frequent with the initial infusion. The rate of serious infections was similar between groups. However, there was an imbalance in the number of neoplasms detected, occurring in 2.3% of OCR-treated patients versus 0.8% of controls [40]. Key inclusion criteria for the study included a relatively young age (the mean age was 44 years, enrollment allowed 18–55 years) and moderate levels of disability [median baseline EDSS of 4.5, enrollment allowed EDSS 3–6.5 (requiring a walker or 2 canes for ambulation)]. The median disease duration (time since onset of MS symptoms) was 6 years, and median time since MS diagnosis was 1.6 years. The US Food and Drug Administration approved OCR for PPMS in March 2017 under a breakthrough therapy designation with priority review.
Comparing the RTX and OCR PPMS trials, key differences include an older mean age (50.1 years vs 44 years), a longer disease duration (9 years since onset vs 6.7 years for OCR) and slightly higher median EDSS at enrollment (5.0 vs 4.5) in the RTX trial. The primary endpoint for the RTX PPMS trial was measured at 96 weeks with a reduced hazard of CDP12 of 0.77 (30.2% for RTX vs 38.5% for placebo) versus a hazard of 0.75 for OCR at both 12 and 24 weeks (96 week data for OCR has not yet been reported). While the benefits in reducing disability progression in progressive MS with OCR are statistically significant, the effect size was modest in this relatively young sample of patients with PPMS, many of whom had evidence of acute inflammatory activity on MRI. Whether the incomplete protection observed was due to the inadequacy of currently available clinical and imaging endpoints to detect meaningful change in PPMS to the limited penetration of drug into B-cell-rich lymphoid follicles in the CNS or to other disease mechanisms driving PPMS biology is unknown, but it is likely that all of these possibilities were contributors to the incomplete effects of OCR on measures of clinical progression and neurodegeneration. A trial of intrathecal RTX for secondary progressive MS was stopped early for futility following an interim biomarker analysis, in part owing to inadequate depletion of intrathecal B cells, despite profound circulating B-cell depletion in blood [41].
Although the availability of OCR for patients represents a long-awaited, transformational change in what is possible for patients with PPMS, clearly more research is needed to identify even more effective treatment options and approaches for this progressive and disabling form of the disease. Furthermore, whether the positive effects of OCR can be generalized to patients with PPMS who would not have been candidates for the pivotal study (because of age, disability, or clinical trajectory) remains to be determined.
Practical Safety Considerations When Prescribing Anti-CD20 B-Cell-Depleting Therapies in MS
Infusion Reactions
Infusion reactions are the most common adverse event reported with anti-CD20 mAbs, whether chimeric, humanized, or human. The relative frequencies of infusion-related reactions with OCR, RTX, and OFA have not been studied in head-to-head fashion, and in the MS trials differences in protocols (e.g., premedication with glucocorticoids in the OCR and OFA, but not RTX, studies) make any cross-trial comparisons difficult. OCR has greater relative ADCC than CDC activity, whereas the opposite is true for RTX and OFA. As CDC activity is believed to play an important role in triggering infusion-related reactions [42], OCR might be expected to have a more favorable mechanism of action in this regard.
Infusion reactions can manifest in many ways, including itching, rash, fever, chills, rigors, throat irritation, nausea, headache, cough, tiredness, dizziness, headache, hypotension, bronchospasms, or angioedema. For intravenously administrated mAbs, it is essential to have established safety, surveillance, and stop protocols in place as part of the infusion practice and systems in place to handle severe, emergent infusion reactions. Premedication with an antihistamine, such as diphenhydramine, as well as an antipyretic such as acetaminophen, can be helpful in reducing the risk of infusion reactions. Premedication with glucocorticoids, typically intravenous methylprednisolone at a dose of 100 mg can help reduce the risk of serious infusion reactions. Premedication with intravenous methylprednisolone (or an equivalent glucocorticoid) is recommended in the RTX label for rheumatoid arthritis and the OCR label for MS [43].
Infection
While rates of serious adverse infections for RTX monotherapy and OCR monotherapy in clinical trials were not significantly different than the immunomodulatory or placebo control arms, serious infections, including opportunistic infections, have been reported with anti-CD20 mAbs. Clinical trials are too short and their sample sizes are too small to measure such rare or longer-term risks. For non-MS indications, anti-CD20 mAbs are often used in combination with other immunosuppressants, including with methotrexate for rheumatoid arthritis, with steroids for granulomatosis with polyangiitis, and with polychemotherapy for lymphoma, whereas anti-CD20 mAbs are widely used as monotherapy in MS. Tuberculosis reactivation is a specific consideration, especially in endemically affected areas or populations, and prescreening and active surveillance with latent tuberculosis testing is recommended. Owing to the occurrence of fulminant hepatitis from hepatitis B reactivation following B-cell depletion, screening should also include hepatitis B surface antigen, surface antibody, and core antibody, with testing for hepatitis B DNA by polymerase chain reaction if the surface antigen or core antibody are positive [44]. There is concern about the use of B-cell-depleting therapy in patients who are hepatitis B core antibody-positive but surface antibody, antigen, and polymerase chain reaction negative [45, 46]; hepatology consultation and prophylaxis with antiviral therapy such as entecavir or lamivudine should be considered in such cases. The risk of progressive multifocal leukoencephalopathy (PML) with RTX therapy for nonmalignant indications appears to be relatively low—estimated to be about 1 case per 25,000 individuals treated with rheumatoid arthritis, a condition that is associated with PML independent of any therapy [47]—but ongoing vigilance and monitoring is advised, in particular with real-world and potentially longer-term use of OCR and RTX in MS and especially in patients who switch from PML-associated disease-modifying therapies to anti-CD20 mAbs.
Vaccination
Patients should be advised to complete any required vaccinations at least 6 weeks prior to treatment initiation. Live-attenuated or live vaccines are not recommended during treatment and until B-cell recovery.
Malignancy
There was an observed imbalance in malignancies with OCR versus IFN-β1a in RRMS and placebo with PPMS in the MS clinical trials [27, 40]. Breast cancer occurred in 6 of 781 females treated with OCR and none of 668 females treated with IFN-β1a or placebo. There are a number of reasons the observed imbalance may not be biologically significant: the total numbers of patients with breast or other cancers in the OCR-treated populations were not higher than epidemiological expectations, the incidence has fallen during the open-label extension studies, and no increase in the risk of breast cancer or other malignancies has been noted with RTX, now in use for 20 years and with more than 4 million total infusions. Patients receiving OCR should follow standard breast cancer screening guidelines, and further research in the MS population will be needed to clarify risk of malignancy and guide strategies for risk mitigation. Because there is currently no risk-mitigation program associated with OCR, determination as to whether longer-term exposure to this medication is associated with an increased malignancy risk will be dependent on voluntary reporting.
Pregnancy
Because many patients living with MS are in their reproductive years, pregnancy poses particular challenges in regard to MS disease-modifying therapies [48]. Exposure to small molecules or biologics is associated with potential risk to the fetus. Therefore, agents that have pharmacodynamic effects that exceed their pharmacokinetic elimination have the potential to provide relief from MS disease activity for the mother and carry low risk for the fetus. Although not formally studied in this special population, B-cell-depleting mAbs may fit this profile in that their effect on B-cell depletion appears to greatly exceed their pharmacologic half-life. It is conceivable that with appropriate pregnancy planning, B-cell-depleting mAbs could be used for prevention of MS disease activity during pregnancy if administered sufficiently far enough in advance so that the maternal serum concentration of the mAb would be low enough not to affect the gestating fetus. Administration of B-cell-depleting mAbs during pregnancy is relatively contraindicated because of potential harm to the developing fetus and infant [43]. Transient B-cell depletion and peripheral lymphocytopenia were reported in infants born of mothers treated with B-cell-depleting mAbs during pregnancy. Furthermore, pregnancy studies in primates showed severe decreases in B cells in the neonates, renal toxicity, testicular toxicity, lymphoid follicle formation in the bone marrow, and perinatal deaths. It is recommended that women of childbearing potential use contraception while receiving OCR and for 6 months after the last infusion.
Future Directions
Anti-CD20 B-cell-depleting therapies represent a breakthrough for treating RRMS and PPMS. For patients with relapsing forms of MS, including secondary progressive MS with ongoing relapses, the benefits of anti-CD20 therapy on clinical and MRI measures of focal inflammatory disease activity are dramatic, and for patients with PPMS at long last a partially effective therapy is now available. Further work will be needed to determine if the benefits observed in the pivotal trials might be further augmented by earlier treatment; the extent to which these short-term effects translate into disability protection longer-term in both relapsing and progressive forms of MS and via what mechanisms; the optimal duration of treatment with B-cell-depleting therapies; and when or if such treatment can be safely stopped. Understanding mechanisms by why B-cell depletion is so effective in MS may also promote a better understanding of MS pathogenesis. Monitoring and measuring the real-world, long-term safety of B-cell-depleting mAbs in MS will be essential to help position these therapies within the greater context of available MS disease-modifying therapies.
References
Lassmann H, Bradl M. Multiple sclerosis: experimental models and reality. Acta Neuropathol 2017;133(2):223–244.
Bruck W, Gold R, Lund BT, et al. Therapeutic decisions in multiple sclerosis: moving beyond efficacy. JAMA Neurol 2013;70(10):1315–1324.
Longbrake EE, Cross AH. Effect of multiple sclerosis disease-modifying therapies on B cells and humoral immunity. JAMA Neurol 2016;73(2):219–225.
van Oosten BW, Lai M, Hodgkinson S, et al. Treatment of multiple sclerosis with the monoclonal anti-CD4 antibody cM-T412: results of a randomized, double-blind, placebo-controlled, MR-monitored phase II trial. Neurology 1997;49(2):351–357.
Li R, Rezk A, Miyazaki Y, et al. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci Transl Med 2015;7(310):310ra166.
Dobson R, Ramagopalan S, Davis A, Giovannoni G. Cerebrospinal fluid oligoclonal bands in multiple sclerosis and clinically isolated syndromes: a meta-analysis of prevalence, prognosis and effect of latitude. J Neurol Neurosurg Psychiatry 2013;84(8):909–914.
Howell OW, Reeves CA, Nicholas R, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011;134(Pt 9):2755–2771.
Magliozzi R, Howell OW, Reeves C, et al. A Gradient of neuronal loss and meningeal inflammation in multiple sclerosis. Ann Neurol 2010;68(4):477–493.
Bar-Or A, Fawaz L, Fan B, et al. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol 2010;67(4):452–461.
Kinnunen T, Chamberlain N, Morbach H, et al. Specific peripheral B cell tolerance defects in patients with multiple sclerosis. J Clin Invest 2013;123:2737–2741.
International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium 2, Sawcer S, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011;476(7359):214–219.
Stashenko P, Nadler LM, Hardy R, Schlossman SF. Characterization of a human B lymphocyte-specific antigen. J Immunol 1980;125(4):1678–1685.
Krumbholz M, Derfuss T, Hohlfeld R, Meinl E. B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat Rev Neurol 2012;8(11):613–623.
Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 2008;358(7):676–688.
Hauser SL, Li D, Calabresi P, et al. Week 144 results of a phase II, randomized, multicenter trial assessing the safety and efficacy of ocrelizumab in patients with relapsing–remitting multiple sclerosis (RRMS). Neurology 2013;80(7 Supplement, S31.004).
Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011;378(9805):1779–1787.
Palanichamy A, Jahn S, Nickles D, et al. Rituximab efficiently depletes increased CD20-expressing T cells in multiple sclerosis patients. J Immunol 2014;193(2):580–586.
Cree BA, Lamb S, Morgan K, Chen A, Waubant E, Genain C. An open label study of the effects of rituximab in neuromyelitis optica. Neurology 2005;64(7):1270–1272.
Bar-Or A, Calabresi PA, Arnold D, et al. Rituximab in relapsing-remitting multiple sclerosis: a 72-week, open-label, phase I trial. Ann Neurol 2008;63(3):395–400.
Salzer J, Svenningsson R, Alping P, et al. Rituximab in multiple sclerosis: a retrospective observational study on safety and efficacy. Neurology 2016;87(20):2074–2081.
Rubenstein JL, Combs D, Rosenberg J, et al. Rituximab therapy for CNS lymphomas: targeting the leptomeningeal compartment. Blood 2003;101(2):466–468.
Ruhstaller TW, Amsler U, Cerny T. Rituximab: active treatment of central nervous system involvement by non-Hodgkin's lymphoma? Ann Oncol 2000;11(3):374–375.
Naismith RT, Piccio L, Lyons JA, et al. Rituximab add-on therapy for breakthrough relapsing multiple sclerosis: a 52-week phase II trial. Neurology 2010;74(23):1860–1867.
Alping P, Frisell T, Novakova L, et al. Rituximab versus fingolimod after natalizumab in multiple sclerosis patients. Ann Neurol 2016;79(6):950–958.
Rommer PS, Dorner T, Freivogel K, et al. Safety and clinical outcomes of rituximab treatment in patients with multiple sclerosis and neuromyelitis optica: experience from a national online registry (GRAID). J Neuroimmune Pharmacol 2016;11(1):1–8.
Dale RC, Brilot F, Duffy LV, et al. Utility and safety of rituximab in pediatric autoimmune and inflammatory CNS disease. Neurology 2014;83(2):142–150.
Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus Interferon beta-1a in relapsing multiple sclerosis. N Engl J Med 2017;376(3):221–234.
Klein C, Lammens A, Schafer W, et al. Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. mAbs 2013;5(1):22–33.
Giovannoni G, Turner B, Gnanapavan S, Offiah C, Schmierer K, Marta M. Is it time to target no evident disease activity (NEDA) in multiple sclerosis? Mult Scler Relat Disord 2015;4(4):329–333.
Bevan CJ, Cree BA. Disease activity free status: a new end point for a new era in multiple sclerosis clinical research? JAMA Neurol 2014;71(3):269–270.
Sorensen PS, Lisby S, Grove R, et al. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: a phase 2 study. Neurology 2014;82(7):573–581.
Bar-Or A, Grove R, Austin D, et al. The MIRROR Study: a randomized, double-blind, placebo-controlled, parallel-group, dose-ranging study to investigate the safety and MRI efficacy of subcutaneous ofatumumab in subjects with relapsing-remitting multiple sclerosis. Neurology 2014;82(10):S23.006.
clinicaltrials.gov. Efficacy and Safety of Ofatumumab Compared to Teriflunomide in Patients With Relapsing Multiple Sclerosis (ASCLEPIOS I) [Available from: https://clinicaltrials.gov/ct2/show/NCT02792218]; Efficacy and Safety of Ofatumumab Compared to Teriflunomide in Patients With Relapsing Multiple Sclerosis. (ASCLEPIOS II) [Available from: https://clinicaltrials.gov/ct2/show/NCT02792231]. Accessed May 31, 2017.
Weinshenker BG, Bass B, Rice GP, et al. The natural history of multiple sclerosis: a geographically based study. I. Clinical course and disability. Brain 1989;112 ( Pt 1):133–146.
Tremlett H, Zhao Y, Devonshire V. Natural history comparisons of primary and secondary progressive multiple sclerosis reveals differences and similarities. J Neurol 2009;256(3):374–381.
Confavreux C, Vukusic S, Moreau T, Adeleine P. Relapses and progression of disability in multiple sclerosis. N Engl J Med 2000;343(20):1430–1438.
Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011;69(2):292–302.
Ontaneda D, Thompson AJ, Fox RJ, Cohen JA. Progressive multiple sclerosis: prospects for disease therapy, repair, and restoration of function. Lancet 2017;389:1357–1366.
Hawker K, O'Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol 2009;66(4):460–471.
Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 2017;376(3):209–220.
Komori M, Lin YC, Cortese I, et al. Insufficient disease inhibition by intrathecal rituximab in progressive multiple sclerosis. Ann Clin Transl Neurol 2016;3(3):166–179.
van der Kolk LE, Grillo-Lopez AJ, Baars JW, Hack CE, van Oers MH. Complement activation plays a key role in the side-effects of rituximab treatment. Br J Haematol 2001;115(4):807–811.
RTX & OCR Labels. Rituxan Prescribing Information [Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/103705s5311lbl.pdf]; Ocrevus Prescribing Information [Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761053lbl.pdf]. Accessed May 31, 2017.
Dyson JK, Jopson L, Ng S, et al. Improving testing for hepatitis B before treatment with rituximab. Eur J Gastroenterol Hepatol 2016;28(10):1172–1178.
Lee J, Park JY, Huh KH, et al. Rituximab and hepatitis B reactivation in HBsAg-negative/ anti-HBc-positive kidney transplant recipients. Nephrol Dialysis Transplant 2017;32(4):722–729.
Seto WK, Wong DH, Chan TY, et al. Association of hepatitis B core-related antigen with hepatitis B virus reactivation in occult viral carriers undergoing high-risk immunosuppressive therapy. Am J Gastroenterol 2016;111(12):1788–1795.
Clifford DB, Ances B, Costello C, , et al. Rituximab-associated progressive multifocal leukoencephalopathy in rheumatoid arthritis. Arch Neurol 2011;68(9):1156–1164.
Bove R, Alwan S, Friedman JM, et al. Management of multiple sclerosis during pregnancy and the reproductive years: a systematic review. Obstetr Gynecol 2014;124(6):1157–1168.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest Statement
J.M.G. has received compensation for consulting for Genentech and Medimmune. He has received research support from Genentech, MedDay, and Quest Diagnostics. He has also received compensation for medical legal consulting. B.A.C.C. has received compensation for consulting from AbbVie, Biogen, EMD Serono, Novartis, and Shire. S.L.H. serves on the Scientific Advisory Boards of Annexon, Bionure, Symbiotix, and Molecular Stethoscope, and on the Board of Trustees of Neurona; and also reports receiving travel reimbursement and writing assistance from F. Hoffmann-La Roche for CD20-related meetings and presentations.
Funding
S.L.H.: R01NS026799; R01NS049477; NMSS RG 2899; The Conrad N. Hilton Foundation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 498 kb)
Rights and permissions
About this article

Cite this article
Gelfand, J.M., Cree, B.A.C. & Hauser, S.L. Ocrelizumab and Other CD20+ B-Cell-Depleting Therapies in Multiple Sclerosis. Neurotherapeutics 14, 835–841 (2017). https://doi.org/10.1007/s13311-017-0557-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-017-0557-4