
Abstract
Demyelinating forms of Charcot-Marie-Tooth disease (CMT) are genetically and phenotypically heterogeneous and result from highly diverse biological mechanisms including gain of function (including dominant negative effects) and loss of function. While no definitive treatment is currently available, rapid advances in defining the pathomechanisms of demyelinating CMT have led to promising pre-clinical studies, as well as emerging clinical trials. Especially promising are the recently completed pre-clinical genetic therapy studies in PMP-22, GJB1, and SH3TC2-associated neuropathies, particularly given the success of similar approaches in humans with spinal muscular atrophy and transthyretin familial polyneuropathy. This article focuses on neuropathies related to mutations in PMP-22, MPZ, and GJB1, which together comprise the most common forms of demyelinating CMT, as well as on select rarer forms for which promising treatment targets have been identified. Clinical characteristics and pathomechanisms are reviewed in detail, with emphasis on therapeutically targetable biological pathways. Also discussed are the challenges facing the CMT research community in its efforts to advance the rapidly evolving biological insights to effective clinical trials. These considerations include the limitations of currently available animal models, the need for personalized medicine approaches/allele-specific interventions for select forms of demyelinating CMT, and the increasing demand for optimal clinical outcome assessments and objective biomarkers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction to Demyelinating CMT
Charcot-Marie-Tooth disease (CMT) refers to a heterogeneous set of genetic peripheral nerve disorders that collectively comprise the most common inherited neurological disease, with an estimated prevalence of 1:2500 individuals [1, 2]. The varied forms of CMT span the phenotypic spectrum from subclinical neuropathy to that resulting in early loss of ambulation in the setting of severe weakness and sensory loss. Most commonly, CMT presents as a sensory motor neuropathy, though primarily sensory and motor forms (hereditary sensory autonomic neuropathy and distal hereditary motor neuropathy, respectively) can also occur. Disease onset can also vary widely, with some patients showing signs in infancy and others developing symptoms in their elderly years.
Historically, the classification of CMT has anchored on the mode of inheritance and the primary pathology observed in nerve, as reflected in nerve conduction studies. Demyelinating CMT, which is the focus of this review, is defined by upper extremity motor conduction velocities (CV) of less than 38 m/s, resulting from homogeneous demyelination of large, myelinated axons. Pathological evidence of demyelination can also be observed on nerve biopsy, though with the increasing yield of genetic testing, biopsy is generally reserved only for cases that pose a particular diagnostic challenge.
Demyelinating forms of CMT are characterized as CMT1 (autosomal dominant (AD) inheritance), CMT4-demyelinating (autosomal recessive (AR) inheritance), and X-linked forms (Table 1 and Fig. 1). In the era of increased genetic testing, the phenotypic spectrum associated with specific genes in CMT has broadened significantly, and the classification of CMT is gradually evolving to incorporate the specific gene that is mutated in individuals with CMT [3]. It should also be emphasized that while the term “demyelinating” CMT has historically been used, there is evidence to suggest that some forms of CMT actually involve a developmental abnormality of myelin formation or “dysmyelination,” which may both cause abnormal nerve function and predispose nerves to demyelination throughout the course of disease [4,5,6].

Demyelinating CMT genes
Many of the forms of CMT result in similar clinical manifestations, which can pose a challenge in establishing a precise diagnosis at the bedside. While select subtypes do have distinguishing phenotypic features, the majority of patients with CMT present with foot deformities, including pes cavus, pes planus, or hammertoes, length dependent muscle weakness and atrophy, and length-dependent sensory loss. Deep tendon reflexes are typically diminished or absent however can also be preserved or rarely even heightened in select forms of CMT. While genetic testing in CMT continues to be guided by the clinical examination, family history, and electrophysiology, testing is increasingly being accomplished through the use of next-generation sequencing (NGS) panels containing multiple disease relevant genes, rather than through sequential gene testing [7, 8]. Given the high prevalence of CMT1A, however, it is still reasonable to exclude this possibility in patients presenting with the classic demyelinating CMT phenotype as a first step in the diagnostic evaluation [9].
With the increasing use of next-generation sequencing, gene discovery in CMT has rapidly accelerated over the past two decades, with over 120 causative genes identified to date [10]. New mutations in these genes are also continuously being discovered as captured in the inherited neuropathy variant browser (http://hihg.med.miami.edu/code/http/cmt/public_html/index.html#/) [11]. While this genetic diversity can be daunting in both the clinical and research settings, it should be emphasized that of the ~ 65% of patients in whom a genetic diagnosis can be defined, ~90% have a mutation in one the four most common genes known to underlie CMT, namely PMP-22, MPZ, GJB1, and MFN2 [9, 12,13,14]. For this reason, this review will primarily focus on PMP-22, MPZ, and GJB1, which together comprise the most common forms of demyelinating CMT.
Disease-causing mutations in CMT can result in clinical neuropathy through both loss and gain of function, with the latter being more common. Gain of function mutations can cause cellular derangements that are unrelated to the primary role of the encoded protein, whereas the loss of function mutations leads to either reduced levels or abnormal function of the protein. The majority of CMT1 is mediated by gain of function mechanisms, whereas the AR CMT-demyelinating forms are more commonly caused by loss of function [15]. In the heterozygous state, AD gain of function mutations also tend to cause a more severe neuropathy than those resulting in a loss of function [16]. These distinctions have important implications for therapeutic development in these disorders.
Despite rapid advances in understanding the biology of CMT, no definitive treatment is currently available, and a few pre-clinical studies have progressed to clinical trials. As reviewed by Juneja et al., there are several critical obstacles in identifying effective treatments in CMT [10]. These obstacles include the rarity of many of the forms of CMT, the genetic and phenotypic heterogeneity of these disorders, and the challenge of developing optimal animal models and translating candidate treatments to humans. Many of the treatments investigated thus far in CMT also target pathways downstream from the initial biological insult that results from a given genetic mutation. Another important challenge therefore lies in defining ways to target upstream biological derangements as early as possible in the disease in order to minimize neurodegeneration.
As the CMT research community works to overcome these challenges, there is also increasing reason for optimism. Recent developments in the treatment of spinal muscular atrophy and transthyretin familial polyneuropathy have been transformative in paving the way for effective genetic therapy approaches for neuromuscular disorders; and the emergence of genetic therapies for select forms of CMT now offer tangible hope for people with these conditions [17,18,19].
Introduction to Myelin Biology
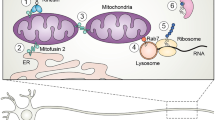
The myelin sheath plays a critical role in enabling rapid conduction of nerve impulses within the peripheral nervous system. Peripheral nerve myelin is a multi-layer structure composed of Schwann cells, which surround single axons in a one-to-one ratio (Fig. 2). The myelin sheath includes compact regions of myelin (which are predominant) as well as non-compact regions, which include the paranode and juxtaparanode and are adjacent to the nodes of Ranvier (gaps between two myelin segments that enable the influx of ions) [20]. Outward potassium currents take place at the juxtaparanode and internode. This molecular architecture allows for preservation of the depolarization current by the high internodal capacitance and enables its efficient propagation until the next node of Ranvier, where a new action potential is triggered. This allows for a higher conduction velocity in myelinated nerve fibers than that observed in unmyelinated fibers. In addition to facilitating rapid conduction, myelin also protects axons, supports signaling and communication between axons and Schwann cells, and provides metabolic support to axons [21]. The compact and non-compact regions of myelin include distinct proteins, such as PMP-22, MPZ, and myelin basic protein within compact myelin [16]. The expression of these proteins is highly regulated within Schwann cells, with even slight changes resulting in abnormal development or maintenance of the myelin sheath [22, 23].

Schematic of Schwann Cell
In humans, myelin formation occurs in the postnatal period; however, Schwann cells continue to maintain myelin integrity into adulthood. Importantly, in addition to causing demyelinating CMT, mutations in genes encoding myelin proteins can sometimes result in primarily axonal neuropathies [24,25,26]. As mentioned, it is also important to distinguish abnormal myelin development, or “dysmyelination,” from the disruption of normally formed myelin or “demyelination,” as the nature and timing of myelin injury are integral to the varied pathomechanisms of demyelinating CMT and have important implications for the development of treatments.
PMP-22- Associated Neuropathy: Epidemiology and Clinical Features
CMT1A is the most common hereditary neuropathy, accounting for ~50% of all CMT and 70–80% of CMT1 [9, 13, 14]. CMT1A has a de novo mutation rate of 10%, and patients therefore may report the absence of relevant family history [27]. The disease most commonly manifests with a “classic CMT phenotype,” namely pes cavus, length-dependent weakness and sensory loss resulting in gait difficulty, and hyporeflexia. The majority of patients present in the first decade of life, though later presentations are not uncommon. As CMT1A progresses, patients commonly require ankle foot orthotics but rarely lose ambulation. While CMT1A has historically been perceived as primarily impacting large, myelinated nerve fibers, several studies have confirmed injury to small unmyelinated fibers, which may explain the high prevalence of neuropathic pain observed in the disease (~20% of patients) [28, 29].
Nerve conduction studies in CMT1A reveal reduced sensory and motor compound amplitudes (SNAPs and CMAPs) and uniform slowing of motor conduction velocities in the demyelinating range. This uniformity of CV slowing (meaning that there is a similar degree of slowing in each nerve) contrasts with the patchy and heterogeneous slowing of CVs observed in acquired immune neuropathies. Regardless of the degree of conduction velocity slowing, it is the reduction in CMAPs that reflects the degeneration of motor axons and correlates with disability [30, 31]. Nerve biopsy, which is no longer routinely performed for diagnosis, demonstrates the presence of onion bulbs [32, 33]. While onion bulb formation in CMT1A has traditionally been attributed to recurrent demyelination and remyelination, this assumption has recently been put into question. A large study of electrophysiological data from patients with CMT1A revealed that both motor and sensory nerve conduction velocities increase with age and confirmed the absence of any acquired demyelinating features (i.e., partial conduction block) [4]. Given the striking uniformity of motor conduction velocities, the presence of slow conduction velocities in the first year of life, and the stability of the velocities over time, it has been suggested that CMT1A is primarily dysmyelinating rather than demyelinating and that onion bulb formation actually reflects abnormal organization of Schwann cells around their axons [4,5,6, 34]. Examination of dermal myelinated nerve fibers in CMT1A has also revealed uniformly shortened internodes, potentially related to a developmental defect in internodal lengthening, which may account for the uniformity of conduction velocity slowing [35]. The possibility of a primary developmental defect in myelin formation in CMT1A underscores the need to treat early in the disease course in order to minimize disability.
It is important to emphasize that despite the relative genetic homogeneity of CMT1A as compared to other forms of CMT, the disease results in notable inter- and intra-familial variability, which may relate to the influence of genetic modifiers, as well as other epigenetic or environmental factors [36, 37].
PMP-22 Biology and Pathomechanisms
CMT1A most commonly results from a 1.4 Mb tandem duplication on chromosome 17 p11.2, following an unequal crossing over event in meiosis [38]. In contrast, depletion of the protein resulting from deletions in PMP-22 causes hereditary neuropathy with liability to pressure palsies (HNPP). HNPP manifests with a distinct clinical phenotype of recurrent compressive neuropathies superimposed on a slowly progressive polyneuropathy [39, 40]. CMT1, resulting from point mutations in PMP-22 (termed CMT1E), can also be phenotypically distinct, with an earlier disease onset, more notable disability, and more severe slowing of motor conduction velocities [41]. Rarely, CMT1E can also resemble HNPP [42]. Pathologically, CMT1E is associated with the aggregation of PMP22 in the cytoplasm of Schwann cells, a finding not observed in CMT1A [43].
PMP-22 encodes peripheral myelin protein 22, a 22 kDa hydrophobic transmembrane glycoprotein that accounts for approximately 2–5% of peripheral nerve myelin protein [44]. PMP-22 is expressed in Schwann cells during myelination and is thought to affect the organization of lipids in compact myelin [45,46,47,48]. The majority of PMP-22 is degraded immediately following translation due to inefficient folding, while the remaining ~10% of PMP-22 is folded in the endoplasmic reticulum (ER), glycosylated in the Golgi, and incorporated into the compacted regions of myelin [45, 49]. PMP-22 is critical for both the synthesis and maintenance of myelin and is believed to play a structural role in the myelin sheath, though the exact mechanisms by which the protein functions within myelin remain somewhat elusive [9, 50]. It has also been hypothesized that PMP-22 binds tetramers of myelin protein zero, thereby helping to compact and stabilize myelin [39, 51, 52].
The precise mechanisms by which PMP-22 duplications impact Schwann cell development and function have also not been determined; however, many studies have suggested the increased dose of PMP-22 resulting from the duplication underlies the development of neuropathy. Overexpression of PMP-22 in rodent models results in a demyelinating neuropathy, whereas reducing PMP-22 transcription in these models improves both myelination and neuropathy severity [19, 53,54,55,56,57,58]. Further underscoring that gene dosage of PMP-22 is critical for nerve health are the observations that patients harboring a PMP-22 triplication have a more severe neuropathy phenotype and those with a PMP-22 duplication on one chromosome and a deletion on the other do not develop neuropathy [55, 59, 60].
While the dose of PMP-22 is clearly important to nerve health, pre-clinical work also suggests a straightforward dosage mechanism cannot fully explain the pathogenesis of CMT1A. Rodent models have shown elevated levels of PMP-22 mRNA; however, PMP-22 protein levels are highly variable and can even fall into the normal range [49, 55, 61]. Examination of the skin and sural nerve biopsies in humans with CMT1A also have not consistently demonstrated elevations in PMP-22 relative to controls [61, 62], and no clear correlation between the amount of PMP-22 expressed in intact myelin and disease severity has been established [39, 55]. This contrasts with the uniform reduction in dermal PMP-22 seen in patients with HNPP [63]. The levels of PMP-22 expression in CMT1A also fluctuate over time. It is increasingly recognized therefore that in addition to downregulating the expression of the protein, effective treatments for CMT1A will also need to prevent the excessive fluctuations in PMP-22 [5, 39, 64].
PMP-22 expression is tightly regulated by two promoters (P1 and P2), which are tissue specific. The P1 promoter is expressed only in Schwann cells, and the P2 promoter is expressed in non-peripheral nervous system tissues [65]. In addition, a late myelinating Schwann cell enhancer (LMSE) has been identified upstream of the P1 promoter. LMSE is important in the later stages of myelination, as well as in remyelination following injury [66, 67], and Pantera et al. have shown that deleting the LMSE significantly reduces the expression of PMP-22 by disproportionately impacting the P1 promoter [68, 69]. Small duplications containing LMSE can also result in mild forms of CMT1A, suggesting that the additional copy of the super-enhancer region can be disease causing independently of PMP-22 [55].
PMP-22 is additionally regulated by multiple transcription factors, some of which have been evaluated as therapeutic targets in pre-clinical studies. Among these are EGR2/Krox20 (Early growth response protein 2) and SOX10 (SRY sex determining region Y-box 10), which bind PMP-22 within an intronic regulatory element and induce its expression by mechanisms that thus far are not well defined [55, 70]. Other important regulators include YAP/TAZ and TEAD [71]. Makoukji et al. also demonstrated that oxysterols (molecules formed from the oxidation of cholesterol) inhibit the expression of both PMP-22 and MPZ in Schwann cells and that this inhibition is mediated by liver X receptors (LXRs) [72]. Furthermore, a selective LXR agonist (TO901317) successfully downregulated the expression of both PMP-22 and MPZ suggesting a potential therapeutic approach [72].
Post-transcriptionally, PMP-22 is regulated by select microRNAs (miRNAs), small regulatory molecules that target the 3′UTR of mRNA and inhibit its function [73,74,75]. Specifically, Verrier et al. demonstrated that miR-29a inhibits PMP22 reporter expression [74], and Lee et al. found that miR-381 is downregulated in the C22 mouse model of CMT1A (see section entitled “Biological Models of PMP-22-Associated Neuropathy” below for a detailed description of the C22 mouse model) (76). Furthermore, administration of an miR-381 expressing lentiviral vector into the sciatic nerves of C22 mice resulted in an improvement in both clinical and electrophysiological measures [76].
The notable intra-familial variability observed in CMT1A has led to a search for genetic modifier genes that can be targeted to ameliorate the CMT1A phenotype [77, 78]. Tao et al. performed a genome-wide analysis in 330 patients with CMT1A of European ancestry on the phenotypic extremes and identified four single nucleotide polymorphisms in the signal-induced proliferation-associated 1 like 2 (SIPA1L2) gene, which was associated with foot dorsiflexion strength [78]. The authors further demonstrated that SIPA1L2 is a part of a co-expressed network of key myelination genes under the regulation of SOX10 and that knockdown of SIPA1L2 in Schwann cells results in reduced PMP-22 expression. Variants in other CMT-causing genes have also shown associations with the severity of CMT1A. Earlier and more severe manifestations of CMT1A were reported with a co-existing I92V variant in LITAF/SIMPLE [79, 80] and several SNP alleles in the SH3TC2 gene associated with phenotypic differences in CMT1A [81]. As mentioned previously, miRNAs play an important role in the regulation of PMP-22 expression, and a variant in the miR-149 was closely associated with neuropathy severity in a Korean CMT1A cohort [82].
As is true of several forms of CMT, the aggregation of misfolded protein is believed to play a role in CMT1A [83,84,85,86,87]. Overexpression of PMP-22 (which is inefficiently folded even in a non-pathogenic state) can result in the accumulation of misfolded protein in the ER, with subsequent activation of the unfolded protein response (UPR), reduced protein translation, and potentially apoptosis of Schwann cells (see section titled “Intracellular Changes in Response to MPZ Derangement” and Fig. 4 for detailed discussion of the UPR). The aggregation of PMP-22 is observed more commonly in patients with point mutations in PMP-22 than in those harboring the duplication [43] and is recapitulated in the C22 and trembler J mouse models, in which protein aggregates are seen in the cytoplasm of Schwann cells [86, 88]. Importantly, in vitro studies suggest that the retention of protein in the ER can affect the amount of PMP-22 present in the plasma membrane [86]. Furthermore, there is evidence to suggest that it is possible to reduce the impact of misfolded protein and prevent the formation of protein aggregates through modulation of chaperones that support effective protein trafficking [85, 89]. This approach has been explored in rodent models with promising results. Specifically, treatment of DRG explants from C22 mice with small-molecule inhibitors of heat shock protein 90 (HSP90) resulted in improved trafficking of PMP-22 and in myelination [90].
Schwann cells play an important role in axonal regeneration, and duplications in PMP-22 have been shown to impair the regeneration of large diameter axons [91]. Targeting of denervated Schwann cells to increase the efficiency of axonal regeneration has therefore been explored as a therapeutic avenue in CMT1A. Specifically, deficiency in NT-3 (a neurotrophic factor that plays a role in the Schwann cell autocrine loop and stimulates myelination) has been shown to impair nerve regeneration, and NT-3 knockout mice manifest a progressive motor neuropathy [92,93,94]. A small clinical trial assessing the efficacy of subcutaneously administered NT-3 did show clinical improvement; however, the short half-life of the drug limited further investigation [95]. As discussed later in this review, gene therapy studies using adeno-associated virus (AAV)-mediated neurotrophin 3 (NT-3) have since shown promise [96].
Multiple studies have suggested excess PMP-22 interferes with Schwann cell differentiation, and thereby with myelination, as evidenced by the abnormal expression of genes associated with immature Schwann cells, such as Sox2 and c-Jun [62]. This abnormal gene expression is particularly evident in vitro when Schwann cells are exposed to neurons, underscoring the role of Schwann cell axon interactions in the setting of abnormal PMP-22 dosing [97]. Fledrich et al. also found that in transgenic CMT1A rats, Schwann cells develop a persistent differentiation defect resulting from an imbalance of the phosphatidylinositol 4,5-bisphosphate 3-kinase (PI3K)-Akt and the mitogen-activated protein kinase 1 (Mek)-Erk intracellular signaling pathways, the latter of which is known to play a role in Schwann cell plasticity and nerve regeneration [62, 98]. The authors further demonstrated that enhancing PI3K-Akt signaling with epidermal growth factor (EGF)-like growth factor neuregulin-1 (NRG1) type I promoted Schwann cell differentiation. In contrast, Fornasari et al. found that different isoforms of NRG1 are actually strongly overexpressed in the nerves of transgenic CMT1A rats, suggesting that NRG1 may not be a viable treatment for CMT1A [99].
Another proposed mechanism for PMP-22 induced defects in Schwann cell differentiation is a rise in the influx of extracellular [Ca2 +] into Schwann cells, which was shown to be related to an overexpression of the purinergic receptor P2X7 [100,101,102]. This was further explored by Vanoye and colleagues who demonstrated that PMP-22 specifically increases calcium influx through store-operated calcium channels, which help replenish [Ca2 +] in the endoplasmic reticulum. The authors hypothesize that PMP-22 accumulation in the endoplasmic reticulum may therefore result in elevated intracellular [Ca2 +] and subsequent demyelination [101, 103]. As discussed in more detail later in this review, inhibition of P2X7 was also found to be a viable therapeutic target in animal models of CMT1A [100, 104].
The importance of PMP-22 for effective intracellular lipid metabolism has recently been elucidated, presenting a new angle of the pathogenesis of CMT1A. Seventy percent of the myelin membrane is composed of lipids, including phospholipids, cholesterol, and glycosphingolipids [105]. Furthermore, cholesterol synthesis in Schwann cells is required for de novo synthesis of myelin. Fledrich et al. previously examined sciatic nerve and skin tissue mRNA extracts in CMT1A rats and demonstrated differential dysregulation of lipid metabolism-associated genes in mildly versus severely affected animals [106]. PMP-22 also interacts with cholesterol [107], and alterations in PMP-22 appear to impact cholesterol metabolism. Nerves from Trembler (Tr) mice were found to have reduced cholesterol synthesis [107,108,109,110,111], and both Schwann cells and nerves from PMP22 knockout mice showed an abnormal cholesterol distribution [112]. Zhou et al. also recently found that PMP22, through interaction with the cholesterol efflux regulatory protein ABCA1, facilitates the efflux of cholesterol from Schwann cells [113]. In Schwann cells from homozygous Trembler J (TrJ) mice, cholesterol is retained in the Golgi along with PMP-22 and diminished in the plasma membrane [107], and it has been demonstrated that the cholesterol-binding motif known as CRAC of PMP-22 plays a particularly important role in PMP-22-mediated cholesterol localization within Schwann cells [107]. Taken together, these studies suggest that restoring cholesterol metabolism within Schwann cells could potentially offer therapeutic benefit in CMT1A.
Biological Models of PMP-22-Associated Neuropathy
The first animal models used to study myelin abnormalities associated with the PMP22 gene were the naturally occurring mouse mutant Trembler (Tr) [114, 115] and Trembler J (Tr-J) [116]. The Trembler mice carry an autosomal dominant missense mutation that substitutes an aspartic acid residue for a glycine in residue 150 (G150D) and present a severe phenotype characterized by spastic paresis, generalized tremor, and transient tonic–clonic seizures at an early age (after 10–14 days of age). Trembler-J (Tr-J) was produced by the Jackson Laboratory from the C57BL/6 J strain of Tr mice. Tr-J carries a missense mutation replacing a leucine with a proline residue at position 16 (L16P) in the first transmembrane domain of PMP22 [116]. Tr-J mice present progressive limb weakness and tremor. It is important to note that despite their historical importance in understanding the connection between PMP22 and demyelinating neuropathies, Tr and Tr-J mice are models of missense mutations in PMP22 and therefore emulate the human disease CMT1E and not CMT1A, as they don’t carry extra PMP22 copies. Other Trembler lines include the Trembler-m1H, tr-m2H, and tr-m3H, all of which present a more severe phenotype than expected for a CMT1A model [117, 118].
To better recapitulate the biology of PMP22 copy number variation characteristics of CMT1A, different PMP22 overexpression models have been created since the late 1990s. These include transgenic mice lines C22 (carrying seven supplementary human PMP22 transgene copies), C61 (carrying eight supplementary human PMP22 transgene copies [119]), and C3 (carrying 3 to 4 PMP22 transgene copies [119,120,121]. Despite the closer genotype and phenotype to CMT1A when compared to the Trembler models, several issues still remain regarding the reliability of these transgenic PMP22 overexpressing mouse models. C22 and C61 models have to carry an excessive number of PMP22 copies to demonstrate a phenotype and therefore do not adequately recapitulate the PMP22 duplication characteristic of the human disease. Even when considering the more similar C3 model, which carries between 3 to 4 PMP22 transgene copies, another issue common to all of these models is their inability to recapitulate genomic and epigenomic effects of the 1.4 Mb tandem duplication, including changes in microRNAs and long noncoding RNA, as well as possible modifier genes that may have important roles in disease biology and phenotypic expression. Other transgenic mice used in the study and modeling of CMT1A also include the JP18 and JP18/JY13 mouse models, which carry one and two extra copies of PMP22, respectively. It is important to note that the large duplicated 1.4 Mb DNA segment characteristic of CMT1A is beyond the limit of current cloning techniques. Therefore, none of the current animal models contains this large mutation and truly recapitulates the genetics of human CMT1A.
A PMP22-transgenic rat established in the late 1990s has become one of the most used rodent CMT1A models [122]. This transgenic rat carries three copies of a 43 kb restriction fragment that contains the PMP22 transcription unit, including 7 kb upstream of exon 1A and 4 kb downstream of exon 5, and presents behavioral, electrophysiological, and pathological features consistent with CMT1A in humans. This model has been used in several studies evaluating different treatments for CMT1A, including Onapristone [57], PXT3003 [123], AAV2/9 shRNA targeting PMP-22 mRNA [124], and antisense oligonucleotides against PMP-22 mRNA [19].
Cellular systems are also commonly used in pre-clinical studies for CMT1A, usually utilizing lines derived from the above-mentioned rodent models. A commonly used cell line is the S16 rat SC line, previously shown to sustain high levels of PMP-22 expression comparable to those in myelinating Schwann cells and demonstrating transcription factor binding patterns similar to rat sciatic nerve [70, 71, 125, 126]. These lines have been genetically engineered with different reporter systems and used in high-throughput screening assays to identify candidate compounds capable of reducing PMP-22 mRNA expression [15].
Unfortunately, due to the basic differences on a genetic level between rodent models of CMT1A and the human disease, the translation of pre-clinical studies into successful human trials have been challenging. Therefore, models that better recapitulate the genomic network involved in human CMT1A are urgently needed. As an alternative, human-induced pluripotent stem cells derived from patients with CMT1A could provide a superior model to study CMT1A in an authentic genetic background [10, 127]. However, challenges in fully differentiating these stem cells into mature myelinating Schwann cells capable of in vitro myelination still limit the use of this strategy in pre-clinical therapy development studies for demyelinating CMT.
Therapeutic Targets in PMP-22-Associated Neuropathy (Table 2 and Fig. 3)

CMT1 treatment targets and therapies
Lowering PMP-22 in CMT1A
Since pre-clinical studies have underscored the importance of PMP-22 dosing in the pathogenesis of CMT1A, treatment efforts have focused on reducing the expression of PMP-22 and supporting effective myelination. One of the first candidate therapies examined was ascorbic acid (AA), an antioxidant with pro-myelinating effects that successfully reduced PMP-22 expression in Schwann cells via inhibition of adenylate cyclase and reduction of cyclic AMP levels [128, 129]. In vivo studies in C22 mice subsequently confirmed that AA suppresses PMP-22 expression and showed improved motor function in treated animals [56]. These promising results, together with the known safety profile of AA, motivated multiple clinical trials. Unfortunately, these trials failed to demonstrate clinical efficacy of AA in people with CMT1A [130,131,132,133].
The inhibition of progesterone, a neuroactive steroid involved in the myelination program of Schwann cells, similarly offered promise in pre-clinical studies of CMT1A [134]. In vitro studies in rat Schwann cells demonstrated that progesterone and its derivatives activate the P1 PMP-22 promoter and increase the expression of transcription factors SOX-10 and KROX-20, thereby further driving the expression of PMP-22 [134,135,136]. Sereda et al. targeted the action of progesterone in male transgenic CMT1A rats with onapristone, a progesterone/glucocorticoid receptor antagonist, and demonstrated reduced expression of PMP-22 and an improvement in the clinical phenotype [57, 137]. Due to concerns regarding their potential toxicity, progesterone inhibitors have not progressed to clinical trials.
A treatment that has more recently shown promise in CMT1A is PXT3003, a combination of compounds identified using a systems biology approach focused on pathways that promote myelination, while simultaneously downregulating PMP-22. PXT3003 combines three repurposed drugs including (1) baclofen, a GABA receptor agonist that reduces PMP22 transcription in Schwann cells by reducing adenylate cyclase activity [138]; (2) naltrexone, an opioid receptor antagonist believed to potentiate Baclofen’s mechanism of action; and (3) D-sorbitol, a natural metabolite involved in the polyol pathway postulated to stabilize misfolded proteins [139].
PXT3003 was demonstrated to successfully downregulate PMP-22 via the PI3K-AKT/MEK-ERK signaling pathway in transgenic CMT1A rats, with an improvement in myelination and in the clinical phenotype [123]. Combination therapy proved superior to treatment with the individual components of PXT3003 [139]. Prukop et al. also demonstrated that brief treatment of transgenic CMT1A rats with PXT3003 during early development delays disease onset in adulthood, with a dose-dependent improvement in limb strength [140]. Interestingly, despite the clinical improvement observed, the only electrophysiological measure that improved was distal motor latency. Furthermore, while treatment resulted in a shift towards large-caliber axons, there was no change in the total number of myelinated axons or in myelin thickness. The authors hypothesized the clinical benefit of PXT3003 may result in part from enhanced muscle innervation at the neuromuscular junction, uncoupled from the effects of demyelination. Recent in vitro and in vivo work has offered support for this hypothesis [139]. Specifically, transgenic CMT1A rats treated with PXT3003 showed an increased number of innervated neuromuscular junctions. Interestingly, PMP-22 expression did not decline in response to treatment, contrary to prior studies [139]. While the precise biological mechanisms underlying PXT3003-associated improvement in CMT1A will require further study, early clinical trials are promising. Notably, a randomized, double blind, placebo-controlled, phase 2 study including 80 CMT1A patients confirmed the safety and tolerability of PXT3003 and showed a significant improvement in the Charcot-Marie-Tooth Neuropathy Score (CMTNS) and the Overall Neuropathy Limitations Scale (ONLS) in those receiving the highest treatment dose for 12 months (n = 19) [141]. Results from a Phase III clinical trial (ClinicalTrials.gov identifier NCT02579759) have also shown a significant improvement in the 10-m walk test and ONLS in the higher dose group, with no serious adverse events [142].
PMP-22 Independent Targets in CMT1A
The identification of P2X7 channel activation and subsequent accumulation of Ca + in Schwann cells as a potential pathomechanism in CMT1A has led to pre-clinical studies of P2X7 inhibitors. Nobbio et al. found that antagonizing P2X7 using pharmacological inhibitors and small-interfering RNA (siRNA) rescues the phenotype of CMT1A Schwann cells [100]. Sociali et al. then demonstrated improved myelination in DRG cultures in response to a P2X7 antagonist (A438079) [104]. In vivo studies in transgenic CMT1A rats also showed improvement in hind limb strength in response to treatment with A438079 and further supported the hypothesis that P2X7 inhibition promotes Schwann cell differentiation. While the authors observed an increase in myelinated axons, there was no change in CMAP amplitudes in treated animals. These studies also underscored a potential adverse impact of P2X7 inhibition, namely muscle weakness resulting from higher doses of A438079.
Reduction of oxidative stress has also been examined as a therapeutic strategy in CMT1A. In this regard, curcumin, which is safe and possesses both neuroprotective and antioxidant effects, is an appealing candidate treatment. Curcumin is also a known sarcoplasmic/endoplasmic reticulum calcium pump (SERCA) inhibitor and can therefore alleviate the accumulation of misfolded proteins in the ER, thereby reducing excessive UPR activation in Schwann cells [143,144,145]. To overcome the inefficient pharmacokinetic properties of curcumin, Caillaud et al. developed curcumin-cyclodextrin/cellulose nanocrystals (Nano-Cur) and examined their effects in transgenic CMT1A rats [146]. Treatment resulted in improvement in the clinical phenotype, as well as in electrophysiological findings (motor and sensory conduction velocities) and in myelin thickness. The authors also observed an associated reduction in markers of oxidative stress in both nerve and muscle, and in vitro proof of concept experiments confirmed a reduction in reactive oxygen species and improved mitochondrial membrane potential in CMT1A Schwann cells [146].
Another approach to minimizing impact of protein misfolding in CMT1A has been to activate the heat shock pathway, an intracellular stress response mediated by chaperones that facilitate protein folding and trafficking, thereby reducing protein aggregation [85, 147]. Specifically, the heat shock pathway can be activated through the inhibition of HSP90, a molecular chaperone protein, and HSP90 inhibitors have previously shown benefit in disorders related to protein misfolding [148]. Chittoor-Vinod et al. demonstrated that select HSP90 inhibitors enhance myelin synthesis in vitro and found that intraperitoneal treatment in C22 mice improved peak muscle force and slowed decline in rotarod performance [147]. Because HSP inhibitors can result in cellular toxicity, further study will be needed prior to their potential use in humans.
HSP90 also proved to be targetable in a recent pre-clinical study evaluating Histone deacetylase 6 (HDAC6) inhibition in CMT1A [149]. Histone deacetylase 6 (HDAC6) is an enzyme that controls the acetylation of cytosolic proteins, including α-tubulin, and plays a role in microtubule stability and axonal transport [149]. HDAC6 inhibitors were previously found to improve the CMT2 phenotype in animal models via the acetylation of tubulin [150]. Ha et al. recently evaluated the effect of CKD-504, an HDAC6 inhibitor, on mesenchymal stem cell-derived Schwann cells from CMT1A patients and demonstrated reduced PMP22 protein expression and induction of Schwann cell differentiation in response to treatment [149]. In C22 mice, treatment resulted in the acetylation of α-tubulin and reduced PMP-22 protein in the sciatic nerve, with improved myelination, motor function, and electrophysiological features. HDAC6 also induced the acetylation, and thereby inhibition, of HSP90, which was hypothesized to increase folding and reduce the aggregation of excess protein. While the significant toxicity associated with HDAC6 inhibitors has limited their use in patients, CKD-504 is currently in Phase 1 clinical trials for use in Huntington’s disease (NCT0371389) [149].
As previously discussed, alterations in both the metabolism and distribution of lipids have been observed in association with excess PMP-22, and studies have begun to examine whether ameliorating these lipid derangements can improve myelination in CMT1A. Fledrich et al. showed that substitution of phosphatidylcholine and phosphatidylethanolamine in the diet increased the number of myelinated axons in peripheral nerves (without changes in the thickness of the myelin sheaths), prevented axonal loss, and improved the clinical phenotype in CMT1A rats [105]. The clinical benefit did not persist beyond the treatment period, however. Zhou et al. also treated TrJ mice with advanced neuropathy with a lipid enriched, high fat diet and identified improvements in the maintenance of myelinated axons [112]. As mentioned, PMP-22 is also important for cholesterol metabolism in Schwann cells, and alterations in cholesterol trafficking have been observed in CMT1A. Future studies may explore whether cholesterol supplementation or manipulation of cholesterol transport can offer benefit in CMT1A.
Emerging Genetic Therapies in CMT1A
The recent success of genetic therapies, including antisense oligonucleotides (ASOs) and small-interfering RNA (siRNA)-based treatments, in other neuromuscular disorders has greatly informed the current approaches to the treatment of CMT1A and offered promise for a disease modifying treatment [17, 18, 151]. Genetic therapy studies in CMT1A began when Sahenk et al. introduced adeno-associated virus (AAV)-mediated neurotrophin 3 (NT-3) gene therapy in the TrJ mouse model [152]. NT-3 is an autocrine-derived factor expressed by Schwann cells that promotes both myelination and axonal regeneration [95, 152]. The authors demonstrated that rAAV1.NT-3 gene transfer into muscle allows for NT-3 secretion, with an increase in serum levels and associated improvement in clinical, pathological, and electrophysiological features in treated animals. Follow-up work by the same group revealed that AAV.NT-3 gene therapy in TrJ mice affects muscle enzyme metabolism and activates the mammalian target of rapamycin complex 1 (mTOR) pathway, resulting in an increase in muscle fiber size [96]. NT-3 may therefore have a synergistic effect on both nerve and muscle. Interestingly, the aforementioned effects of AAV.NT-3 were not observed in the WT animals, suggesting a specific predilection for pathological nerve fibers. While these results are encouraging, it is important to note the TrJ mouse strain, which harbors a naturally occurring point mutation in PMP-22, is not a model of CMT1A but rather better represents CMT1E. An ongoing Phase I/II clinical trial is evaluating the effect of AAV.NT-3 gene therapy delivered via a single intramuscular injection in humans with CMT1A (NCT03520751).
AAV vectors allow for stable expression, do not integrate into the host genome, and have low immunogenicity. Due to these favorable properties, they have served as the preferred delivery mechanism in neurologic disorders. There are barriers to the use of AAV-mediated gene therapies, however, including their potential for off-target effects as well as concerns regarding ectopic gene expression [153]. Targeted delivery to Schwann cells could help offset these adverse effects. To this end, Gautier et al. recently reported successful intraneural delivery of a recombinant adeno-associated viral vector serotype 9 (AAV2/9) expressing a small hairpin inhibitory RNA (shRNA) directed against PMP-22 mRNA, to transgenic CMT1A rats, and demonstrated efficient transduction [124]. Early treatment also prevented myelination defects, as well as motor and sensory impairments. Given that treatment induced a reduction of PMP-22 protein expression, without reducing PMP-22 mRNA, the authors postulate that the disruption occurred at the level of translation machinery rather than via direct targeting of mRNA [124]. Importantly, intraneural delivery did restrict biodistribution of the vector, suggesting that direct delivery to Schwann cells could help minimize off-target effects.
As mentioned, a particularly exciting recent development in the treatment of genetic neuromuscular disorders has been the use of antisense oligonucleotides (ASOs). ASOs are single-stranded synthetic nucleic acids that can target specific cell types and bind target mRNA, leading to its degradation [154]. Zhao et al. demonstrated that ASOs successfully suppress PMP-22 mRNA in the nerves of both the C22 mouse and the transgenic CMT1A rat models [19]. Treatment of C22 mice with weekly subcutaneous injections of the PMP-22 ASO after disease onset resulted in a dose-dependent reduction in PMP-22 mRNA. Furthermore, treatment improved motor function, electrophysiology (with motor conduction velocities approaching normal levels and an increase in motor amplitudes) and pathological features, as evidenced by increased numbers of myelinated axons and reduced onion bulb formation. The authors also demonstrated a treatment-induced reduction in PMP-22 mRNA levels in Schwann cells from skin biopsies of CMT1A rats, suggesting that this could serve as a treatment-specific biomarker in future studies. The finding that ASOs can cross the blood nerve barrier to target Schwann cells and favorably affect PMP-22 gene expression and myelination opens an exciting new pathway for therapeutic development in CMT1A [19]. At the same time, important challenges remain. Specifically, the timing of ASO treatment, as well as mode of administration and dose, will all require further investigation [155]. Furthermore, concerns about off-target effects, and the known side effects of ASOs including thrombocytopenia, could outweigh the benefit of treatment for a slowly progressive neuropathy such as CMT1A [156]. Finally, as maintaining the right degree of PMP-22 expression is critical to nerve health, excessive suppression could result in an HNPP phenotype, which is associated with patient morbidity comparable to that of CMT1A [157, 158].
Another gene therapy approach that has shown promising results in pre-clinical studies of CMT1A is the use of small-interfering RNA (siRNA). siRNAs are small double-stranded RNAs that can selectively silence the expression of a targeted gene by degrading its mRNA [159]. Intraperitoneal injection of siRNAs reduced mutant PMP-22 expression, improved myelination, and alleviated the clinical phenotype in the Tr-J mouse model [160]. Boutary et al. recently tested i.v administration of siRNAs in the JP18 and JP18/JY13 mouse models, which carry one and two extra copies of PMP22, respectively [161]. To achieve successful delivery of siRNAs to Schwann cells, the authors used a nanoparticle-stabilized siRNA (siRNA PMP22-SQ NPs). Treatment resulted in a rapid and dramatic improvement of the clinical phenotype with improved limb strength and locomotor function, as well as normalization of CMAP amplitudes and sensory conduction velocities, with positive effects persisting for 3 weeks beyond the treatment period [161]. The clinical improvement was accompanied by normalization of sciatic nerve levels of transcription factors Krox20 and Sox10, as well as heavy neurofilament levels, consistent with the recovery of both the myelin and axons. Importantly, in the aforementioned studies, the expression of other myelin proteins was not impacted by siRNA PMP-22, underscoring the potential of this treatment to limit off-target effects [159, 160].
A recent study evaluated CRISPR-based gene editing in CMT1A. In contrast to other treatment strategies, this approach offers the potential for a single-dose therapy. In a proof-of-concept study, Lee and colleagues delivered CRISPR/Cas9 intraneurally to C22 mice in order to target the TATA-box of the P1 promoter in Schwann cells [162]. Treatment prior to disease onset resulted in downregulation of PMP-22 expression and improvement in both electrophysiological features and in myelination. As discussed by the authors, various challenges, including the potential for immunogenicity and off-target effects, will need to be closely examined in future studies.
While the recent advances in genetic therapies for CMT1A are encouraging, it is important to emphasize that we are still in the early stages of understanding the impacts of gene therapy in humans and that many obstacles, both expected and unanticipated, likely lie ahead. This is exemplified by the recent discovery that treatment of SMA mouse models with adeno-associated virus serotype 9 (AAV9)—SMN gene therapy—can result in toxic gain of function injury to motor neurons due to aggregation of the overexpressed protein [163].
Myelin Protein Zero-Associated Neuropathy: Epidemiology and Clinical Features
MPZ neuropathies account for 5% of all of CMT and 10% of all demyelinating forms of CMT [9, 13, 164]. While MPZ neuropathy is an AD disorder, de novo mutations are common, and the absence of a family history should not preclude consideration of this diagnosis [165]. To date, there have been over 200 disease-causing mutations identified in the MPZ gene, with 76 new mutations reported between 2005 and 2018 [24]. MPZ neuropathies span a wide phenotypic spectrum from severe infantile onset demyelinating neuropathy to milder adult-onset axonal forms [165]. This striking genotypic and phenotypic heterogeneity poses challenges both for accurately identifying patients with MPZ neuropathy and for designing effective clinical trials.
The nomenclature used to describe MPZ neuropathies has evolved over the years leading to some confusion. Traditionally, demyelinating neuropathy, with upper extremity CV < 38 m/s resulting from MPZ mutations, has been referred to as CMT1B and the axonal forms as CMT2I. Other descriptors have included “Dejerine-Sottas,” in reference to infantile onset neuropathy, and “congenital hypomyelination,” in reference to severe, early-onset neuropathies with pathological evidence of myelination failure [166,167,168,169]. Increasingly, MPZ neuropathy phenotypes are being classified by the patient’s age at presentation, the primary nerve pathology, and the specific genetic mutation [170].
The clinical features of most MPZ-associated neuropathies are similar to those seen with other forms of CMT, namely foot deformities, distal muscle weakness and atrophy, and length-dependent sensory loss. Additional features can include scoliosis, and hip dysplasia, which are more common in patients with the infantile onset demyelinating forms [170]. In contrast, tonic pupils, dysphagia, and neuropathic pain are distinguishing features that occur more commonly with the axonal forms [171]. Hearing loss can also occur, with both the early and adult-onset forms of MPZ neuropathy [170]. Electrophysiological findings in MPZ neuropathy are diverse. Motor conduction velocities in the demyelinating, axonal, and intermediate ranges can be seen, and rare cases of partial conduction block have been reported, leading to suspicion for immune-mediated rather than hereditary nerve disease [172]. Similarly, nerve pathology can include findings of either dysmyelination or demyelination, as well as of primary axonal degeneration [173,174,175,176,177,178,179].
Genotype–phenotype correlation studies have identified three distinct phenotypic groups in MPZ neuropathy, including infantile, childhood, and adult-onset CMT [30, 33, 170, 180]. Patients in the infantile-onset group develop symptoms prior to 3 years of age and have severely slowed motor CVs (ulnar motor CV < 15 m/s) and more difficulty with ambulation than the other groups (19% wheelchair dependent in one series) [170]. The childhood-onset group demonstrates higher ulnar motor CVs (15–35 m/s) and presents similarly to patients with CMT1A with symptoms emerging in the second decade, and the adult-onset group is distinguished by axonal range CVs [165, 170]. Importantly, the majority of MPZ mutations consistently manifest with one of the three distinct clinical phenotypes [15, 24, 165, 171]. Examples include the His10Pro and Thr95Met mutations, which result in adult-onset neuropathy, versus Ser34del, which manifests with early-onset, demyelinating CMT [165]. Why individual MPZ mutations result in specific clinical phenotypes is not known [170]. The clinical progression of MPZ neuropathies is also highly variable. Early-onset, severe neuropathies tend to cause notable disability in childhood, with slower rates of progression beyond adolescence [181]. In contrast, select axonal forms can present in adulthood and progress rapidly leading to a loss of ambulation in later life [25, 181, 182].
While this review focuses on demyelinating forms of CMT, it is worth noting that the preponderance of recently discovered MPZ mutations is responsible for adult onset, axonal neuropathies; the prevalence of which likely continues to be underestimated [24]. Defining the pathomechanisms of axonal forms of MPZ neuropathy is especially important, given that MPZ is expressed exclusively in myelinating Schwann cells, and yet minimal pathological evidence of demyelination is observed in patients with axonal neuropathy related to MPZ [26, 183]. It has been hypothesized that the axonal injury may result from disruptions in the signaling pathways between the myelin and the axon, though the detailed nature of this disruption at the cellular level is not well understood [170, 177, 178].
MPZ Biology and Pathomechanisms
MPZ, also termed P0, is the major protein in peripheral nerve myelin and a member of the immunoglobulin (Ig) supergene family [26]. The protein plays an important role both in the formation of myelin and in the maintenance of myelin homeostasis and stability throughout adulthood [184]. The protein is encoded by the MPZ gene on chromosome 1q22-q23 and is only expressed in myelinating Schwann cells [185]. MPZ consists of three structural domains: a 124 amino acid immunoglobulin-like extracellular domain, a 26 amino acid transmembrane domain, and a 69 amino acid intracellular domain. The protein is synthesized in the ER of Schwann cells, trafficked through the Golgi compartment, and ultimately sorted into vesicles and incorporated into the myelin sheath [186,187,188]. Of the 248 amino acids encoded by MPZ, the first 29 comprise a signaling protein that targets MPZ to the myelin sheath and is cleaved prior to the protein’s incorporation into myelin [26]. MPZ additionally undergoes post-translational modification by the addition of an N-linked oligosaccharide, as well as sulfate, acyl, and phosphate groups [189, 190]. While MPZ largely localizes to compact myelin, it is also found in the paranode and node of Ranvier, where it helps maintain nodal structure through interactions with neurofascins [191].
Once incorporated into the myelin sheath, MPZ behaves as a homophilic adhesion molecule, facilitating the compaction of myelin. Compaction is achieved when the extracellular MPZ domains on opposing myelin wraps form homotetramers that interact in-trans, thereby adhering the opposing myelin wraps to each other [165, 189, 192,193,194]. The critical role of MPZ in myelin compaction is evidenced by the presence of thin and uncompacted myelin, and severe neuropathy in MPZ knockout mice, as well as in transgenic mice containing extra copies of MPZ [195, 196].
MPZ neuropathy results from diverse mechanisms, including numerous gain of function and loss of function mechanisms [197, 198]. Some structural changes to the protein result in retention of MPZ in the ER, whereas other derangements allow the protein to successfully incorporate into the myelin sheath but disrupt interactions with the wild-type protein, thereby impairing myelin adhesion [199]. While early-onset demyelinating forms of MPZ neuropathy more commonly impede successful compaction, later onset forms tend to disrupt MPZ-mediated signal transduction and Schwann cell-axonal interactions. Importantly, the cellular mechanisms of MPZ do not reliably predict the clinical phenotype in MPZ neuropathy, as exemplified by the R198S mutation, which fully prevents myelin adhesion but causes a late-onset neuropathy [197, 198]. Given the varied gain of function mechanisms of MPZ neuropathy, it is increasingly being recognized that treatments will likely be diverse with focus on allele-specific gene silencing approaches [200].
MPZ Mutations Resulting in Altered Protein Structure and Functionality
Mutations in MPZ can alter normal function at varied intracellular locations. The majority of pathogenic mutations are located in the extracellular domain; however, both the extracellular and the cytoplasmic domains are necessary for the effective compaction of myelin [173, 174, 198, 201]. Particularly disruptive changes to MPZ include the addition of a charged amino acid, the alteration of a cysteine residue in the extracellular domain, the truncation of the cytoplasmic MPZ domain, and the alteration of an evolutionarily conserved amino acid [165]. Packing defects in the myelin intra-period line also result from several mutations in the extracellular domain [202]. Additionally, mutations can disrupt the post-translational modification of the protein. For example, increased glycosylation resulting from a second glycosylation site in the D23N mutant protein results in a severe, early-onset demyelinating neuropathy [203]. In contrast, mutations that prevent glycosylation of MPZ do not appear to interfere with myelination but may disrupt axon-Schwann cell interactions leading to the development of late-onset axonal neuropathies [177, 197, 204]. Lastly, mutations that alter MPZ’s ability to interact with the node and paranode underlie select adult-onset forms of MPZ neuropathy [191].
As mentioned, mutations in the cytoplasmic domain of MPZ can also be disease causing, and the truncation of the cytoplasmic domain has specifically been shown to prevent myelin adhesion [198, 205]. The cytoplasmic domain is believed to contribute to myelin compaction through an adhesion-mediated signal transduction cascade that enables interactions with the cytoskeleton [165, 206,207,208]. Gaboreanu and colleagues specifically demonstrated phosphorylation of the cytoplasmic domain, which is mediated by PKCα, and the receptor for activated C kinase 1 (RACK1) is important in the regulation of MPZ-mediated adhesion [206]. Changes in the cytoplasmic domain additionally impact effective MPZ targeting at the pre-myelinating stage. Fratta and colleagues used knock-in mice with the nonsense Q215X mutation (a cause of congenital hypomyelinating neuropathy in humans) to demonstrate that eliminating the last 33 amino acids of the cytoplasmic domain results in altered trafficking of MPZ to non-myelin plasma membranes and alters radial axonal sorting by Schwann cells [200].
Depolarization changes in Schwann cells may also contribute to the pathogenesis of MPZ neuropathy. Sural nerve biopsies from a patient with the R69C mutation demonstrated a switch to the subtype 1.8 voltage-gated sodium channels at the demyelinating/remyelinating internodes [181]. Additionally, Moldovan and colleagues examined homozygous mice deficient in MPZ with severe, demyelinating neuropathy and found abnormal potassium ion currents and ectopic Na(V)1.8 channels in unmyelinated nerve segments, which disrupted axon excitability [209]. Follow-up work examining a family harboring an MPZ frameshift mutation (Asp104ThrfsTer14) suggested that axonal depolarization resulting from abnormal voltage-gated sodium channels may precede axonal degeneration, mirroring the prior findings in mouse models [210].
Intracellular Changes in Response to MPZ Derangement
The Unfolded Protein Response
A major focus in the study of MPZ neuropathy pathogenesis has been that of the unfolded protein response (UPR), which has also been implicated in other forms of CMT, including PMP-22 and GJB1-associated neuropathies [199]. Because Schwann cells produce large amounts of protein, they are particularly vulnerable to potential endoplasmic reticulum stress resulting from protein misfolding [211,212,213,214]. The UPR serves as an adaptive mechanism employed by the cell to handle the accumulation of misfolded proteins and acts by upregulating transcription of chaperones, reducing translation of proteins, and increasing proteasomal protein degradation [215, 216]. While adaptive under normal circumstances, at excessively high levels of ER stress, the UPR can alter the phenotype of the cell in a way that impedes its normal function or potentially leads to apoptosis [215, 217].
In humans, three transducers mediate the UPR: inositol requiring enzyme (IRE1), activating transcription factor (ATF6), and protein kinase RNA-like endoplasmic reticulum kinase (PERK), all of which are located in the ER membrane (Fig. 4). The UPR cascade is activated by BiP, an ER chaperone that in normal circumstances binds IRE1 and PERK but in the presence of misfolded protein dissociates from the transducers rendering them active [215]. Once activated, IRE1 promotes the activation of genes involved in ER-associated degradation (ERAD) through the spliced X box binding protein (XBP1) transcription factor [211, 218, 219]. ATF6 promotes ER-resident chaperones, thereby supporting folding within the ER [220,221,222,223,224]. The PERK arm of the UPR is particularly important to Schwann cell survival in the setting of increased ER stress [225, 226]. PERK phosphorylates the α subunit of eukaryotic initiation factor 2 alpha (eIF2alpha) leading to a reduction in the translation of messenger RNAs. In addition, PERK increases the translation of activating transcription factor 4 (ATF4), which in turn upregulates the CCAAT/enhancer-binding protein homologous gene (CHOP) [227,228,229,230]. CHOP is a transcription factor associated with apoptosis related to ER stress and is a key regulator of cell death. Paradoxically CHOP also upregulates DNA damage-inducible protein 34 (GADD34). The Gadd34 gene encodes a regulatory subunit of protein phosphatase 1 (PP1) holophosphatase, which dephosphorylates eIF2alpha and thereby reactivates protein translation, enabling protein translation to resume [226]. Surprisingly, Musner and colleagues found that PERK haploinsufficiency actually improves myelin defects in vitro and in vivo, despite reduced levels of P-eIF2alpha, suggesting that PERK has effects on neuropathy that are unrelated to the UPR [211, 231].

Unfolded Protein Response
A large number of MPZ mutations activate the UPR in a dose-dependent fashion, resulting in Schwann cell dysfunction and ultimately in demyelination [212, 229, 232,233,234]. The most extensively studied mutations that result in the retention of misfolded MPZ in the ER and subsequent UPR activation are R98C and S63del, both of which are found in the extracellular domain [144, 197, 199, 217]. In S63del Schwann cells, globally misfolded mutant protein triggers the canonical UPR by exposing a hydrophobic surface of MPZ and promoting BiP binding with downstream activation of CHOP [229]. Increased CHOP expression results in growth arrest, demyelination, and secondary Schwann cell death. This is distinct from the immediate CHOP-induced cell death seen in many other disorders and suggests a unique function of CHOP in Schwann cells [184, 217, 229].
It is important to emphasize that MPZ mutations that activate the UPR do so by varied mechanisms. While the PERK pathway appears to underlie nerve injury in S63del, it is the IRE and ATF6 arms of the UPR that are implicated in R98C [145]. This difference may contribute in part to the two neuropathies being pathologically distinct, with R98C primarily causing dysmyelination or hypomyelination, and S63del resulting primarily in demyelination [184]. It has been suggested that in addition to activating the UPR, S63del mutant protein also negatively impacts wild-type MPZ, causing it to be retained in the ER and reducing its levels in the myelin sheath [235].
The precise mechanism by which R98C impedes myelination is not well understood but is believed to involve an elevation in transcription factor C-Jun (a negative regular that inhibits myelination) and a reduction in Krox-20. Importantly, UPR activity resulting from MPZ mutations does not clearly correlate with the clinical onset or severity of the neuropathy [232], with activation correlating with an infantile onset neuropathy with Arg98Cys but a childhood onset neuropathy with Ser63Del [229, 236]. It is also worth emphasizing that not all MPZ mutants retained in the ER actually activate the UPR in animal models, a phenomenon that may be related to the varying proteasomal capacity and ability to eliminate misfolded protein in Schwann cells [197].
Biological Models of MPZ Neuropathy
The two most commonly examined mouse models of MPZ have been the hemizygous S63del and heterozygous R98C transgenic mice [181, 184, 199, 217, 236,237,238]. ParallelingMPZ neuropathy in humans, the R98C knock-in mice demonstrate a more severe and earlier onset demyelinating neuropathy, whereas the S63del animals present later and do not show the same degree of developmental hypomyelination. Phenotypically, S63del mice demonstrate motor impairment, uniformly slowed NCS velocities, pathological evidence of demyelination with onion bulb formation, and clinical progression with age [199]. The model does not manifest the axonal loss seen in human disease, perhaps owing to the limited lifespan and the reduced nerve length in the animals [199, 239]. Both the heterozygous (R98C/ +) and homozygous (R98C/R98C) mice demonstrate weakness, abnormal nerve conduction velocities, and pathologically abnormal myelin, with the homozygous animals being more severely affected [217]. Both models also demonstrate retention of mutant protein in the ER with a resulting increase in UPR activation and CHOP expression; however, while CHOP ablation fully rescues the motor phenotype in S63del mouse models, it does not improve neuropathy in R98C mice [199, 217, 229, 230, 236]. This underscores the observation that different arms of the UPR are likely involved in nerve injury in the two mutants, namely the PERK pathway in S63del, versus IRE1 and ATF6 in R98C. A model that does not involve activation of the UPR is the Q215X mouse model of congenital hypomyelination, which has been used to examine aberrant MPZ trafficking [200, 232].
Methods that evaluate the degree of activation in the three arms of the UPR pathway (i.e., determination of CHOP levels to gauge activity in the PERK pathway) are increasingly being employed to examine UPR-mediated treatments in animal models [240]. In addition, in vitro assays, such as those assessing XBP1 splicing as a measure of IRE1 activation, have been used to identify UPR-activating MPZ mutants and to evaluate the effect of pharmacological agents [145, 232].
Therapeutic Targets in MPZ Neuropathy (Table 3 and Fig. 3)
Targeting the UPR to Treat MPZ Neuropathy
Interventional studies targeting the UPR in animal models have led to important insights into MPZ neuropathy pathogenesis and have offered promise for future therapies. Specifically, CHOP ablation in S63del mice rescued the motor deficits and reduced active demyelination two-fold [229]. Prolonging eIF2a phosphorylation and further attenuating protein translation by manipulating the PERK arm of the UPR is hypothesized to reduce the translation of mutant MPZ and thereby enhance the delivery of wild-type protein to the myelin sheath [232]. Furthermore, genetic and pharmacological inhibition of GADD34 reduced mutant protein retention in the ER and ameliorated the clinical phenotype in S63del, even more effectively than CHOP ablation [230]. Salubrinol, a molecule that inhibits the dephosphorylation of eIF2 by Gadd34, also reduced the accumulation of mutant MPZ in the ER and improved myelination in S63del nerves [230, 241]. Finally, Das et al. showed Sephin1, a selective inhibitor of the Gadd34 holophosphatase, effectively prolongs eIF2a phosphorylation, and ameliorates neuropathy in Ser63del mice, as evidenced by clinical and pathological measures [242]. Selectively correcting protein homeostasis and delaying recovery of protein translation may therefore offer an important therapeutic avenue for MPZ neuropathy, as well as other protein misfolding disorders.
Another approach to reducing UPR activation in MPZ neuropathy has been to employ sarcoplasmic/endoplasmic reticulum calcium pump (SERCA) inhibitors. SERCA inhibitors reduce ER stress and UPR activation by inhibiting calcium binding and disrupting calnexin function and have been shown to improve the phenotype of MPZ mutants in vitro [144]. Patzko and colleagues treated R98C mice with curcumin, a low affinity SERCA inhibitor, and found that phosphatidylcholine curcumin administration, designed to increase bioavailability, resulted in improvements in rotarod performance, CMAP amplitudes, and in the number of large diameter axons in the treated animals [145]. Treatment attenuated the IRE and ATF6 arms of the UPR, but did not alter the PERK pathway. Interestingly, treatment was also associated with changes in the NMJ, including an increase in the percentage of fully myelinated preterminal internodes and a decrease in the length of demyelinated segments approaching the NMJ [145, 240]. The authors hypothesized that the reduction in the “toxic gain of function” caused by mutant MPZ enabled Schwann cells to remain in a pro-myelinating state and suggested that improvement to haploinsufficiency could have a substantial clinical impact in MPZ neuropathy [145]. Despite these promising pre-clinical results, curcumin is not likely to be examined in clinical trials given its suboptimal pharmacokinetic properties [10].
The potential to manipulate the ER-associated degradation (ERAD) pathway to minimize the burden of misfolded protein in MPZ neuropathy was underscored by the work of Volpi and colleagues. ERAD facilitates the targeting of misfolded proteins to proteasomes for degradation, and the authors showed that Schwan cell-specific ablation of the ERAD factor Derlin-2 in S63del nerves increased myelin defects and the UPR in vivo. In contrast, treatment with N-Acetyl-D-glucosamine (GlcNAc) (an ERAD enhancing metabolite) of S63del dorsal-root-ganglia (DRG) explants improved nerve myelination [243]. These findings suggest that the ERAD has a protective role in MPZ neuropathy and that variations in ERAD may, in part, explain the phenotypic variability seen in neuropathies related to UPR activation.
UPR-Independent Treatments of MPZ Neuropathy
A non-UPR-mediated therapeutic target previously examined in MPZ neuropathy is axonal neuregulin 1 type III (Nrg1TIII), which activates the signaling pathways that lead to the expression of myelination genes, as well as increases in cholesterol and fatty acids [244]. Both overexpressing Nrg1TIII and suppressing the Nrg1TIII inhibitor tumor necrosis factor-alpha-converting enzyme (TACE/ADAM17) improved the clinical phenotype of the S63del mouse model without increasing ER stress [244]. As mentioned previously, ectopic expression of NaV1.8 sodium channels has been identified on motor axons in animal models of MPZ neuropathy. Rosberg et al. therefore evaluated the effects of an oral NaV1.8 blocker and demonstrated improved membrane dysfunction and motor performance in mice deficient in MPZ [245]. Lastly, as discussed in the next section on the treatment of connexin 32-associated neuropathy, reducing cytokine-activated macrophages and low-grade inflammation in P0Het mice (a model mimicking a heterozygous P0 loss-of-function mutation in humans) using a colony-stimulating factor 1 (CSF-1) receptor kinase inhibitor led to improved preservation of myelin, increased muscle action potential amplitudes, improved nerve conduction velocities, and improved muscle strength [246]. This suggests a potential role for immunomodulation of the secondary inflammatory response seen in some genetic demyelinating neuropathies.
Connexin 32-Associated Neuropathy: Epidemiology and Clinical Features
CMT1X, the most common form of X-linked CMT, is caused by mutations in GJB1, which encodes the gap junction protein connexin 32 (Cx32) [247]. CMT1X represents between 10 and 15% of CMT cases with a defined molecular diagnosis, and 5 to 10% of all CMT cases [9, 14, 248]. Over 400 mutations in GJB1 have been linked to CMT1X, and they span across all domains of Cx32. Most mutations are missense variants and are believed to cause predominantly loss of function phenotypes [249]. Interestingly, several variants in the non-coding regions of GJB1 have been demonstrated to cause CMT1X [250,251,252], at least in some cases due to abnormal splicing of GJB1 [253]. Furthermore, copy number variations in GJB1 have also been identified in patients with CMT1X [254, 255]. Therefore, care should be taken when interpreting results from commercial genetic testing, as non-coding variants as well as copy number variations may be missed. Of note, there is no specific correlation between phenotype and specific GJB1 mutations [256].
As in other dominant X-linked diseases, male patients with CMT1X present with a more severe phenotype, and women are usually only mildly affected; however, moderately to severely affected female patients with CMT1X are seen in approximately one-third of cases as a consequence of skewed X-inactivation of the nonmutated allele [257]. Most men will have symptoms in childhood, though about 20% have a later age of onset [9]. Clinically, CMT1X has distinctive features when compared to other demyelinating CMT subtypes. A split hand syndrome (abductor pollicis brevis more wasted and weaker than the first dorsal interosseous) can often be observed, as well as marked atrophy of all compartments of the calf muscles. Asymmetrical (non-uniform) slowing of nerve conduction velocities, with conduction block and temporal dispersion, which are characteristic of true segmental demyelination and also seen in hereditary neuropathy with liability to pressure palsy (HNPP), CMT4J, HSAN1C, and acquired inflammatory neuropathies, may be found in patients with missense mutations in GJB1, leading to misdiagnosis as an inflammatory neuropathy and unnecessary immunosuppressive treatment [258, 259]. CMT1X is also a common cause of intermediate nerve conduction velocities, with men usually presenting motor nerve conduction velocities (MNCV) between 25 and 45 m/s and women usually having MNCV greater than 35 m/s [9]. Another unique feature of CMT1X, predominantly in men, is the occurrence of transient stroke-like episodes with MRI changes following stressors, such as infection or fever, travel to high altitude, and intensive exercise [260].
Biological Models of Connexin 32 Neuropathy
Based on the hypothesis that most cases of CMT1X are due to loss of function of connexin 32, several of the early studies addressing the biology of CMT1X used mice knockdown for Gjb1 (Gjb1 − / − or Cx32 null mice) [261]. As genome editing technology advanced, several knock-in Gjb1 mice models have been created including the R142W [262], T55I, R75W, and N175D [263], which allowed investigators to study the trafficking properties of these different Cx32 mutants. Other models systems used in mechanistic studies of CMT1X include Xenopus oocytes [264] and N2A cells [265], which are used to evaluate the expression level and biophysical parameters of mutant forms of Cx32 in regard to their ability to form functional gap junctions. HeLa cells co-expressing wild type and mutant Cx32 have been used to study trafficking and interactions between different mutant forms of Cx32 [266].
Connexin 32 Biology and Pathomechanisms
Connexins are a group of membrane-spanning proteins that interact to form gap junction channels, allowing for the passage of ions and small molecules between cellular membranes. In the peripheral nervous system, Cx32 is found in the paranodal myelin loops and Schmidt-Lanterman incisures of myelinating Schwann cells where they form hexameric hemichannels. The docking of two hemichannels forms intracellular gap junctions between folds of Schwann cell cytoplasm, allowing the transfer of ions and molecules across the span of this highly polarized cell.
Abrams et al. demonstrated in paired Xenopus oocytes expressing seven distinct CMT1X-associated Cx32 mutants (G12S, R15Q, R15W, S85C, H94Q, H94Y, and V139M) that all mutants resulted in reduced or no conductance across the resulting gap junctions, albeit through different biophysical mechanisms. The authors concluded that a large number of CMT1X are due to loss of function of Cx32 [264] Using a similar approach in N2A cells, Wang and colleagues evaluated 22 CMT1X mutant Cx32 proteins for their ability to traffic to the cell membrane and form functional channels. Ten mutant Cx32 proteins either assembled dysfunctional junctional channels (Y65C, V95M, R107W, L156R, R164W, and G199R) or failed to form gap junctions (G12S, S182T, E208K, and Y211stop). Most mutant proteins were localized in the cell membrane despite their impaired ability to form functional gap junctions. Interestingly, 12 CMT1X mutants (V13L, R15Q, R22Q, I30N, V35M, V63I, R75Q, Q80R, W133R, P158A, P172S, and N205S) did not affect the ability of Cx32 to form homotypic gap junctions, suggesting that other mechanisms besides impaired gap junction formation should play a role in CMT1X. Abrams et al. proposed a possible mechanism to explain this discrepancy when studying the S85C Cx32 mutant [267]. This mutant Cx32 protein forms functional cell–cell channels in paired Xenopus oocytes but have a higher open probability compared to wild-type Cx32. Open hemichannels may render Schwann cells exposed to increased influx of calcium and loss of ionic gradients and metabolites, which can be damaging to the cells. Interestingly, the same group demonstrated differences in the ability of a Cx32 mutant to form gap junctions and produce at least some degree of junctional coupling that may determine whether a CMT1X patient is at risk of presenting central nervous system (CNS) manifestations. By comparing 10 Cx32 mutations associated with CNS involvement with 4 “neuropathy-exclusive” mutations, Abrams et al. found that all 10 CNS mutations formed no morphological gap junction plaques or, if they did, produced little or no detectable junctional coupling. In contrast, all four neuropathy mutations formed gap junction plaques and produced levels of junctional coupling similar to those for wild-type Cx32 [268].
Mutant Cx32 proteins also differ in their trafficking and subcellular localization. Yum et al. investigated the distribution of several mutant Cx32 proteins in HeLa cells and demonstrated preferential subcellular localization including the endoplasmic reticulum (M34K, N205I, and Y211x), the Golgi apparatus without reaching the cell membrane (M34T, V38M, A40V, R75Q, R75P, R75W, and C217x), the Golgi apparatus but also forming rare small gap junction-like plaques (M34I, M34V, and V37M), or mainly on the cell membrane, forming gap junction-like plaques (V35M, I213V, R219C, R219H, R220G, R230C, R230L, R238H, L239I, and S281x) [269]. They confirmed their findings in cultured rat Schwann cells for some of the mutants. Similar differences in trafficking were also identified by Jeng et al. [262] and Matsuyama et al. [270]. Kyriakoudi and colleagues also demonstrated using HeLa cells co-expressing wild-type and different mutant Cx32 proteins, that Golgi-retained mutants hinder gap junction assembly by wild type Cx32. Confirming these findings, in vivo intraneural delivery of wild-type Cx32 in mice bearing a Golgi-retained mutant (R75W) did not traffic normally, while the same virally delivered protein was correctly localized in mice expressing an endoplasmic reticulum-retained mutant (T55I). This work suggests that patients with CMT1X may respond differently to gene replacement therapy depending on their Cx32 mutation [266]. It is important to mention that although useful as a research tool to catalogue the different Cx32 mutations, impaired gap junction gating only explains a limited portion of the CMT1X pathogenesis and has not been correlated well with clinical disability. Therefore, care should be taken when using this parameter to predict disease severity or potential treatment response.
A possible second mechanism involved in the pathophysiology of CMT1X is the role of immune-mediated damage to Cx32 mutant peripheral nerves. Kobsar and colleagues identified an age-related increase in the number of macrophages in demyelinating nerves of Cx32-null mice [271] and were able to reduce endoneurial macrophages and both myelin and axonal degeneration by cross breeding Cx32-null mice with mice deficient for the recombination activating gene-1 (RAG-1), which lack mature T and B lymphocytes [272]. Groh et al. further demonstrated the connection between macrophages and peripheral nerve damage in CMT1X by cross breeding Cx32 null mice with monocyte chemoattractant protein-1 (MCP-1) knockout mice [273]. MCP-1 is a chemokine involved in the recruitment of macrophages to the peripheral nerves and has been shown to mediate macrophage-related neural damage in other models of inherited neuropathies [274]. The resulting double knockout mice still displayed increased endoneurial macrophages due to compensatory proliferation of resident macrophages. However, heterozygous deletion of MCP-1 led to reduced numbers of phagocytosing macrophages, transient improvement in myelination, and persistent improvement of axonal degeneration, with robust axonal sprouting lasting up to 12 months. This improvement in nerve pathology also translated into improved electrophysiological parameters, reduced muscle denervation and atrophy, and increased muscle strength. This study also implicated the MEK-ERK signaling pathway as mediating MCP-1 expression in Cx32-deficient Schwann cells. The authors concluded that preventing MCP-1 upregulation by inhibiting ERK phosphorylation may be a promising approach to treat CMT1X [273]. Groh et al. also explored the role of macrophage activation and low-grade inflammation in the dedifferentiation of Schwann cells, a typical feature of nerve fiber damage associated with several forms of demyelinating CMT. The authors determined that dedifferentiation of Cx32-deficient Schwann cells was strictly dependent on macrophage activation by the fibroblast-borne cytokine colony-stimulating factor-1(CSF-1), as CSF1/Cx32 double knockout Schwann cells demonstrated improvement in myelin preservation and did not upregulate dedifferentiation markers NCAM and L1. Importantly, this effect of CSF-1 was independent of the ERK signaling pathway [275, 276]. Taken together, these results provided proof of concept for the role of a secondary immune-mediated damage leading to Schwann cell dedifferentiation as part of the pathophysiology of CMT1X.
Cx32 mutations have been shown to cause polyploidy and an increase in nuclear volume due to mitotic instability causing centrosome overduplication [277]. This abnormal mitotic activity has been shown to be mediated through increased CamKII (Ca2+/calmodulin-dependent protein kinase II) activity and to result in perturbation in cell division of cell lines from transgenic CMT1X mice [278]. Abnormal Schwann cell division has been hypothesized to cause defects in myelination in experimental models as well as in patient nerves. However, further work in patient cells is needed to confirm this mechanism is also present in the human disease.
Therapeutic Targets in Connexin 32-Associated Neuropathy (Table 4 and Fig. 3)
Gene Replacement Therapy for Connexin 32-Associated Neuropathy
As loss of function is considered to be the main disease mechanism in CMT1X, gene replacement therapy has been in the forefront of therapy development for Cx32-related CMT. Accordingly, Sargiannidou and colleagues first demonstrated delivery of GJB1 under a Schwann cell-specific promoter (myelin protein zero promoter) by direct lentiviral intraneural injection in Cx32 null mice resulted in expression of the construct throughout the length of the sciatic nerve in up to 50% of Schwann cells starting 2 weeks after injection and remaining stable for up to 16 weeks as measured by eGFP expression. Cx32 expression was detected at the expected physiologic locations (non-compact myelin of the paranodal and Schmidt-Lanterman incisures) and resulted in the formation of gap junctions. Of note, a gene therapy trial in 2-month-old Cx32 null mice significantly improved myelination and reduced secondary inflammation at 6 months of age when compared to control, mock-treated animals [279]. Following up on these results, Kagiava and colleagues investigated the therapeutic effect of intrathecal delivery of the same lentiviral vector through a single lumbar injection in Cx32-null mice. A similar, widespread, and stable expression of Cx32 was observed in up to 50% of Schwann cells in multiple lumbar spinal roots and peripheral nerves. Importantly, this study demonstrated not only pathological rescue, but also behavioral improvement, with treated mice showing significantly improved motor performance, quadriceps muscle contractility, and sciatic nerve motor conduction velocities. As observed for the Cx32 mice treated with the intraneural injection, treated mice also exhibited reduced numbers of demyelinated and remyelinated fibers and fewer inflammatory cells in lumbar motor roots, as well as in the femoral motor and sciatic nerves [280].
To answer whether different Cx32 mutants would respond differently to gene replacement therapy, a question was raised by the identification of differentially trafficked Cx32 mutants causing dominant-negative effects by direct interactions with WT Cx32 (266, 269); Kagiava and colleagues treated transgenic Cx32 KO mice expressing the T55I (ER-retained), R75W, or N175D (Golgi-retained) CMT1X mutations. Following intrathecal delivery of the GJB1 gene, virally delivered wild-type (WT) Cx32 was observed in non-compact myelin of T55I KO mice, but only rarely in N175D KO or R75W KO mice, confirming the dominant-negative effects of the Golgi-retained mutants. GJB1-treated T55I mice also showed behavioral and pathological improvement with better motor performance, lower ratios of abnormally myelinated fibers, and a reduction of inflammatory cells in spinal roots and peripheral nerves compared with mock-treated littermates. For the Golgi-retained mutants, only partial (N175D) or no (R75W) improvement was observed. The authors concluded that certain CMT1X mutants may interfere with gene therapy for CMT1X, and further studies will be needed to determine the best strategy to treat patients who carry such mutations. Recently, Kagiava et al. moved their work closer to future human application by using adeno-associated virus (AAV) as their vector system. AAV are not known to cause human disease and have been the delivery system of choice for several FDA-approved gene replacement therapies and for ongoing clinical trials. Using AAV-9 to deliver a similar GJB1/Cx32 gene under the myelin protein zero (Mpz) promoter for targeted expression in Schwann cells, the authors observed widespread distribution in the peripheral nervous system, including lumbar roots, sciatic, and femoral nerves after intrathecal lumbar injection. This gene replacement strategy was successful in rescuing the normal Cx32 expression in non-compact myelin and improved behavioral, neurophysiological, and pathological features of both pre- and post-onset-treated Cx32-null mice [281]. Of note, neurofilament light chain (NEFL), a biomarker of axonal degeneration, also improved in response to gene replacement therapy. These results were in keeping with a similar study using their lentiviral vector and suggest that gene replacement therapy may be beneficial even in patients with established disease [282].
Immune Regulation as a Treatment Strategy for Connexin 32-Associated Neuropathy
Cross breeding of Cx32-null mice with mice deficient for MCP1 or CSF-1 has demonstrated the role of cytokine-activated macrophages and low-grade inflammation in Schwann cell dedifferentiation and demyelination in CMT1X and provided a target for therapy development for connexin 32-associated neuropathy [273, 275, 276]. Klein et al. targeted this pathway by orally administering a CSF-1 receptor kinase inhibitor (PLX5622) to Cx32-null mice [246]. CSF-1 receptors are expressed by macrophages and activated by fibroblast-released CSF-1. PLX5622 led to a significant reduction in nerve macrophage numbers and fewer abnormal axons while increasing the number of axonal sprouts (an indication of axonal reinnervation). PLX5622 treatment also increased hindlimb grip strength and compound muscle action potential amplitudes in the intrinsic foot muscles. Interestingly, this approach did not change the number of abnormally myelinated axons [246].
Ca2+/Calmodulin-Dependent Protein Kinase II (CamKII) Inhibitor to Treat Mitotic Instability in Connexin 32-Associated Neuropathy
Transgenic mice expressing CMT1X-associated Cx32 mutants displayed evidence of mitotic instability, such as polyploidy, nuclear volume, and centrosome overduplication likely due to overexpression of CamKII. Mones et al. used a CamKII inhibitor, KN 93, to treat transgenic GJB1 mice carrying the G12S or S26L mutations [277]. KN 93 treatment resulted in slower progression of muscle weakness as determined by rotarod analysis, as well as improvement in mitotic instability in fibroblasts derived from the transgenic mice [278].
Adeno-Associated Virus/Neurothrophin-3 Gene Therapy for CMT1X
Similar to work done for CMT1A, Ozes and colleagues treated Cx32 null mice using a scAAV1.tMCK.NT-3 vector to induce expression of neurotrophin-3 (NT-3). Intramuscular delivery of NT-3 in 3-month-old Cx32 null mice resulted in significant improvement in myelin thickness, nerve conduction velocity, and muscle fiber diameter, with normalized compound muscle action potential amplitudes, and no functional decline at 9 months of age [283]. An increase in the number of Schmidt-Lanterman incisures was also observed, although the functional meaning of this finding is uncertain.
Pathomechanisms and Emerging Treatments in Select Forms of AR Demyelinating CMT (Table 5 and Fig. 3)
The most prevalent of the recessively inherited demyelinating CMTs is CMT4C, which results from loss of function mutations in the SH3TC2 gene on chromosome 5q32. Over 100 pathogenic mutations have been identified in SH3TC2 to date [14, 284, 285]. In addition to the typical features seen with CMT, SH3TC2 neuropathy is distinguished by the presence of scoliosis and cranial neuropathies (manifesting as hearing loss, sluggish pupils, and tongue fasciculations) [285,286,287,288,289]. Nerve conduction studies reflect demyelinating neuropathy, and nerve biopsy reveals an increase in basal membranes around both myelinated and unmyelinated axons, as well as characteristic large cytoplasmic Schwann cell extensions [290,291,292]. The Sh3tc2 − / − mouse model recapitulates the clinical phenotype of progressive hypomyelinating and demyelinating polyneuropathy [293, 294].
The SH3TC2 protein contains two Src homology 3 (SH3) and 10 tetratricopeptide repeat (TPR) domains, and based on observations in the Sh3tc2 − / − mouse model, it is believed to serve as a scaffolding protein and to play a role in the formation of the nodes of Ranvier [153, 292, 293]. In normal circumstances, SH3TC2 is involved in the endocytic pathway; however, mutations in Sh3tc2 can lead to the mistargeting of the protein away from endosomes [153, 295].
Schiza at el. developed a lentiviral vector under control of the MPZ promoter to drive the expression of SH3TC2 cDNA in Schwann cells [296]. Intrathecal injection of the vector into Sh3tc2 − / − mice at 3 weeks of age resulted in effective localization of SH3TC2 to the Schwann cell cytoplasm and resulted in partial improvement of motor function and motor conduction velocities, as well as the numbers of myelinated fibers in the treated animals [296]. Treatment also resulted in reduced blood levels of neurofilament light chain, a biomarker of axonal degeneration that was previously evaluated in CMT [297]. This study serves as an important proof of principal for viral gene replacement therapy in SH3TC2 though further work will be needed to ensure safety, while also optimizing gene expression.
Genes associated with endo-lysosomal membrane trafficking have been implicated in several recessives forms of CMT. Most of these genes are phosphatases involved in phosphoinositide metabolism and include the myotubularin-related (MTMR) protein family (MTMR2, 13 and 5) and FIG4 [298,299,300,301].
MTMR2, MTMR13, and MTMR5/SBF1, the genetic causes of CMT4B1, CMT4B2, and CMT4B3, respectively, are associated with abnormal myelin formation characterized by myelin outfoldings, and redundant loops of myelin lamellae, which can be seen in patients’ nerve and skin biopsies. Interestingly, while MTMR2 encodes for an active phosphate, MTMR13 and MTMR5 express inactive enzymes (also known as a pseudophosphatases). Nonetheless, both MTMR2 and MTMR13 have been demonstrated to interfere with PI 3-kinase-Akt signaling pathway activation, causing dys/demyelination [302]. Experimental work on the biology of the MTMR protein family has demonstrated that this group of phosphatases regulates the metabolism of phosphoinositides, signaling molecules that regulate membrane trafficking by controlling membrane dynamics and vesicle movement, tethering, and fusion. These cellular processes are key mediators of axonal transport and myelination in the nervous system.
A recent multicenter, retrospective study of patients with MTMR-related neuropathy characterized the phenotype associated with this group of neuropathies in a total of 50 patients, including 26 with CMT4B1 (MTMR2), 19 with CMT4B2 (MTMR13), and 5 with CMT4B3 (MTMR5). CMT4B1 patients demonstrated a significantly more severe phenotype compared to CMT4B2, with earlier onset, motor milestone delay, wheelchair use, and respiratory involvement. Vocal cord involvement is a feature of both subtypes, but glaucoma occurred in CMT4B2 only [298, 303]. CMT4B3 patients demonstrated a distinct phenotype, with a slowly progressive, pure demyelinating neuropathy with myelin outfoldings and, in some cases, a syndromic neuropathy characterized by axonal degeneration, multiple cranial nerve involvement, intellectual disability, microcephaly, and dysmorphic features. Nerve conduction velocities were similarly slowed in all subtypes [303].
The advances in understanding the biology of MTMR phosphatases have opened new therapy development avenues for this group of autosomal recessive neuropathies. Vaccari and colleagues demonstrated that inhibition of the kinase PIKfyve (also known as PIP5K3) by small molecule kinase inhibitors rebalances the levels of PtdIns (3,5) P2 phospholipids, which have been demonstrated to be elevated in the absence of MTMR2 activity. The use of such kinase inhibitors was able to significantly reduce the occurrence of myelin outfoldings in vitro, in a co-culture system using dorsal root ganglia-derived neurons and Schwann cells from Mtmr2-null mice [304]. It is important to note that this approach relies on the precise dosing of PIKfyve as reduction of PtdIns(3,5)P2 phospholipids may be detrimental and cause severe toxicity.
A second approach focuses on the neuregulin pathway, one of the main signaling pathways regulating Axon-Schwann cell interactions, Schwann cell development, and myelination [305]. Neuregulin 1 (NRG1) type III is expressed in the surface of axons and interacts with ErbB2/3 receptors in the Schwann cell membrane to activate the PI 3-kinase-Akt signaling pathway, which promotes myelination. Inhibition of the NRG1 type III signaling therefore should reduce the amount of abnormal myelin formed in MTMR-related neuropathies. For this purpose, Bolino et al. explored the use of tumor necrosis factor-α converting enzyme (TACE), a negative regulator of the NRG1 type III pathway, as a treatment strategy for MTMR-associated neuropathies. Using Niacin-Niaspan, an FDA-approved TACE activator, the authors demonstrated reduction in NRG1 type III signaling as well as a reduction in myelin outfoldings in the same co-culture system used to validate PIKfyve kinase inhibitors [306].
Another autosomal recessive CMT gene, FIG4, also encodes for a phosphatase involved in phosphoinositide metabolism. FIG4 was first identified as a disease-associated gene in the spontaneously occurring “pale-tremor” (plt) mutant mice. These mice present with severe tremor, abnormal gait, peripheral neuropathy, and diluted pigmentation and were found to carry a transposon insertion in intron 18 of the mouse FIG4 gene, resulting in a loss-of-function allele. Loss of FIG4 function reduces the intracellular concentration of phosphoinositol(3,5)P2- [PtdIns(3,5)P2], leading to impaired vacuole fission, formation of enlarged vacuoles, and reduced retrograde traffic to the late endosome [301]. The FIG4 protein is a [PtdIns(3,5)P2] phosphatase, which forms a complex with other enzymes that generate PI(3,5)P2, including PIKFYVE and VAC14. Loss of FIG4 causes instability of this complex, resulting in the mislocalization of all three proteins and reduced levels of PI (3,5) P2 [307].
Clinically, CMT4J is characterized by a heterogeneous phenotype with significant variability in age of onset and severity of symptoms, ranging from mild symptoms to wheelchair use. Unlike the typical length-dependent and symmetrical presentation of most CMT subtypes, CMT4J can present with both proximal and distal weakness as well as marked asymmetry in clinical and electrophysiological findings [308, 309]. Sensory motor demyelinating polyneuropathy was consistently found in all patients studied by Hu et al. [310] and was associated with non-uniform slowing of conduction velocities, conduction block, and temporal dispersion on nerve conduction studies. These features sometimes cause diagnostic error, with CMT4J patients being frequently misdiagnosed as having acquired inflammatory neuropathies or motor neuron disease. From a genetic perspective, affected individuals are compound heterozygotes carrying the missense allele FIG4I41T in combination with a null allele. Lenk et al. demonstrated, in a mouse model of CMT4J expressing a Fig4I41T cDNA transgene on Fig4 null background, that FIG4I41T is a hypomorphic allele encoding a protein that is unstable in vivo. Expression of FIG4I41T protein at 10% of normal level was shown to be enough to rescue the mouse phenotype, suggesting that increasing the levels of FIG4I41T, by increasing its production or stability, should be a goal of therapies for CMT4J [307].
This hypothesis was recently pursued by Presa et al., by treating pale tremor (plt) mice with a single-stranded AAV9 to deliver a codon-optimized human FIG4 sequence. Compared to untreated plt mice, which have a median survival of approximately 5 weeks, mice treated at postnatal day 1 or 4 survived at least 1 year, with significant rescue of their clinical, electrophysiological, and pathological phenotypes. AAV9 treatment at postnatal day 7 or 11 still increased life span, albeit less than the earlier treatment, and the clinical phenotype was only partially rescued. These results are in keeping with findings from other gene replacement therapies for neurodegenerative genetic disorders, where the earlier the treatment, the better the overall treatment response [311].
Another mechanism identified in the pathophysiology of CMT4J is abnormally reduced lysosomal calcium efflux resulting from impairment in the calcium channel TRPML1. PI(3,5)P2 is an endogenous ligand of TRPML1, and its deficiency in CMT4J leads to deactivation of TRPML1. The consequent reduced efflux of lysosomal calcium leads to its accumulation inside the organelle, impairing lysosome fission and leading to the enlarged vacuoles seen in fibroblasts, neurons, and Schwann cells of patients with CMT4J [301, 308, 312]. This mechanism was explored by Zou et al. by treating plt mice with a synthetic ligand of TRPML1, ML-SA1, to activate this calcium channel and reduce the intralysosomal calcium level. This pharmacological intervention rescued abnormal lysosomal storage in plt mice culture cells and ex vivo DRGs [313].
Abnormal calcium accumulation was also observed in Schwann cells of plt mice, leading to overexpression of c-Jun, a negative regulator of myelination, and consequent Schwann cell de-differentiation and segmental demyelination. Of note, chelation of calcium using BAPTA reduced dedifferentiation and demyelination of Schwann cells in vitro and in vivo, providing another treatment strategy for FIG4-related CMT [310].
Looking Towards the Future: the Evolving Arena of Clinical Outcome Assessments and Biomarkers in Demyelinating CMT
As disease-modifying treatments for select forms of demyelinating CMT become increasingly plausible, it is critical to address the current barriers to successful clinical trials. Most forms of CMT progress slowly and can plateau, or even improve, over the course of the disease, posing a challenge in developing clinical outcome assessments sensitive enough to capture disease progression. Clinical trials evaluating ascorbic acid in CMT1A have also highlighted the limited responsiveness of the Charcot-Marie-Tooth Neuropathy Score (CMTNS), the main clinical outcome assessment in CMT, and revealed a positive placebo effect [131, 314]. The CMNTS was subsequently revised and modified using Rasch analysis, with a resulting increase in sensitivity [315, 316]; however, its use continues to require very large sample sizes to adequately assess therapeutic benefit [317]. The development of new instruments to assess progression in CMT has therefore become an active area of research. Given that the optimal time for the treatment in CMT is in infancy or early childhood, several early-life clinical outcome assessments (including the CMT pediatric, CMT infant scales, and a pediatric CMT-specific quality of life outcome measure) have been developed [318,319,320].
Rather than using a single optimized measure of disease progression, clinical trials in CMT will need to employ a composite of rigorously examined and validated clinical outcomes assessments and biomarkers. Encouragingly, quantitative calf muscle fat accumulation on MRI has proven to be a highly sensitive measure of progression in patients with CMT1A, with a notable increase in fat fraction observed over a 12-month period [321, 322]. MRI is now being increasingly employed in natural history studies of CMT and will play an important role in clinical trials. It is worth emphasizing, however, that in assessing treatment response, MRI will still have to be used alongside optimized CMT-specific functional outcomes.
In regard to plasma biomarkers, Sandelius et al. have demonstrated elevations in the concentration of neurofilament light (NfL) chain (a marker of axonal damage) in patients with two forms of demyelinating CMT (CMT1A and CMT1X) [297]. Additionally, Wang et al. highlighted the TMPRSS5 protein (Transmembrane protease serine 5) as a potential Schwann cell-specific biomarker in patients with CMT1A, with a greater than twofold increase seen in the CMT1A patients as compared to controls [323].
Skin biopsy has also offered promise as a biomarker in CMT, both in regard to assessing neuropathy progression and as a method for examining gene-specific targets [324, 325]. As previously discussed, evaluation of PMP-22 in skin biopsy to test the effect of PMP-22 lowering therapies has proven effective in animal models but showed variable results in human studies as reviewed by Pantera et al. [55]. Interestingly, the authors demonstrate that employing digital methods of PMP-22 analysis and normalizing skin PMP-22 levels to Schwann cell-specific genes may offer more consistency going forward; however, the inter-patient variability in PMP-22 levels remains an ongoing challenge [55].
In summary, the biology underlying demyelinating forms of CMT is complex and varied, and unifying therapeutic approaches are unlikely to be feasible. Rather, treatments will need to be developed with attention not just to gene, but also to allele-specific pathomechanisms. While important challenges lie ahead, this is undoubtedly a very exciting time in CMT research, as years of biological investigation converge towards the development of novel and increasingly attainable disease modifying treatments for several demyelinating CMT subtypes.
References
Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet. 1974;6(2):98-118.
Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980;103(2):259-80.
Magy L, Mathis S, Le Masson G, Goizet C, Tazir M, Vallat JM. Updating the classification of inherited neuropathies: Results of an international survey. Neurology. 2018;90(10):e870-e6.
Manganelli F, Pisciotta C, Reilly MM, Tozza S, Schenone A, Fabrizi GM, et al. Nerve conduction velocity in CMT1A: what else can we tell? Eur J Neurol. 2016;23(10):1566-71.
Li J. Caveats in the Established Understanding of CMT1A. Ann Clin Transl Neurol. 2017;4(8):601-7.
Garcia A, Combarros O, Calleja J, Berciano J. Charcot-Marie-Tooth disease type 1A with 17p duplication in infancy and early childhood: a longitudinal clinical and electrophysiologic study. Neurology. 1998;50(4):1061-7.
Cortese A, Wilcox JE, Polke JM, Poh R, Skorupinska M, Rossor AM, et al. Targeted next-generation sequencing panels in the diagnosis of Charcot-Marie-Tooth disease. Neurology. 2020;94(1):e51-e61.
Dohrn MF, Glockle N, Mulahasanovic L, Heller C, Mohr J, Bauer C, et al. Frequent genes in rare diseases: panel-based next generation sequencing to disclose causal mutations in hereditary neuropathies. J Neurochem. 2017;143(5):507-22.
Saporta AS, Sottile SL, Miller LJ, Feely SM, Siskind CE, Shy ME. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol. 2011;69(1):22-33.
Juneja M, Burns J, Saporta MA, Timmerman V. Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development. J Neurol Neurosurg Psychiatry. 2019;90(1):58-67.
Saghira C, Bis DM, Stanek D, Strickland A, Herrmann DN, Reilly MM, et al. Variant pathogenicity evaluation in the community-driven Inherited Neuropathy Variant Browser. Hum Mutat. 2018;39(5):635-42.
Latour P, Gonnaud PM, Ollagnon E, Chan V, Perelman S, Stojkovic T, et al. SIMPLE mutation analysis in dominant demyelinating Charcot-Marie-Tooth disease: three novel mutations. J Peripher Nerv Syst. 2006;11(2):148-55.
Murphy SM, Laura M, Fawcett K, Pandraud A, Liu YT, Davidson GL, et al. Charcot-Marie-Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry. 2012;83(7):706-10.
Fridman V, Bundy B, Reilly MM, Pareyson D, Bacon C, Burns J, et al. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: a cross-sectional analysis. J Neurol Neurosurg Psychiatry. 2015;86(8):873-8.
Brennan KM, Bai Y, Shy ME. Demyelinating CMT--what's known, what's new and what's in store? Neurosci Lett. 2015;596:14-26.
Jerath NU, Shy ME. Hereditary motor and sensory neuropathies: Understanding molecular pathogenesis could lead to future treatment strategies. Biochim Biophys Acta. 2015;1852(4):667-78.
Finkel RS, Mercuri E, Darras BT, Connolly AM, Kuntz NL, Kirschner J, et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N Engl J Med. 2017;377(18):1723-32.
Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018;379(1):22-31.
Zhao HT, Damle S, Ikeda-Lee K, Kuntz S, Li J, Mohan A, et al. PMP22 antisense oligonucleotides reverse Charcot-Marie-Tooth disease type 1A features in rodent models. J Clin Invest. 2018;128(1):359-68.
Girault JA, Peles E. Development of nodes of Ranvier. Curr Opin Neurobiol. 2002;12(5):476-85.
Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485(7399):517-21.
Niemann A, Berger P, Suter U. Pathomechanisms of mutant proteins in Charcot-Marie-Tooth disease. Neuromolecular Med. 2006;8(1-2):217-42.
Roa BB, Warner LE, Garcia CA, Russo D, Lovelace R, Chance PF, et al. Myelin protein zero (MPZ) gene mutations in nonduplication type 1 Charcot-Marie-Tooth disease. Hum Mutat. 1996;7(1):36-45.
Callegari I, Gemelli C, Geroldi A, Veneri F, Mandich P, D'Antonio M, et al. Mutation update for myelin protein zero-related neuropathies and the increasing role of variants causing a late-onset phenotype. J Neurol. 2019;266(11):2629-45.
Liu L, Li X, Zi X, Huang S, Zhan Y, Jiang M, et al. Two novel MPZ mutations in Chinese CMT patients. J Peripher Nerv Syst. 2013;18(3):256-60.
Lemke G, Lamar E, Patterson J. Isolation and analysis of the gene encoding peripheral myelin protein zero. Neuron. 1988;1(1):73-83.
Blair IP, Nash J, Gordon MJ, Nicholson GA. Prevalence and origin of de novo duplications in Charcot-Marie-Tooth disease type 1A: first report of a de novo duplication with a maternal origin. Am J Hum Genet. 1996;58(3):472-6.
Laura M, Hutton EJ, Blake J, Lunn MP, Fox Z, Pareyson D, et al. Pain and small fiber function in Charcot-Marie-Tooth disease type 1A. Muscle Nerve. 2014;50(3):366-71.
Sommer C. Nerve and skin biopsy in neuropathies. Curr Opin Neurol. 2018;31(5):534-40.
Krajewski KM, Lewis RA, Fuerst DR, Turansky C, Hinderer SR, Garbern J, et al. Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1A. Brain. 2000;123 ( Pt 7):1516-27.
Pareyson D, Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. 2009;8(7):654-67.
Fabrizi GM, Simonati A, Morbin M, Cavallaro T, Taioli F, Benedetti MD, et al. Clinical and pathological correlations in Charcot-Marie-Tooth neuropathy type 1A with the 17p11.2p12 duplication: a cross-sectional morphometric and immunohistochemical study in twenty cases. Muscle Nerve. 1998;21(7):869–77.
Thomas PK, Marques W, Jr., Davis MB, Sweeney MG, King RH, Bradley JL, et al. The phenotypic manifestations of chromosome 17p11.2 duplication. Brain. 1997;120 ( Pt 3):465–78.
Yiu EM, Burns J, Ryan MM, Ouvrier RA. Neurophysiologic abnormalities in children with Charcot-Marie-Tooth disease type 1A. J Peripher Nerv Syst. 2008;13(3):236-41.
Saporta MA, Katona I, Lewis RA, Masse S, Shy ME, Li J. Shortened internodal length of dermal myelinated nerve fibres in Charcot-Marie-Tooth disease type 1A. Brain. 2009;132(Pt 12):3263-73.
Li J, Parker B, Martyn C, Natarajan C, Guo J. The PMP22 gene and its related diseases. Mol Neurobiol. 2013;47(2):673-98.
Tao F, Beecham GW, Rebelo AP, Blanton SH, Moran JJ, Lopez-Anido C, et al. Modifier Gene Candidates in Charcot-Marie-Tooth Disease Type 1A: A Case-Only Genome-Wide Association Study. J Neuromuscul Dis. 2019;6(2):201-11.
Fridman V, Reilly MM. Inherited Neuropathies. Semin Neurol. 2015;35(4):407-23.
Katona I, Wu X, Feely SM, Sottile S, Siskind CE, Miller LJ, et al. PMP22 expression in dermal nerve myelin from patients with CMT1A. Brain. 2009;132(Pt 7):1734-40.
Chance PF, Alderson MK, Leppig KA, Lensch MW, Matsunami N, Smith B, et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72(1):143-51.
Scherer SS, Chance PF. Myelin genes: getting the dosage right. Nat Genet. 1995;11(3):226-8.
Fusco C, Spagnoli C, Salerno GG, Pavlidis E, Frattini D, Pisani F. Hereditary neuropathy with liability to pressure palsy (HNPP): report of a family with a new point mutation in PMP22 gene. Ital J Pediatr. 2017;43(1):97.
Hanemann CO, D'Urso D, Gabreels-Festen AA, Muller HW. Mutation-dependent alteration in cellular distribution of peripheral myelin protein 22 in nerve biopsies from Charcot-Marie-Tooth type 1A. Brain. 2000;123 ( Pt 5):1001-6.
Adlkofer K, Frei R, Neuberg DH, Zielasek J, Toyka KV, Suter U. Heterozygous peripheral myelin protein 22-deficient mice are affected by a progressive demyelinating tomaculous neuropathy. J Neurosci. 1997;17(12):4662-71.
Snipes GJ, Suter U, Welcher AA, Shooter EM. Characterization of a novel peripheral nervous system myelin protein (PMP-22/SR13). J Cell Biol. 1992;117(1):225-38.
Lee S, Amici S, Tavori H, Zeng WM, Freeland S, Fazio S, et al. PMP22 is critical for actin-mediated cellular functions and for establishing lipid rafts. J Neurosci. 2014;34(48):16140-52.
Mittendorf KF, Marinko JT, Hampton CM, Ke Z, Hadziselimovic A, Schlebach JP, et al. Peripheral myelin protein 22 alters membrane architecture. Sci Adv. 2017;3(7):e1700220.
Suter U, Snipes GJ. Peripheral myelin protein 22: facts and hypotheses. J Neurosci Res. 1995;40(2):145-51.
Pareek S, Notterpek L, Snipes GJ, Naef R, Sossin W, Laliberte J, et al. Neurons promote the translocation of peripheral myelin protein 22 into myelin. J Neurosci. 1997;17(20):7754-62.
Garbay B, Heape AM, Sargueil F, Cassagne C. Myelin synthesis in the peripheral nervous system. Prog Neurobiol. 2000;61(3):267-304.
D'Urso D, Ehrhardt P, Muller HW. Peripheral myelin protein 22 and protein zero: a novel association in peripheral nervous system myelin. J Neurosci. 1999;19(9):3396-403.
Hasse B, Bosse F, Hanenberg H, Muller HW. Peripheral myelin protein 22 kDa and protein zero: domain specific trans-interactions. Mol Cell Neurosci. 2004;27(4):370-8.
Magyar JP, Martini R, Ruelicke T, Aguzzi A, Adlkofer K, Dembic Z, et al. Impaired differentiation of Schwann cells in transgenic mice with increased PMP22 gene dosage. J Neurosci. 1996;16(17):5351-60.
Sergeev VM, Bychkov VA. [Diagnosis and therapy of acute purulent destructive pneumonia in children (current state of the problem)]. Grudn Khir. 1977(3):60-8.
Pantera H, Shy ME, Svaren J. Regulating PMP22 expression as a dosage sensitive neuropathy gene. Brain Res. 2020;1726:146491.
Passage E, Norreel JC, Noack-Fraissignes P, Sanguedolce V, Pizant J, Thirion X, et al. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med. 2004;10(4):396-401.
Sereda MW, Meyer zu Horste G, Suter U, Uzma N, Nave KA. Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A). Nat Med. 2003;9(12):1533–7.
Perea J, Robertson A, Tolmachova T, Muddle J, King RH, Ponsford S, et al. Induced myelination and demyelination in a conditional mouse model of Charcot-Marie-Tooth disease type 1A. Hum Mol Genet. 2001;10(10):1007-18.
Liu P, Gelowani V, Zhang F, Drory VE, Ben-Shachar S, Roney E, et al. Mechanism, prevalence, and more severe neuropathy phenotype of the Charcot-Marie-Tooth type 1A triplication. Am J Hum Genet. 2014;94(3):462-9.
Hirt N, Eggermann K, Hyrenbach S, Lambeck J, Busche A, Fischer J, et al. Genetic dosage compensation via co-occurrence of PMP22 duplication and PMP22 deletion. Neurology. 2015;84(15):1605-6.
Nobbio L, Visigalli D, Radice D, Fiorina E, Solari A, Lauria G, et al. PMP22 messenger RNA levels in skin biopsies: testing the effectiveness of a Charcot-Marie-Tooth 1A biomarker. Brain. 2014;137(Pt 6):1614-20.
Fledrich R, Stassart RM, Klink A, Rasch LM, Prukop T, Haag L, et al. Soluble neuregulin-1 modulates disease pathogenesis in rodent models of Charcot-Marie-Tooth disease 1A. Nat Med. 2014;20(9):1055-61.
Li J, Ghandour K, Radovanovic D, Shy RR, Krajewski KM, Shy ME, et al. Stoichiometric alteration of PMP22 protein determines the phenotype of hereditary neuropathy with liability to pressure palsies. Arch Neurol. 2007;64(7):974-8.
Morena J, Gupta A, Hoyle JC. Charcot-Marie-Tooth: From Molecules to Therapy. Int J Mol Sci. 2019;20(14).
Suter U, Snipes GJ, Schoener-Scott R, Welcher AA, Pareek S, Lupski JR, et al. Regulation of tissue-specific expression of alternative peripheral myelin protein-22 (PMP22) gene transcripts by two promoters. J Biol Chem. 1994;269(41):25795-808.
Maier M, Castagner F, Berger P, Suter U. Distinct elements of the peripheral myelin protein 22 (PMP22) promoter regulate expression in Schwann cells and sensory neurons. Mol Cell Neurosci. 2003;24(3):803-17.
Maier M, Berger P, Nave KA, Suter U. Identification of the regulatory region of the peripheral myelin protein 22 (PMP22) gene that directs temporal and spatial expression in development and regeneration of peripheral nerves. Mol Cell Neurosci. 2002;20(1):93-109.
Pantera H, Moran JJ, Hung HA, Pak E, Dutra A, Svaren J. Regulation of the neuropathy-associated Pmp22 gene by a distal super-enhancer. Hum Mol Genet. 2018;27(16):2830-9.
Pantera H, Hu B, Moiseev D, Dunham C, Rashid J, Moran JJ, et al. Pmp22 super-enhancer deletion causes tomacula formation and conduction block in peripheral nerves. Hum Mol Genet. 2020;29(10):1689-99.
Jones EA, Lopez-Anido C, Srinivasan R, Krueger C, Chang LW, Nagarajan R, et al. Regulation of the PMP22 gene through an intronic enhancer. J Neurosci. 2011;31(11):4242-50.
Lopez-Anido C, Poitelon Y, Gopinath C, Moran JJ, Ma KH, Law WD, et al. Tead1 regulates the expression of Peripheral Myelin Protein 22 during Schwann cell development. Hum Mol Genet. 2016;25(14):3055-69.
Makoukji J, Shackleford G, Meffre D, Grenier J, Liere P, Lobaccaro JM, et al. Interplay between LXR and Wnt/beta-catenin signaling in the negative regulation of peripheral myelin genes by oxysterols. J Neurosci. 2011;31(26):9620-9.
Felekkis K, Touvana E, Stefanou C, Deltas C. microRNAs: a newly described class of encoded molecules that play a role in health and disease. Hippokratia. 2010;14(4):236-40.
Verrier JD, Lau P, Hudson L, Murashov AK, Renne R, Notterpek L. Peripheral myelin protein 22 is regulated post-transcriptionally by miRNA-29a. Glia. 2009;57(12):1265-79.
Arthur-Farraj PJ, Morgan CC, Adamowicz M, Gomez-Sanchez JA, Fazal SV, Beucher A, et al. Changes in the Coding and Non-coding Transcriptome and DNA Methylome that Define the Schwann Cell Repair Phenotype after Nerve Injury. Cell Rep. 2017;20(11):2719-34.
Lee JS, Kwak G, Kim HJ, Park HT, Choi BO, Hong YB. miR-381 Attenuates Peripheral Neuropathic Phenotype Caused by Overexpression of PMP22. Exp Neurobiol. 2019;28(2):279-88.
van Paassen BW, van der Kooi AJ, van Spaendonck-Zwarts KY, Verhamme C, Baas F, de Visser M. PMP22 related neuropathies: Charcot-Marie-Tooth disease type 1A and Hereditary Neuropathy with liability to Pressure Palsies. Orphanet J Rare Dis. 2014;9:38.
Tao F, Beecham GW, Rebelo AP, Svaren J, Blanton SH, Moran JJ, et al. Variation in SIPA1L2 is correlated with phenotype modification in Charcot- Marie- Tooth disease type 1A. Ann Neurol. 2019;85(3):316-30.
Sinkiewicz-Darol E, Lacerda AF, Kostera-Pruszczyk A, Potulska-Chromik A, Sokolowska B, Kabzinska D, et al. The LITAF/SIMPLE I92V sequence variant results in an earlier age of onset of CMT1A/HNPP diseases. Neurogenetics. 2015;16(1):27-32.
Meggouh F, de Visser M, Arts WF, De Coo RI, van Schaik IN, Baas F. Early onset neuropathy in a compound form of Charcot-Marie-Tooth disease. Ann Neurol. 2005;57(4):589-91.
Brewer MH, Ma KH, Beecham GW, Gopinath C, Baas F, Choi BO, et al. Haplotype-specific modulation of a SOX10/CREB response element at the Charcot-Marie-Tooth disease type 4C locus SH3TC2. Hum Mol Genet. 2014;23(19):5171-87.
Nam SH, Kanwal S, Nam DE, Lee MH, Kang TH, Jung SC, et al. Association of miR-149 polymorphism with onset age and severity in Charcot-Marie-Tooth disease type 1A. Neuromuscul Disord. 2018;28(6):502-7.
Notterpek L, Snipes GJ, Shooter EM. Temporal expression pattern of peripheral myelin protein 22 during in vivo and in vitro myelination. Glia. 1999;25(4):358-69.
Fortun J, Dunn WA, Jr., Joy S, Li J, Notterpek L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J Neurosci. 2003;23(33):10672-80.
Fortun J, Verrier JD, Go JC, Madorsky I, Dunn WA, Notterpek L. The formation of peripheral myelin protein 22 aggregates is hindered by the enhancement of autophagy and expression of cytoplasmic chaperones. Neurobiol Dis. 2007;25(2):252-65.
Fortun J, Go JC, Li J, Amici SA, Dunn WA, Jr., Notterpek L. Alterations in degradative pathways and protein aggregation in a neuropathy model based on PMP22 overexpression. Neurobiol Dis. 2006;22(1):153-64.
Lee S, Bazick H, Chittoor-Vinod V, Al Salihi MO, Xia G, Notterpek L. Elevated Peripheral Myelin Protein 22, Reduced Mitotic Potential, and Proteasome Impairment in Dermal Fibroblasts from Charcot-Marie-Tooth Disease Type 1A Patients. Am J Pathol. 2018;188(3):728-38.
Tobler AR, Liu N, Mueller L, Shooter EM. Differential aggregation of the Trembler and Trembler J mutants of peripheral myelin protein 22. Proc Natl Acad Sci U S A. 2002;99(1):483-8.
Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6(1):11-22.
Rangaraju S, Madorsky I, Pileggi JG, Kamal A, Notterpek L. Pharmacological induction of the heat shock response improves myelination in a neuropathic model. Neurobiol Dis. 2008;32(1):105-15.
Sahenk Z, Serrano-Munuera C, Chen L, Kakabadze I, Najagara HN. Evidence for impaired axonal regeneration in PMP22 duplication: studies in nerve xenografts. J Peripher Nerv Syst. 2003;8(2):116-27.
Sahenk Z, Ozes B. Gene therapy to promote regeneration in Charcot-Marie-Tooth disease. Brain Res. 2020;1727:146533.
Sahenk Z, Oblinger J, Edwards C. Neurotrophin-3 deficient Schwann cells impair nerve regeneration. Exp Neurol. 2008;212(2):552-6.
Woolley AG, Sheard PW, Duxson MJ. Neurotrophin-3 null mutant mice display a postnatal motor neuropathy. Eur J Neurosci. 2005;21(8):2100-10.
Sahenk Z, Nagaraja HN, McCracken BS, King WM, Freimer ML, Cedarbaum JM, et al. NT-3 promotes nerve regeneration and sensory improvement in CMT1A mouse models and in patients. Neurology. 2005;65(5):681-9.
Yalvac ME, Amornvit J, Chen L, Shontz KM, Lewis S, Sahenk Z. AAV1.NT-3 gene therapy increases muscle fiber diameter through activation of mTOR pathway and metabolic remodeling in a CMT mouse model. Gene Ther. 2018;25(2):129–38.
Nobbio L, Vigo T, Abbruzzese M, Levi G, Brancolini C, Mantero S, et al. Impairment of PMP22 transgenic Schwann cells differentiation in culture: implications for Charcot-Marie-Tooth type 1A disease. Neurobiol Dis. 2004;16(1):263-73.
Napoli I, Noon LA, Ribeiro S, Kerai AP, Parrinello S, Rosenberg LH, et al. A central role for the ERK-signaling pathway in controlling Schwann cell plasticity and peripheral nerve regeneration in vivo. Neuron. 2012;73(4):729-42.
Fornasari BE, Ronchi G, Pascal D, Visigalli D, Capodivento G, Nobbio L, et al. Soluble Neuregulin1 is strongly up-regulated in the rat model of Charcot-Marie-Tooth 1A disease. Exp Biol Med (Maywood). 2018;243(4):370-4.
Nobbio L, Sturla L, Fiorese F, Usai C, Basile G, Moreschi I, et al. P2X7-mediated increased intracellular calcium causes functional derangement in Schwann cells from rats with CMT1A neuropathy. J Biol Chem. 2009;284(34):23146-58.
Smith KJ, Hall SM, Schauf CL. Vesicular demyelination induced by raised intracellular calcium. J Neurol Sci. 1985;71(1):19-37.
Stevens B, Fields RD. Response of Schwann cells to action potentials in development. Science. 2000;287(5461):2267-71.
Vanoye CG, Sakakura M, Follis RM, Trevisan AJ, Narayan M, Li J, et al. Peripheral myelin protein 22 modulates store-operated calcium channel activity, providing insights into Charcot-Marie-Tooth disease etiology. J Biol Chem. 2019;294(32):12054-65.
Sociali G, Visigalli D, Prukop T, Cervellini I, Mannino E, Venturi C, et al. Tolerability and efficacy study of P2X7 inhibition in experimental Charcot-Marie-Tooth type 1A (CMT1A) neuropathy. Neurobiol Dis. 2016;95:145-57.
Fledrich R, Abdelaal T, Rasch L, Bansal V, Schutza V, Brugger B, et al. Targeting myelin lipid metabolism as a potential therapeutic strategy in a model of CMT1A neuropathy. Nat Commun. 2018;9(1):3025.
Fledrich R, Schlotter-Weigel B, Schnizer TJ, Wichert SP, Stassart RM, Meyer zu Horste G, et al. A rat model of Charcot-Marie-Tooth disease 1A recapitulates disease variability and supplies biomarkers of axonal loss in patients. Brain. 2012;135(Pt 1):72–87.
Zhou Y, Borchelt D, Bauson JC, Fazio S, Miles JR, Tavori H, et al. Subcellular diversion of cholesterol by gain- and loss-of-function mutations in PMP22. Glia. 2020;68(11):2300-15.
Bourre JM, Morand O, Dumont O, Boutry JM, Hauw JJ. Lipid metabolism in peripheral nerve cell culture (rich in Schwann cells) from normal and trembler mice. J Neurochem. 1981;37(2):272-5.
Clouet PM, Bourre JM. Ketone body utilization for lipid synthesis in the murine sciatic nerve: alterations in the dysmyelinating trembler mutant. J Neurochem. 1988;50(5):1494-7.
Yao JK, Bourre JM. Metabolic alterations of endoneurial lipids in developing trembler nerve. Brain Res. 1985;325(1-2):21-7.
Heape A, Juguelin H, Fabre M, Boiron F, Cassagne C. A quantitative developmental study of the peripheral nerve lipid composition during myelinogenesis in normal and trembler mice. Brain Res. 1986;390(2):181-9.
Zhou Y, Bazick H, Miles JR, Fethiere AI, Salihi MOA, Fazio S, et al. A neutral lipid-enriched diet improves myelination and alleviates peripheral nerve pathology in neuropathic mice. Exp Neurol. 2019;321:113031.
Zhou Y, Miles JR, Tavori H, Lin M, Khoshbouei H, Borchelt DR, et al. PMP22 Regulates Cholesterol Trafficking and ABCA1-Mediated Cholesterol Efflux. J Neurosci. 2019;39(27):5404-18.
Suter U, Welcher AA, Ozcelik T, Snipes GJ, Kosaras B, Francke U, et al. Trembler mouse carries a point mutation in a myelin gene. Nature. 1992;356(6366):241-4.
Koenig H, Do Thi A, Ferzaz B, Ressouches A. Schwann cell proliferation during postnatal development, Wallerian degeneration and axon regeneration in trembler dysmyelinating mutant. Adv Exp Med Biol. 1991;296:227-38.
Suter U, Moskow JJ, Welcher AA, Snipes GJ, Kosaras B, Sidman RL, et al. A leucine-to-proline mutation in the putative first transmembrane domain of the 22-kDa peripheral myelin protein in the trembler-J mouse. Proc Natl Acad Sci U S A. 1992;89(10):4382-6.
Isaacs AM, Davies KE, Hunter AJ, Nolan PM, Vizor L, Peters J, et al. Identification of two new Pmp22 mouse mutants using large-scale mutagenesis and a novel rapid mapping strategy. Hum Mol Genet. 2000;9(12):1865-71.
Isaacs AM, Jeans A, Oliver PL, Vizor L, Brown SD, Hunter AJ, et al. Identification of a new Pmp22 mouse mutant and trafficking analysis of a Pmp22 allelic series suggesting that protein aggregates may be protective in Pmp22-associated peripheral neuropathy. Mol Cell Neurosci. 2002;21(1):114-25.
Huxley C, Passage E, Manson A, Putzu G, Figarella-Branger D, Pellissier JF, et al. Construction of a mouse model of Charcot-Marie-Tooth disease type 1A by pronuclear injection of human YAC DNA. Hum Mol Genet. 1996;5(5):563-9.
Jouaud M, Mathis S, Richard L, Lia AS, Magy L, Vallat JM. Rodent models with expression of PMP22: Relevance to dysmyelinating CMT and HNPP. J Neurol Sci. 2019;398:79-90.
Verhamme C, King RH, ten Asbroek AL, Muddle JR, Nourallah M, Wolterman R, et al. Myelin and axon pathology in a long-term study of PMP22-overexpressing mice. J Neuropathol Exp Neurol. 2011;70(5):386-98.
Sereda M, Griffiths I, Puhlhofer A, Stewart H, Rossner MJ, Zimmerman F, et al. A transgenic rat model of Charcot-Marie-Tooth disease. Neuron. 1996;16(5):1049-60.
Chumakov I, Milet A, Cholet N, Primas G, Boucard A, Pereira Y, et al. Polytherapy with a combination of three repurposed drugs (PXT3003) down-regulates Pmp22 over-expression and improves myelination, axonal and functional parameters in models of CMT1A neuropathy. Orphanet J Rare Dis. 2014;9:201.
Gautier B, Hajjar H, Soares S, Berthelot J, Deck M, Abbou S, et al. AAV2/9-mediated silencing of PMP22 prevents the development of pathological features in a rat model of Charcot-Marie-Tooth disease 1 A. Nat Commun. 2021;12(1):2356.
Jones EA, Brewer MH, Srinivasan R, Krueger C, Sun G, Charney KN, et al. Distal enhancers upstream of the Charcot-Marie-Tooth type 1A disease gene PMP22. Hum Mol Genet. 2012;21(7):1581-91.
Srinivasan R, Sun G, Keles S, Jones EA, Jang SW, Krueger C, et al. Genome-wide analysis of EGR2/SOX10 binding in myelinating peripheral nerve. Nucleic Acids Res. 2012;40(14):6449-60.
Mukherjee-Clavin B, Mi R, Kern B, Choi IY, Lim H, Oh Y, et al. Comparison of three congruent patient-specific cell types for the modelling of a human genetic Schwann-cell disorder. Nat Biomed Eng. 2019;3(7):571-82.
Kaya F, Belin S, Bourgeois P, Micaleff J, Blin O, Fontes M. Ascorbic acid inhibits PMP22 expression by reducing cAMP levels. Neuromuscul Disord. 2007;17(3):248-53.
Saberan-Djoneidi D, Sanguedolce V, Assouline Z, Levy N, Passage E, Fontes M. Molecular dissection of the Schwann cell specific promoter of the PMP22 gene. Gene. 2000;248(1-2):223-31.
Gess B, Baets J, De Jonghe P, Reilly MM, Pareyson D, Young P. Ascorbic acid for the treatment of Charcot-Marie-Tooth disease. Cochrane Database Syst Rev. 2015(12):CD011952.
Lewis RA, McDermott MP, Herrmann DN, Hoke A, Clawson LL, Siskind C, et al. High-dosage ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A: results of a randomized, double-masked, controlled trial. JAMA Neurol. 2013;70(8):981-7.
Pareyson D, Reilly MM, Schenone A, Fabrizi GM, Cavallaro T, Santoro L, et al. Ascorbic acid in Charcot-Marie-Tooth disease type 1A (CMT-TRIAAL and CMT-TRAUK): a double-blind randomised trial. Lancet Neurol. 2011;10(4):320-8.
Verhamme C, de Haan RJ, Vermeulen M, Baas F, de Visser M, van Schaik IN. Oral high dose ascorbic acid treatment for one year in young CMT1A patients: a randomised, double-blind, placebo-controlled phase II trial. BMC Med. 2009;7:70.
Magnaghi V, Ballabio M, Roglio I, Melcangi RC. Progesterone derivatives increase expression of Krox-20 and Sox-10 in rat Schwann cells. J Mol Neurosci. 2007;31(2):149-57.
Desarnaud F, Do Thi AN, Brown AM, Lemke G, Suter U, Baulieu EE, et al. Progesterone stimulates the activity of the promoters of peripheral myelin protein-22 and protein zero genes in Schwann cells. J Neurochem. 1998;71(4):1765-8.
Melcangi RC, Magnaghi V, Cavarretta I, Zucchi I, Bovolin P, D'Urso D, et al. Progesterone derivatives are able to influence peripheral myelin protein 22 and P0 gene expression: possible mechanisms of action. J Neurosci Res. 1999;56(4):349-57.
Meyer zu Horste G, Prukop T, Liebetanz D, Mobius W, Nave KA, Sereda MW. Antiprogesterone therapy uncouples axonal loss from demyelination in a transgenic rat model of CMT1A neuropathy. Ann Neurol. 2007;61(1):61–72.
Magnaghi V, Ballabio M, Cavarretta IT, Froestl W, Lambert JJ, Zucchi I, et al. GABAB receptors in Schwann cells influence proliferation and myelin protein expression. Eur J Neurosci. 2004;19(10):2641-9.
Prukop T, Wernick S, Boussicault L, Ewers D, Jager K, Adam J, et al. Synergistic PXT3003 therapy uncouples neuromuscular function from dysmyelination in male Charcot-Marie-Tooth disease type 1A (CMT1A) rats. J Neurosci Res. 2020;98(10):1933-52.
Prukop T, Stenzel J, Wernick S, Kungl T, Mroczek M, Adam J, et al. Early short-term PXT3003 combinational therapy delays disease onset in a transgenic rat model of Charcot-Marie-Tooth disease 1A (CMT1A). PLoS One. 2019;14(1):e0209752.
Attarian S, Vallat JM, Magy L, Funalot B, Gonnaud PM, Lacour A, et al. An exploratory randomised double-blind and placebo-controlled phase 2 study of a combination of baclofen, naltrexone and sorbitol (PXT3003) in patients with Charcot-Marie-Tooth disease type 1A. Orphanet J Rare Dis. 2014;9:199.
Vita G, Vita GL, Stancanelli C, Gentile L, Russo M, Mazzeo A. Genetic neuromuscular disorders: living the era of a therapeutic revolution. Part 1: peripheral neuropathies. Neurol Sci. 2019;40(4):661–9.
Okamoto Y, Pehlivan D, Wiszniewski W, Beck CR, Snipes GJ, Lupski JR, et al. Curcumin facilitates a transitory cellular stress response in Trembler-J mice. Hum Mol Genet. 2013;22(23):4698-705.
Khajavi M, Inoue K, Wiszniewski W, Ohyama T, Snipes GJ, Lupski JR. Curcumin treatment abrogates endoplasmic reticulum retention and aggregation-induced apoptosis associated with neuropathy-causing myelin protein zero-truncating mutants. Am J Hum Genet. 2005;77(5):841-50.
Patzkó A, Bai Y, Saporta MA, Katona I, Wu X, Vizzuso D, et al. Curcumin derivatives promote Schwann cell differentiation and improve neuropathy in R98C CMT1B mice. Brain. 2012;135(Pt 12):3551-66.
Caillaud M, Msheik Z, Ndong-Ntoutoume GM, Vignaud L, Richard L, Favreau F, et al. Curcumin-cyclodextrin/cellulose nanocrystals improve the phenotype of Charcot-Marie-Tooth-1A transgenic rats through the reduction of oxidative stress. Free Radic Biol Med. 2020;161:246-62.
Chittoor-Vinod VG, Bazick H, Todd AG, Falk D, Morelli KH, Burgess RW, et al. HSP90 Inhibitor, NVP-AUY922, Improves Myelination in Vitro and Supports the Maintenance of Myelinated Axons in Neuropathic Mice. ACS Chem Neurosci. 2019;10(6):2890-902.
Westerheide SD, Morimoto RI. Heat shock response modulators as therapeutic tools for diseases of protein conformation. J Biol Chem. 2005;280(39):33097-100.
Ha N, Choi YI, Jung N, Song JY, Bae DK, Kim MC, et al. A novel histone deacetylase 6 inhibitor improves myelination of Schwann cells in a model of Charcot-Marie-Tooth disease type 1A. Br J Pharmacol. 2020;177(22):5096-113.
d'Ydewalle C, Krishnan J, Chiheb DM, Van Damme P, Irobi J, Kozikowski AP, et al. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med. 2011;17(8):968-74.
Adams D, Gonzalez-Duarte A, O'Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N Engl J Med. 2018;379(1):11-21.
Sahenk Z, Galloway G, Clark KR, Malik V, Rodino-Klapac LR, Kaspar BK, et al. AAV1.NT-3 gene therapy for charcot-marie-tooth neuropathy. Mol Ther. 2014;22(3):511–21.
Sargiannidou I, Kagiava A, Kleopa KA. Gene therapy approaches targeting Schwann cells for demyelinating neuropathies. Brain Res. 2020;1728:146572.
Cerritelli SM, Crouch RJ. Ribonuclease H: the enzymes in eukaryotes. FEBS J. 2009;276(6):1494-505.
Shy ME. Antisense oligonucleotides offer hope to patients with Charcot-Marie-Tooth disease type 1A. J Clin Invest. 2018;128(1):110-2.
Frazier KS. Antisense oligonucleotide therapies: the promise and the challenges from a toxicologic pathologist's perspective. Toxicol Pathol. 2015;43(1):78-89.
Fritz NE, Chen Y, Waters L, Saba S, Hackett M, Mada FC, et al. Fatigue in patients with hereditary neuropathy with liability to pressure palsies. Ann Clin Transl Neurol. 2020;7(8):1400-9.
Bjelica B, Peric S, Bozovic I, Jankovic M, Brankovic M, Palibrk A, et al. Quality of life in hereditary neuropathy with liability to pressure palsies is as impaired as in Charcot-Marie-Tooth disease type 1A. Acta Neurol Belg. 2020.
Caillaud M, El Madani M, Massaad-Massade L. Small interfering RNA from the lab discovery to patients' recovery. J Control Release. 2020;321:616-28.
Lee JS, Chang EH, Koo OJ, Jwa DH, Mo WM, Kwak G, et al. Pmp22 mutant allele-specific siRNA alleviates demyelinating neuropathic phenotype in vivo. Neurobiol Dis. 2017;100:99-107.
Boutary S, Caillaud M, El Madani M, Vallat JM, Loisel-Duwattez J, Rouyer A, et al. Squalenoyl siRNA PMP22 nanoparticles are effective in treating mouse models of Charcot-Marie-Tooth disease type 1 A. Commun Biol. 2021;4(1):317.
Lee JS, Lee JY, Song DW, Bae HS, Doo HM, Yu HS, et al. Targeted PMP22 TATA-box editing by CRISPR/Cas9 reduces demyelinating neuropathy of Charcot-Marie-Tooth disease type 1A in mice. Nucleic Acids Res. 2020;48(1):130-40.
Van Alstyne M, Tattoli I, Delestree N, Recinos Y, Workman E, Shihabuddin LS, et al. Gain of toxic function by long-term AAV9-mediated SMN overexpression in the sensorimotor circuit. Nat Neurosci. 2021;24(7):930-40.
Nelis E, Van Broeckhoven C, De Jonghe P, Lofgren A, Vandenberghe A, Latour P, et al. Estimation of the mutation frequencies in Charcot-Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur J Hum Genet. 1996;4(1):25-33.
Shy ME, Jani A, Krajewski K, Grandis M, Lewis RA, Li J, et al. Phenotypic clustering in MPZ mutations. Brain. 2004;127(Pt 2):371-84.
Lyon G. Ultrastructural study of a nerve biopsy from a case of early infantile chronic neuropathy. Acta Neuropathol. 1969;13(2):131-42.
Karch SB, Urich H. Infantile polyneuropathy with defective myelination: an autopsy study. Dev Med Child Neurol. 1975;17(4):504-11.
Kennedy WR, Sung JH, Berry JF. A case of congenital hypomyelination neuropathy. Clinical, morphological, and chemical studies. Arch Neurol. 1977;34(6):337–45.
Cox MJ. Progressive familial hypertrophic polyneuritis (Dejerine-Sottas Syndrome, 1893). Proc R Soc Med. 1956;49(4):183-4.
Sanmaneechai O, Feely S, Scherer SS, Herrmann DN, Burns J, Muntoni F, et al. Genotype-phenotype characteristics and baseline natural history of heritable neuropathies caused by mutations in the MPZ gene. Brain. 2015;138(Pt 11):3180-92.
Hattori N, Yamamoto M, Yoshihara T, Koike H, Nakagawa M, Yoshikawa H, et al. Demyelinating and axonal features of Charcot-Marie-Tooth disease with mutations of myelin-related proteins (PMP22, MPZ and Cx32): a clinicopathological study of 205 Japanese patients. Brain. 2003;126(Pt 1):134-51.
Murphy SM, Laurá M, Blake J, Polke J, Bremner F, Reilly MM. Conduction block and tonic pupils in Charcot-Marie-Tooth disease caused by a myelin protein zero p.Ile112Thr mutation. Neuromuscul Disord. 2011;21(3):223–6.
Mandich P, Mancardi GL, Varese A, Soriani S, Di Maria E, Bellone E, et al. Congenital hypomyelination due to myelin protein zero Q215X mutation. Ann Neurol. 1999;45(5):676-8.
Warner LE, Hilz MJ, Appel SH, Killian JM, Kolodry EH, Karpati G, et al. Clinical phenotypes of different MPZ (P0) mutations may include Charcot-Marie-Tooth type 1B, Dejerine-Sottas, and congenital hypomyelination. Neuron. 1996;17(3):451-60.
Mersiyanova IV, Ismailov SM, Polyakov AV, Dadali EL, Fedotov VP, Nelis E, et al. Screening for mutations in the peripheral myelin genes PMP22, MPZ and Cx32 (GJB1) in Russian Charcot-Marie-Tooth neuropathy patients. Hum Mutat. 2000;15(4):340-7.
Eggers SD, Keswani SC, Melli G, Cornblath DR. Clinical and genetic description of a family with Charcot-Marie-Tooth disease type 1B from a transmembrane MPZ mutation. Muscle Nerve. 2004;29(6):867-9.
De Jonghe P, Timmerman V, Ceuterick C, Nelis E, De Vriendt E, Lofgren A, et al. The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype. Brain. 1999;122 ( Pt 2):281-90.
Li J, Bai Y, Ianakova E, Grandis M, Uchwat F, Trostinskaia A, et al. Major myelin protein gene (P0) mutation causes a novel form of axonal degeneration. J Comp Neurol. 2006;498(2):252-65.
Nelis E, Haites N, Van Broeckhoven C. Mutations in the peripheral myelin genes and associated genes in inherited peripheral neuropathies. Hum Mutat. 1999;13(1):11-28.
Bird TD, Kraft GH, Lipe HP, Kenney KL, Sumi SM. Clinical and pathological phenotype of the original family with Charcot-Marie-Tooth type 1B: a 20-year study. Ann Neurol. 1997;41(4):463-9.
Bai Y, Ianokova E, Pu Q, Ghandour K, Levinson R, Martin JJ, et al. Effect of an R69C mutation in the myelin protein zero gene on myelination and ion channel subtypes. Arch Neurol. 2006;63(12):1787-94.
Dacci P, Taroni F, Bella ED, Milani M, Pareyson D, Morbin M, et al. Myelin protein zero Arg36Gly mutation with very late onset and rapidly progressive painful neuropathy. J Peripher Nerv Syst. 2012;17(4):422-5.
Lemke G, Axel R. Isolation and sequence of a cDNA encoding the major structural protein of peripheral myelin. Cell. 1985;40(3):501-8.
Volpi VG, Touvier T, D'Antonio M. Endoplasmic Reticulum Protein Quality Control Failure in Myelin Disorders. Front Mol Neurosci. 2016;9:162.
Hayasaka K, Himoro M, Sato W, Takada G, Uyemura K, Shimizu N, et al. Charcot-Marie-Tooth neuropathy type 1B is associated with mutations of the myelin P0 gene. Nat Genet. 1993;5(1):31-4.
Trapp BD, Itoyama Y, Sternberger NH, Quarles RH, Webster H. Immunocytochemical localization of P0 protein in Golgi complex membranes and myelin of developing rat Schwann cells. J Cell Biol. 1981;90(1):1-6.
Trapp BD, Kidd GJ, Hauer P, Mulrenin E, Haney CA, Andrews SB. Polarization of myelinating Schwann cell surface membranes: role of microtubules and the trans-Golgi network. J Neurosci. 1995;15(3 Pt 1):1797-807.
Brunden KR, Ding Y, Hennington BS. Myelin protein expression in dissociated superior cervical ganglia and dorsal root ganglia cultures. J Neurosci Res. 1992;32(4):507-15.
D'Urso D, Brophy PJ, Staugaitis SM, Gillespie CS, Frey AB, Stempak JG, et al. Protein zero of peripheral nerve myelin: biosynthesis, membrane insertion, and evidence for homotypic interaction. Neuron. 1990;4(3):449-60.
Eichberg J. Myelin P0: new knowledge and new roles. Neurochem Res. 2002;27(11):1331-40.
Brügger V, Engler S, Pereira JA, Ruff S, Horn M, Welzl H, et al. HDAC1/2-Dependent P0 Expression Maintains Paranodal and Nodal Integrity Independently of Myelin Stability through Interactions with Neurofascins. PLoS Biol. 2015;13(9):e1002258.
Filbin MT, Walsh FS, Trapp BD, Pizzey JA, Tennekoon GI. Role of myelin P0 protein as a homophilic adhesion molecule. Nature. 1990;344(6269):871-2.
Greenfield S, Brostoff S, Eylar EH, Morell P. Protein composition of myelin of the peripheral nervous system. J Neurochem. 1973;20(4):1207-16.
Shapiro L, Doyle JP, Hensley P, Colman DR, Hendrickson WA. Crystal structure of the extracellular domain from P0, the major structural protein of peripheral nerve myelin. Neuron. 1996;17(3):435-49.
Wrabetz L, Feltri ML, Quattrini A, Imperiale D, Previtali S, D'Antonio M, et al. P(0) glycoprotein overexpression causes congenital hypomyelination of peripheral nerves. J Cell Biol. 2000;148(5):1021-34.
Giese KP, Martini R, Lemke G, Soriano P, Schachner M. Mouse P0 gene disruption leads to hypomyelination, abnormal expression of recognition molecules, and degeneration of myelin and axons. Cell. 1992;71(4):565-76.
Grandis M, Vigo T, Passalacqua M, Jain M, Scazzola S, La Padula V, et al. Different cellular and molecular mechanisms for early and late-onset myelin protein zero mutations. Hum Mol Genet. 2008;17(13):1877-89.
Xu W, Shy M, Kamholz J, Elferink L, Xu G, Lilien J, et al. Mutations in the cytoplasmic domain of P0 reveal a role for PKC-mediated phosphorylation in adhesion and myelination. J Cell Biol. 2001;155(3):439-46.
Wrabetz L, D'Antonio M, Pennuto M, Dati G, Tinelli E, Fratta P, et al. Different intracellular pathomechanisms produce diverse Myelin Protein Zero neuropathies in transgenic mice. J Neurosci. 2006;26(8):2358-68.
Fratta P, Ornaghi F, Dati G, Zambroni D, Saveri P, Belin S, et al. A nonsense mutation in myelin protein zero causes congenital hypomyelination neuropathy through altered P0 membrane targeting and gain of abnormal function. Hum Mol Genet. 2019;28(1):124-32.
Filbin MT, Zhang K, Li W, Gao Y. Characterization of the Effect on Adhesion of Different Mutations in Myelin P(0) Protein. Ann N Y Acad Sci. 1999;883(1):160-7.
Kirschner DA, Szumowski K, Gabreëls-Festen AA, Hoogendijk JE, Bolhuis PA. Inherited demyelinating peripheral neuropathies: relating myelin packing abnormalities to P0 molecular defects. J Neurosci Res. 1996;46(4):502-8.
Prada V, Passalacqua M, Bono M, Luzzi P, Scazzola S, Nobbio LA, et al. Gain of glycosylation: a new pathomechanism of myelin protein zero mutations. Ann Neurol. 2012;71(3):427-31.
Blanquet-Grossard F, Pham-Dinh D, Dautigny A, Latour P, Bonnebouche C, Diraison P, et al. Charcot-Marie-Tooth type 1B neuropathy: a mutation at the single glycosylation site in the major peripheral myelin glycoprotein Po. Hum Mutat. 1996;8(2):185-6.
Wong MH, Filbin MT. The cytoplasmic domain of the myelin P0 protein influences the adhesive interactions of its extracellular domain. J Cell Biol. 1994;126(4):1089-97.
Gaboreanu AM, Hrstka R, Xu W, Shy M, Kamholz J, Lilien J, et al. Myelin protein zero/P0 phosphorylation and function require an adaptor protein linking it to RACK1 and PKC alpha. J Cell Biol. 2007;177(4):707-16.
Xu W, Manichella D, Jiang H, Vallat JM, Lilien J, Baron P, et al. Absence of P0 leads to the dysregulation of myelin gene expression and myelin morphogenesis. J Neurosci Res. 2000;60(6):714-24.
Menichella DM, Arroyo EJ, Awatramani R, Xu T, Baron P, Vallat JM, et al. Protein zero is necessary for E-cadherin-mediated adherens junction formation in Schwann cells. Mol Cell Neurosci. 2001;18(6):606-18.
Moldovan M, Alvarez S, Pinchenko V, Klein D, Nielsen FC, Wood JN, et al. Na(v)1.8 channelopathy in mutant mice deficient for myelin protein zero is detrimental to motor axons. Brain. 2011;134(Pt 2):585–601.
Moldovan M, Pisciotta C, Pareyson D, Krarup C. Myelin protein zero gene dose dependent axonal ion-channel dysfunction in a family with Charcot-Marie-Tooth disease. Clin Neurophysiol. 2020;131(10):2440-51.
Musner N, Sidoli M, Zambroni D, Del Carro U, Ungaro D, D'Antonio M, et al. Perk Ablation Ameliorates Myelination in S63del-Charcot-Marie-Tooth 1B Neuropathy. ASN Neuro. 2016;8(2).
D'Antonio M, Feltri ML, Wrabetz L. Myelin under stress. J Neurosci Res. 2009;87(15):3241-9.
Gow A, Wrabetz L. CHOP and the endoplasmic reticulum stress response in myelinating glia. Curr Opin Neurobiol. 2009;19(5):505-10.
Matus S, Glimcher LH, Hetz C. Protein folding stress in neurodegenerative diseases: a glimpse into the ER. Curr Opin Cell Biol. 2011;23(2):239-52.
Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519-29.
Hetz C, Chevet E, Oakes SA. Proteostasis control by the unfolded protein response. Nat Cell Biol. 2015;17(7):829-38.
Saporta MA, Shy BR, Patzko A, Bai Y, Pennuto M, Ferri C, et al. MpzR98C arrests Schwann cell development in a mouse model of early-onset Charcot-Marie-Tooth disease type 1B. Brain. 2012;135(Pt 7):2032-47.
Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol. 2000;2(7):379-84.
Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101(3):249-58.
Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10(11):3787-99.
Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol. 2000;20(18):6755-67.
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881-91.
Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3(1):99-111.
Yamamoto S, Fujii S, Yoshimoto N, Akbarzadehlaleh P. Effects of protein conformational changes on separation performance in electrostatic interaction chromatography: unfolded proteins and PEGylated proteins. J Biotechnol. 2007;132(2):196-201.
Harding HP, Novoa I, Bertolotti A, Zeng H, Zhang Y, Urano F, et al. Translational regulation in the cellular response to biosynthetic load on the endoplasmic reticulum. Cold Spring Harb Symp Quant Biol. 2001;66:499-508.
Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153(5):1011-22.
Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12(7):982-95.
Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6(5):1099-108.
Pennuto M, Tinelli E, Malaguti M, Del Carro U, D'Antonio M, Ron D, et al. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron. 2008;57(3):393-405.
D'Antonio M, Musner N, Scapin C, Ungaro D, Del Carro U, Ron D, et al. Resetting translational homeostasis restores myelination in Charcot-Marie-Tooth disease type 1B mice. J Exp Med. 2013;210(4):821-38.
Sidoli M, Musner N, Silvestri N, Ungaro D, D'Antonio M, Cavener DR, et al. Ablation of Perk in Schwann Cells Improves Myelination in the S63del Charcot-Marie-Tooth 1B Mouse. J Neurosci. 2016;36(44):11350-61.
Bai Y, Wu X, Brennan KM, Wang DS, D'Antonio M, Moran J, et al. Myelin protein zero mutations and the unfolded protein response in Charcot Marie Tooth disease type 1B. Ann Clin Transl Neurol. 2018;5(4):445-55.
Lin W, Popko B. Endoplasmic reticulum stress in disorders of myelinating cells. Nat Neurosci. 2009;12(4):379-85.
Clayton BLL, Popko B. Endoplasmic reticulum stress and the unfolded protein response in disorders of myelinating glia. Brain Res. 2016;1648(Pt B):594-602.
Fratta P, Saveri P, Zambroni D, Ferri C, Tinelli E, Messing A, et al. P0S63del impedes the arrival of wild-type P0 glycoprotein to myelin in CMT1B mice. Hum Mol Genet. 2011;20(11):2081-90.
Miller LJ, Patzko A, Lewis RA, Shy ME. Phenotypic presentation of the Ser63Del MPZ mutation. J Peripher Nerv Syst. 2012;17(2):197-200.
Gabreëls-Festen AA, Hoogendijk JE, Meijerink PH, Gabreëls FJ, Bolhuis PA, van Beersum S, et al. Two divergent types of nerve pathology in patients with different P0 mutations in Charcot-Marie-Tooth disease. Neurology. 1996;47(3):761-5.
Kulkens T, Bolhuis PA, Wolterman RA, Kemp S, te Nijenhuis S, Valentijn LJ, et al. Deletion of the serine 34 codon from the major peripheral myelin protein P0 gene in Charcot-Marie-Tooth disease type 1B. Nat Genet. 1993;5(1):35-9.
Frei R, Mötzing S, Kinkelin I, Schachner M, Koltzenburg M, Martini R. Loss of distal axons and sensory Merkel cells and features indicative of muscle denervation in hindlimbs of P0-deficient mice. J Neurosci. 1999;19(14):6058-67.
Bai Y, Patzko A, Shy ME. Unfolded protein response, treatment and CMT1B. Rare Dis. 2013;1:e24049.
Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307(5711):935-9.
Das I, Krzyzosiak A, Schneider K, Wrabetz L, D'Antonio M, Barry N, et al. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science. 2015;348(6231):239-42.
Volpi VG, Ferri C, Fregno I, Del Carro U, Bianchi F, Scapin C, et al. Schwann cells ER-associated degradation contributes to myelin maintenance in adult nerves and limits demyelination in CMT1B mice. PLoS Genet. 2019;15(4):e1008069.
Scapin C, Ferri C, Pettinato E, Zambroni D, Bianchi F, Del Carro U, et al. Enhanced axonal neuregulin-1 type-III signaling ameliorates neurophysiology and hypomyelination in a Charcot-Marie-Tooth type 1B mouse model. Hum Mol Genet. 2019;28(6):992-1006.
Rosberg MR, Alvarez S, Krarup C, Moldovan M. An oral NaV1.8 blocker improves motor function in mice completely deficient of myelin protein P0. Neurosci Lett. 2016;632:33–8.
Klein D, Patzko A, Schreiber D, van Hauwermeiren A, Baier M, Groh J, et al. Targeting the colony stimulating factor 1 receptor alleviates two forms of Charcot-Marie-Tooth disease in mice. Brain. 2015;138(Pt 11):3193-205.
Ionasescu V, Searby C, Ionasescu R. Point mutations of the connexin32 (GJB1) gene in X-linked dominant Charcot-Marie-Tooth neuropathy. Hum Mol Genet. 1994;3(2):355-8.
DiVincenzo C, Elzinga CD, Medeiros AC, Karbassi I, Jones JR, Evans MC, et al. The allelic spectrum of Charcot-Marie-Tooth disease in over 17,000 individuals with neuropathy. Mol Genet Genomic Med. 2014;2(6):522-9.
Shy ME, Siskind C, Swan ER, Krajewski KM, Doherty T, Fuerst DR, et al. CMT1X phenotypes represent loss of GJB1 gene function. Neurology. 2007;68(11):849-55.
Tomaselli PJ, Rossor AM, Horga A, Jaunmuktane Z, Carr A, Saveri P, et al. Mutations in noncoding regions of GJB1 are a major cause of X-linked CMT. Neurology. 2017;88(15):1445-53.
Kulshrestha R, Burton-Jones S, Antoniadi T, Rogers M, Jaunmuktane Z, Brandner S, et al. Deletion of P2 promoter of GJB1 gene a cause of Charcot-Marie-Tooth disease. Neuromuscul Disord. 2017;27(8):766-70.
Luo S, Jin H, Chen J, Zhang L. A Novel Variant in Non-coding Region of GJB1 Is Associated With X-Linked Charcot-Marie-Tooth Disease Type 1 and Transient CNS Symptoms. Front Neurol. 2019;10:413.
Boso F, Taioli F, Cabrini I, Cavallaro T, Fabrizi GM. Aberrant Splicing in GJB1 and the Relevance of 5' UTR in CMTX1 Pathogenesis. Brain Sci. 2020;11(1).
Gonzaga-Jauregui C, Zhang F, Towne CF, Batish SD, Lupski JR. GJB1/Connexin 32 whole gene deletions in patients with X-linked Charcot-Marie-Tooth disease. Neurogenetics. 2010;11(4):465-70.
Capponi S, Geroldi A, Pezzini I, Gulli R, Ciotti P, Ursino G, et al. Contribution of copy number variations in CMT1X: a retrospective study. Eur J Neurol. 2015;22(2):406-9.
Panosyan FB, Laura M, Rossor AM, Pisciotta C, Piscosquito G, Burns J, et al. Cross-sectional analysis of a large cohort with X-linked Charcot-Marie-Tooth disease (CMTX1). Neurology. 2017;89(9):927-35.
Siskind CE, Murphy SM, Ovens R, Polke J, Reilly MM, Shy ME. Phenotype expression in women with CMT1X. J Peripher Nerv Syst. 2011;16(2):102-7.
Campagnolo M, Taioli F, Cacciavillani M, Ruiz M, Luigetti M, Salvalaggio A, et al. Sporadic hereditary neuropathies misdiagnosed as chronic inflammatory demyelinating polyradiculoneuropathy: Pitfalls and red flags. J Peripher Nerv Syst. 2020;25(1):19-26.
Michell AW, Laura M, Blake J, Lunn MP, Cox A, Gibbons VS, et al. GJB1 gene mutations in suspected inflammatory demyelinating neuropathies not responding to treatment. J Neurol Neurosurg Psychiatry. 2009;80(6):699-700.
Tian D, Zhao Y, Zhu R, Li Q, Liu X. Systematic review of CMTX1 patients with episodic neurological dysfunction. Ann Clin Transl Neurol. 2021;8(1):213-23.
Scherer SS, Xu YT, Nelles E, Fischbeck K, Willecke K, Bone LJ. Connexin32-null mice develop demyelinating peripheral neuropathy. Glia. 1998;24(1):8-20.
Jeng LJ, Balice-Gordon RJ, Messing A, Fischbeck KH, Scherer SS. The effects of a dominant connexin32 mutant in myelinating Schwann cells. Mol Cell Neurosci. 2006;32(3):283-98.
Kagiava A, Karaiskos C, Richter J, Tryfonos C, Lapathitis G, Sargiannidou I, et al. Intrathecal gene therapy in mouse models expressing CMT1X mutations. Hum Mol Genet. 2018;27(8):1460-73.
Abrams CK, Freidin MM, Verselis VK, Bennett MV, Bargiello TA. Functional alterations in gap junction channels formed by mutant forms of connexin 32: evidence for loss of function as a pathogenic mechanism in the X-linked form of Charcot-Marie-Tooth disease. Brain Res. 2001;900(1):9-25.
Wang HL, Chang WT, Yeh TH, Wu T, Chen MS, Wu CY. Functional analysis of connexin-32 mutants associated with X-linked dominant Charcot-Marie-Tooth disease. Neurobiol Dis. 2004;15(2):361-70.
Kyriakoudi S, Sargiannidou I, Kagiava A, Olympiou M, Kleopa KA. Golgi-retained Cx32 mutants interfere with gene addition therapy for CMT1X. Hum Mol Genet. 2017;26(9):1622-33.
Abrams CK, Bennett MV, Verselis VK, Bargiello TA. Voltage opens unopposed gap junction hemichannels formed by a connexin 32 mutant associated with X-linked Charcot-Marie-Tooth disease. Proc Natl Acad Sci U S A. 2002;99(6):3980-4.
Abrams CK, Goman M, Wong S, Scherer SS, Kleopa KA, Peinado A, et al. Loss of Coupling Distinguishes GJB1 Mutations Associated with CNS Manifestations of CMT1X from Those Without CNS Manifestations. Sci Rep. 2017;7:40166.
Yum SW, Kleopa KA, Shumas S, Scherer SS. Diverse trafficking abnormalities of connexin32 mutants causing CMTX. Neurobiol Dis. 2002;11(1):43-52.
Matsuyama W, Nakagawa M, Moritoyo T, Takashima H, Umehara F, Hirata K, et al. Phenotypes of X-linked Charcot-Marie-Tooth disease and altered trafficking of mutant connexin 32 (GJB1). J Hum Genet. 2001;46(6):307-13.
Kobsar I, Maurer M, Ott T, Martini R. Macrophage-related demyelination in peripheral nerves of mice deficient in the gap junction protein connexin 32. Neurosci Lett. 2002;320(1-2):17-20.
Kobsar I, Berghoff M, Samsam M, Wessig C, Maurer M, Toyka KV, et al. Preserved myelin integrity and reduced axonopathy in connexin32-deficient mice lacking the recombination activating gene-1. Brain. 2003;126(Pt 4):804-13.
Groh J, Heinl K, Kohl B, Wessig C, Greeske J, Fischer S, et al. Attenuation of MCP-1/CCL2 expression ameliorates neuropathy in a mouse model for Charcot-Marie-Tooth 1X. Hum Mol Genet. 2010;19(18):3530-43.
Fischer S, Kleinschnitz C, Muller M, Kobsar I, Ip CW, Rollins B, et al. Monocyte chemoattractant protein-1 is a pathogenic component in a model for a hereditary peripheral neuropathy. Mol Cell Neurosci. 2008;37(2):359-66.
Groh J, Klein I, Hollmann C, Wettmarshausen J, Klein D, Martini R. CSF-1-activated macrophages are target-directed and essential mediators of Schwann cell dedifferentiation and dysfunction in Cx32-deficient mice. Glia. 2015;63(6):977-86.
Groh J, Weis J, Zieger H, Stanley ER, Heuer H, Martini R. Colony-stimulating factor-1 mediates macrophage-related neural damage in a model for Charcot-Marie-Tooth disease type 1X. Brain. 2012;135(Pt 1):88-104.
Mones S, Bordignon B, Fontes M. Connexin 32 is involved in mitosis. Glia. 2012;60(3):457-64.
Mones S, Bordignon B, Peiretti F, Landrier JF, Gess B, Bourguignon JJ, et al. CamKII inhibitors reduce mitotic instability, connexon anomalies and progression of the in vivo behavioral phenotype in transgenic animals expressing a mutated Gjb1 gene. Front Neurosci. 2014;8:151.
Sargiannidou I, Kagiava A, Bashiardes S, Richter J, Christodoulou C, Scherer SS, et al. Intraneural GJB1 gene delivery improves nerve pathology in a model of X-linked Charcot-Marie-Tooth disease. Ann Neurol. 2015;78(2):303-16.
Kagiava A, Sargiannidou I, Theophilidis G, Karaiskos C, Richter J, Bashiardes S, et al. Intrathecal gene therapy rescues a model of demyelinating peripheral neuropathy. Proc Natl Acad Sci U S A. 2016;113(17):E2421-9.
Kagiava A, Karaiskos C, Richter J, Tryfonos C, Jennings MJ, Heslegrave AJ, et al. AAV9-mediated Schwann cell-targeted gene therapy rescues a model of demyelinating neuropathy. Gene Ther. 2021.
Kagiava A, Richter J, Tryfonos C, Karaiskos C, Heslegrave AJ, Sargiannidou I, et al. Gene replacement therapy after neuropathy onset provides therapeutic benefit in a model of CMT1X. Hum Mol Genet. 2019;28(21):3528-42.
Ozes B, Myers M, Moss K, McKinney J, Ridgley A, Chen L, et al. AAV1.NT-3 gene therapy for X-linked Charcot-Marie-Tooth neuropathy type 1. Gene Ther. 2021.
Azzedine H, Salih MA. SH3TC2-Related Hereditary Motor and Sensory Neuropathy. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, et al., editors. GeneReviews((R)). Seattle (WA)1993.
Piscosquito G, Saveri P, Magri S, Ciano C, Gandioli C, Morbin M, et al. Screening for SH3TC2 gene mutations in a series of demyelinating recessive Charcot-Marie-Tooth disease (CMT4). J Peripher Nerv Syst. 2016;21(3):142-9.
Colomer J, Gooding R, Angelicheva D, King RH, Guillen-Navarro E, Parman Y, et al. Clinical spectrum of CMT4C disease in patients homozygous for the p.Arg1109X mutation in SH3TC2. Neuromuscul Disord. 2006;16(7):449–53.
Gooding R, Colomer J, King R, Angelicheva D, Marns L, Parman Y, et al. A novel Gypsy founder mutation, p.Arg1109X in the CMT4C gene, causes variable peripheral neuropathy phenotypes. J Med Genet. 2005;42(12):e69.
Kontogeorgiou Z, Nikolaou K, Kartanou C, Breza M, Panas M, Karadima G, et al. Mutational screening of the SH3TC2 gene in Greek patients with suspected demyelinating recessive Charcot-Marie-Tooth disease reveals a varied and unusual phenotypic spectrum. J Peripher Nerv Syst. 2019;24(1):125-30.
Varley TL, Bourque PR, Baker SK. Phenotypic variability of CMT4C in a French-Canadian kindred. Muscle Nerve. 2015;52(3):444-9.
Kessali M, Zemmouri R, Guilbot A, Maisonobe T, Brice A, LeGuern E, et al. A clinical, electrophysiologic, neuropathologic, and genetic study of two large Algerian families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease. Neurology. 1997;48(4):867-73.
Yger M, Stojkovic T, Tardieu S, Maisonobe T, Brice A, Echaniz-Laguna A, et al. Characteristics of clinical and electrophysiological pattern of Charcot-Marie-Tooth 4C. J Peripher Nerv Syst. 2012;17(1):112-22.
Senderek J, Bergmann C, Stendel C, Kirfel J, Verpoorten N, De Jonghe P, et al. Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy. Am J Hum Genet. 2003;73(5):1106-19.
Arnaud E, Zenker J, de Preux Charles AS, Stendel C, Roos A, Medard JJ, et al. SH3TC2/KIAA1985 protein is required for proper myelination and the integrity of the node of Ranvier in the peripheral nervous system. Proc Natl Acad Sci U S A. 2009;106(41):17528-33.
Gouttenoire EA, Lupo V, Calpena E, Bartesaghi L, Schupfer F, Medard JJ, et al. Sh3tc2 deficiency affects neuregulin-1/ErbB signaling. Glia. 2013;61(7):1041-51.
Roberts RC, Peden AA, Buss F, Bright NA, Latouche M, Reilly MM, et al. Mistargeting of SH3TC2 away from the recycling endosome causes Charcot-Marie-Tooth disease type 4C. Hum Mol Genet. 2010;19(6):1009-18.
Schiza N, Georgiou E, Kagiava A, Medard JJ, Richter J, Tryfonos C, et al. Gene replacement therapy in a model of Charcot-Marie-Tooth 4C neuropathy. Brain. 2019;142(5):1227-41.
Sandelius A, Zetterberg H, Blennow K, Adiutori R, Malaspina A, Laura M, et al. Plasma neurofilament light chain concentration in the inherited peripheral neuropathies. Neurology. 2018;90(6):e518-e24.
Azzedine H, Bolino A, Taieb T, Birouk N, Di Duca M, Bouhouche A, et al. Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of Charcot-Marie-Tooth disease associated with early-onset glaucoma. Am J Hum Genet. 2003;72(5):1141-53.
Bolino A, Muglia M, Conforti FL, LeGuern E, Salih MA, Georgiou DM, et al. Charcot-Marie-Tooth type 4B is caused by mutations in the gene encoding myotubularin-related protein-2. Nat Genet. 2000;25(1):17-9.
Nakhro K, Park JM, Hong YB, Park JH, Nam SH, Yoon BR, et al. SET binding factor 1 (SBF1) mutation causes Charcot-Marie-Tooth disease type 4B3. Neurology. 2013;81(2):165-73.
Chow CY, Zhang Y, Dowling JJ, Jin N, Adamska M, Shiga K, et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448(7149):68-72.
Berger P, Tersar K, Ballmer-Hofer K, Suter U. The CMT4B disease-causing proteins MTMR2 and MTMR13/SBF2 regulate AKT signalling. J Cell Mol Med. 2011;15(2):307-15.
Pareyson D, Stojkovic T, Reilly MM, Leonard-Louis S, Laura M, Blake J, et al. A multicenter retrospective study of charcot-marie-tooth disease type 4B (CMT4B) associated with mutations in myotubularin-related proteins (MTMRs). Ann Neurol. 2019;86(1):55-67.
Vaccari I, Dina G, Tronchere H, Kaufman E, Chicanne G, Cerri F, et al. Genetic interaction between MTMR2 and FIG4 phospholipid phosphatases involved in Charcot-Marie-Tooth neuropathies. PLoS Genet. 2011;7(10):e1002319.
Taveggia C, Zanazzi G, Petrylak A, Yano H, Rosenbluth J, Einheber S, et al. Neuregulin-1 type III determines the ensheathment fate of axons. Neuron. 2005;47(5):681-94.
Bolino A, Piguet F, Alberizzi V, Pellegatta M, Rivellini C, Guerrero-Valero M, et al. Niacin-mediated Tace activation ameliorates CMT neuropathies with focal hypermyelination. EMBO Mol Med. 2016;8(12):1438-54.
Lenk GM, Ferguson CJ, Chow CY, Jin N, Jones JM, Grant AE, et al. Pathogenic mechanism of the FIG4 mutation responsible for Charcot-Marie-Tooth disease CMT4J. PLoS Genet. 2011;7(6):e1002104.
Zhang X, Chow CY, Sahenk Z, Shy ME, Meisler MH, Li J. Mutation of FIG4 causes a rapidly progressive, asymmetric neuronal degeneration. Brain. 2008;131(Pt 8):1990-2001.
Nicholson G, Lenk GM, Reddel SW, Grant AE, Towne CF, Ferguson CJ, et al. Distinctive genetic and clinical features of CMT4J: a severe neuropathy caused by mutations in the PI(3,5)P(2) phosphatase FIG4. Brain. 2011;134(Pt 7):1959-71.
Hu B, McCollum M, Ravi V, Arpag S, Moiseev D, Castoro R, et al. Myelin abnormality in Charcot-Marie-Tooth type 4J recapitulates features of acquired demyelination. Ann Neurol. 2018;83(4):756-70.
Presa M, Bailey RM, Davis C, Murphy T, Cook J, Walls R, et al. AAV9-mediated FIG4 delivery prolongs life span in Charcot Marie Tooth disease type 4J mouse model. J Clin Invest. 2021.
Ferguson CJ, Lenk GM, Jones JM, Grant AE, Winters JJ, Dowling JJ, et al. Neuronal expression of Fig4 is both necessary and sufficient to prevent spongiform neurodegeneration. Hum Mol Genet. 2012;21(16):3525-34.
Zou J, Hu B, Arpag S, Yan Q, Hamilton A, Zeng YS, et al. Reactivation of Lysosomal Ca2+ Efflux Rescues Abnormal Lysosomal Storage in FIG4-Deficient Cells. J Neurosci. 2015;35(17):6801-12.
Reilly MM, de Jonghe P, Pareyson D. 136th ENMC International Workshop: Charcot-Marie-Tooth disease type 1A (CMT1A)8-10 April 2005, Naarden, The Netherlands. Neuromuscul Disord. 2006;16(6):396-402.
Murphy SM, Herrmann DN, McDermott MP, Scherer SS, Shy ME, Reilly MM, et al. Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J Peripher Nerv Syst. 2011;16(3):191-8.
Sadjadi R, Reilly MM, Shy ME, Pareyson D, Laura M, Murphy S, et al. Psychometrics evaluation of Charcot-Marie-Tooth Neuropathy Score (CMTNSv2) second version, using Rasch analysis. J Peripher Nerv Syst. 2014;19(3):192-6.
Fridman V, Sillau S, Acsadi G, Bacon C, Dooley K, Burns J, et al. A longitudinal study of CMT1A using Rasch analysis based CMT neuropathy and examination scores. Neurology. 2020;94(9):e884-e96.
Burns J, Ouvrier R, Estilow T, Shy R, Laura M, Pallant JF, et al. Validation of the Charcot-Marie-Tooth disease pediatric scale as an outcome measure of disability. Ann Neurol. 2012;71(5):642-52.
Mandarakas MR, Menezes MP, Rose KJ, Shy R, Eichinger K, Foscan M, et al. Development and validation of the Charcot-Marie-Tooth Disease Infant Scale. Brain. 2018;141(12):3319-30.
Ramchandren S, Wu TT, Finkel RS, Siskind CE, Feely SME, Burns J, et al. Development and Validation of the Pediatric Charcot-Marie-Tooth Disease Quality of Life Outcome Measure. Ann Neurol. 2021;89(2):369-79.
Morrow JM, Sinclair CD, Fischmann A, Machado PM, Reilly MM, Yousry TA, et al. MRI biomarker assessment of neuromuscular disease progression: a prospective observational cohort study. Lancet Neurol. 2016;15(1):65-77.
Morrow JM, Evans MRB, Grider T, Sinclair CDJ, Thedens D, Shah S, et al. Validation of MRC Centre MRI calf muscle fat fraction protocol as an outcome measure in CMT1A. Neurology. 2018;91(12):e1125-e9.
Wang H, Davison M, Wang K, Xia TH, Kramer M, Call K, et al. Transmembrane protease serine 5: a novel Schwann cell plasma marker for CMT1A. Ann Clin Transl Neurol. 2020;7(1):69-82.
Fledrich R, Mannil M, Leha A, Ehbrecht C, Solari A, Pelayo-Negro AL, et al. Biomarkers predict outcome in Charcot-Marie-Tooth disease 1A. J Neurol Neurosurg Psychiatry. 2017;88(11):941-52.
Hartmannsberger B, Doppler K, Stauber J, Schlotter-Weigel B, Young P, Sereda MW, et al. Intraepidermal nerve fibre density as biomarker in Charcot-Marie-Tooth disease type 1A. Brain Commun. 2020;2(1):fcaa012.
Acknowledgements
We would like to thank Cimmaron Retzik-Stahr and Jacob Bockhorst for their contributions in collecting bibliography, creating figures and tables, and helping with manuscript preparation. We would also like to express our gratitude to all of the patients who have participated in clinical trials of demyelinating CMT, as well as those who have partnered with the CMT research community to advance our understanding of these conditions.
Funding
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Fridman, V., Saporta, M.A. Mechanisms and Treatments in Demyelinating CMT. Neurotherapeutics 18, 2236–2268 (2021). https://doi.org/10.1007/s13311-021-01145-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-021-01145-z