
Abstract
Inherited peripheral neuropathies are a genetically and phenotypically diverse group of disorders that lead to degeneration of peripheral neurons with resulting sensory and motor dysfunction. Genetic neuropathies that primarily cause axonal degeneration, as opposed to demyelination, are most often classified as Charcot-Marie-Tooth disease type 2 (CMT2) and are the focus of this review. Gene identification efforts over the past three decades have dramatically expanded the genetic landscape of CMT and revealed several common pathological mechanisms among various forms of the disease. In some cases, identification of the precise genetic defect and/or the downstream pathological consequences of disease mutations have yielded promising therapeutic opportunities. In this review, we discuss evidence for pathogenic overlap among multiple forms of inherited neuropathy, highlighting genetic defects in axonal transport, mitochondrial dynamics, organelle-organelle contacts, and local axonal protein translation as recurrent pathological processes in inherited axonal neuropathies. We also discuss how these insights have informed emerging treatment strategies, including specific approaches for single forms of neuropathy, as well as more general approaches that have the potential to treat multiple types of neuropathy. Such therapeutic opportunities, made possible by improved understanding of molecular and cellular pathogenesis and advances in gene therapy technologies, herald a new and exciting phase in inherited peripheral neuropathy.
Similar content being viewed by others


Introduction
Inherited peripheral neuropathies are a diverse group of disorders caused by dominant, X-linked, recessive, or maternal (mitochondrial) mutations in hundreds of different genes. In addition to their mode of inheritance, each kind of inherited neuropathy also differs according to whether demyelination or axonal loss is the primary pathogenic event, and whether the neuropathy is the principal manifestation of the disorder. There are more than 200 inherited syndromes in which a neuropathy is typically present but overshadowed by other aspects of the disorder; these have been given many different names [1, 2]. When neuropathy is the singular manifestation of the disease, it usually called Charcot-Marie-Tooth disease (CMT). Dominant and recessive demyelinating forms are classified as CMT type 1 (CMT1) and type 4 (CMT4), respectively; dominant and recessive axonal forms are classified as CMT type 2 (CMT2) and autosomal recessive CMT2 (AR-CMT2), respectively [3]. Sensory (with variable autonomic involvement) and motor inherited axonal neuropathies also occur and are usually grouped into hereditary sensory and autonomic neuropathy (HSAN) and hereditary motor neuropathy (HMN), respectively. Finally, mutations in many different genes can cause similar phenotypes, and different mutations in the same gene can cause different phenotypes on an apparent continuum with different patients manifesting variable degrees of sensory and/or motor dysfunction. Together, CMT (including HMN and HSAN) comprises the most common inherited neurological disease [4, 5].
Length-dependent, progressive, sensory and/or motor dysfunction, beginning first in the distal lower limbs and later involving the distal upper limbs is the usual manifestation of CMT and is thought to result from progressive, length-dependent loss of axons [6]. The loss of motor axons results in foot drop and weakness of the intrinsic hand muscles; the loss of sensory axons results in sensory loss in the feet and lower legs and impaired balance. The severity of these impairments can be highly variable, even among patients with the same pathogenic variant in a given gene. The heterogeneity of clinical presentation is not well understood — it may owe to the underlying genetic defect along with genetic susceptibility factors [7] and possibly environmental contributors.
Over the past two to three decades, the genetic landscape of CMT has exploded, with identification of more than 100 different genes, and a steady discovery of new ones [8]. The expansion of genetic defects has been accompanied by experimental evidence illuminating the pathological consequences of these mutations. In CMT1 and CMT4, the causative genes are expressed by myelinating Schwann cells, and their mutations result in dysmyelination or demyelination [9]. The pathogenesis and treatment opportunities for CMT1 are discussed in a separate article in this special issue (Volume 18, issue 4). In CMT2, AR-CMT2, HSAN, and HMN, the causative genes are expressed by PNS neurons, and their mutations lead to axonal degeneration. The relevant functions of these genes are diverse, but at least some genes that cause CMT2 share common pathophysiological mechanisms, which, along with their therapeutic implications, are the subject in this review.
Pathogenesis
In this section, we highlight pathways that are affected by genes whose mutations result in axonal neuropathy. While most of the causative genes in CMT2 encode proteins expressed widely (beyond peripheral sensory and motor neurons), manifestations in other organ systems and even other parts of the nervous system, such as brain and spinal cord, are almost uniformly absent. The basis for this striking peripheral nervous system specificity is not yet understood for the majority of CMT2 forms but likely hints at unique vulnerabilities of peripheral neurons to specific molecular and cellular insults. There also exist numerous examples of hereditary multisystem diseases in which peripheral neuropathy is one of many manifestations, and many of the causative mutations in these multisystem conditions disrupt pathways that are commonly involved in CMT2. These examples further highlight some of the specific metabolic and molecular vulnerabilities of neurons of the peripheral nervous system.
Axonal and Endosomal Trafficking Defects
Axons present unique challenges to the preservation of homeostasis. The volume of a neuronal cell body is dwarfed by the mass of its axon, which can be as long as 1 m. Moreover, the metabolic and energetic support required to maintain the electrochemical gradients that underlie neurotransmission is substantial. Due to the unique demands of axonal physiology, neuronal homeostasis and survival are critically dependent on long-range transport of various cargoes between the cell body and most distal axonal segments. Thus, it is not surprising that many forms of inherited neuropathy are caused by mutations in genes that are involved in axonal transport. Axonal transport is an energy-dependent and highly regulated process of directed movement of various organelles and essential cargo, including mRNA transcripts, translation machinery, and trophic factor signaling complexes, along axonal microtubules [10]. Microtubules are highly dynamic polymers of α and β tubulin dimers that run the length of the axon and serve as tracks for molecular motor protein complexes of the dynein and kinesin families. Transport toward the distal end of the axon is termed “anterograde” and is accomplished by binding of cargo to kinesin motors, while transport toward the cell body is termed “retrograde” and involves dynein motors and the cytoplasmic dynein adaptor protein complex. Diversity of molecular motors as well as motor adaptor proteins imparts specificity to the axonal transport of specific cargoes [11]. In contrast to other cell types, axons contain a large population of long-lived and stable microtubules, and their stability can be regulated by various post-translational modifications, such as reversible acetylation of α tubulin [12].
Genes involved directly with microtubule transport include BICD2 (which binds directly to the retrograde microtubule motor protein complex cytoplasmic dynein and is mutated spinal muscular atrophy lower extremity-predominant; SMALED) [13,14,15], RAB7 (which regulates microtubule trafficking of lysosomes, late endosomes, and autophagosomes within axons and is mutated in CMT2B) [16], DCTN1 (which is incorporated into the cytoplasmic dynein motor complex and is mutated in distal HMN7B) [17], DYNC1H1 (which is also part of the cytoplasmic dynein complex and is mutated in CMT2O, hereditary spastic paraplegia (HSP), and SMALED) [18, 19], KIF1A (which is an anterograde kinesin motor protein and is mutated in HSAN2C) [20], and KIF5A (another anterograde kinesin motor mutated in CMT2, HSP, and amyotrophic lateral sclerosis, ALS) [21,22,23] among others. While often not directly demonstrated experimentally, mutations in these genes are assumed to result in some measure of axonal transport defects. As an example, human DYNC1H1 mutations as well as mutations found in so-called Loa and Cra mice (legs at odd angles and cramping), which harbor distinct missense mutations in Dync1h1, all show reduced motor processivity and slowed retrograde axonal transport [18, 24], directly linking defective transport with peripheral nerve degeneration. In addition, mutations in mitofusin-2 (MFN2), which cause CMT2A, likely impact mitochondrial trafficking within axons, among other aspects of mitochondrial biology, as discussed in more depth below. Moreover, mutations in axonal trafficking proteins not only cause forms of CMT, but are found in some genetic syndromes in which peripheral nerve degeneration is a feature. For example, mutations in the microtubule-severing protein spastin (SPAST) cause HSP, and mutations in the tubulin chaperone TBCE and tubulin isoforms TUBB2A and TUBB3 are mutated in complex syndromic conditions that included axonal neuropathy [2].
Notably, many acquired forms of peripheral neuropathy are also associated with axonal transport defects. Chemotherapy-induced peripheral neuropathy (CIPN) is a prime example, as multiple causative agents either directly or indirectly impact microtubule transport [25, 26]. The chemotherapeutic taxanes, such as paclitaxel and docetaxel, and vinca alkyloids, such as vincristine and vinblastine, interfere with microtubule assembly or disassembly and frequently cause dose-related sensory-predominant axonal neuropathy. These agents directly impair either anterograde and/or retrograde microtubule transport within peripheral sensory neurons [27, 28], which is likely central to the pathogenesis of CIPN [25]. Notably, patients with CMT1A appear to be particularly vulnerable to chemotherapy-induced neuropathy, with descriptions of severe and rapid quadriplegia due to vincristine [29, 30].
Mitochondrial Dynamics
One vitally important cargo transported through axons are mitochondria, which are normally maintained throughout the axonal compartment but are enriched at highly metabolically active sites, including at the axon initiation segment, nodes of Ranvier, and distal terminals such as sensory nerve endings and motor neuron neuromuscular junctions [31]. Proper mitochondrial function requires precise regulation of mitochondrial dynamics—the complex and dynamic processes of mitochondrial fission and fusion as well as trafficking along microtubules. In addition, mitochondria undergo transient interactions with adjacent organelles, including endoplasmic reticulum (ER), late endosomes, and lysosomes, and these contacts are important for regulation of mitochondria function. Thus, mitochondrial homeostasis is dependent on properly regulated transport, fusion, fission, and contacts with other organelles, and genetic and biochemical evidence suggests that perturbation of each of these components of mitochondrial dynamics can contribute to axonal dysfunction in neuropathy.
Mutations in MFN2, a mitochondria-associated dynamin-like GTPase involved in multiple aspects of mitochondrial dynamics, are the best example of how mitochondrial dysfunction can lead to axonal pathology. Dominant MFN2 mutations cause CMT2A, which is the most common form of CMT2, accounting for ~ 20% of all cases and 90% of severe cases [32,33,34,35]. The clinical manifestations of different MFN2 mutations range from severe, infantile-onset to mild, adult-onset neuropathy. Recessive MFN2 mutations also occur, and these typically cause a severe, early-onset axonal neuropathy [35]. MFN2 resides within the mitochondrial outer membrane via two transmembrane domains and contains a GTPase domain as well as two coiled-coil heptad repeat regions, designated HR1 and HR2. The HR2 domains from MFN2 proteins on neighboring mitochondria undergo antiparallel interactions in trans that serve to tether adjacent mitochondria, thereby promoting mitochondrial fusion [36]. MFN2 also supports mitochondrial trafficking through interactions with Miro1, which connects mitochondrial membrane-bound MFN2 to Trak1 and microtubule motor complexes [37].
In addition to MFN2, mutations in other genes involved in mitochondrial dynamics are linked with inherited neuropathies, including ganglioside-induced differentiation-associated protein-1 (GDAP1) and optic atrophy-1 (OPA1) [38,39,40,41]. Like MFN2, GDAP1 localizes to the outer mitochondrial membrane, but unlike MFN2, it interacts with mitochondrial fission machinery. Most GDAP1-associated neuropathy is due to biallelic mutations in GDAP1, causing forms of AR-CMT2, but dominant mutations also occur (designated CMT2K) and generally cause less severe disease [38]. OPA1 is likewise involved in mitochondrial fission as well as other aspects of mitochondrial biology, and dominant mutations appear to lead to loss of function. The clinical presentation in patients with OPA1 mutations is dominated by progressive optic atrophy and is thus not classified as CMT, but about 20% of patients display additional clinical features including hearing loss and peripheral neuropathy [42]. Notably, mutations in the inner mitochondrial membrane protein OPA3 also cause optic atrophy and neuropathy [43], and some patients with MFN2 mutations also develop optic atrophy [35].
While mutations affecting mitochondrial dynamics most often cause forms of CMT2, mutations that impair mitochondrial energy production or disrupt mitochondrial DNA stability cause multisystem disease in which peripheral neuropathy is one of many manifestations [44]. Such mitochondrial diseases can be caused by dominant, recessive, or X-linked mutations of nuclear genes, or through mutations in mitochondrial DNA, resulting in matrilineal inheritance. Additional clinical features in mitochondrial diseases are variable, but often include visual disturbances (as in optic atrophy plus due to mutations in OPA1 and OPA3, and NARP (neuropathy, ataxia, and retinitis pigmentosa) due to mutations in MTATP6), gastrointestinal disturbances (as in MNGIE (mitochondrial neurogastrointestinal encephalopathy) due to TYMP mutations), ataxia (as in SANDO (sensory ataxic neuropathy, dysarthria, and ophthalmoparesis) due to mutations in POLG), myopathy (as in MERRF (myoclonic epilepsy with ragged red fibers) due to mutations in MT-TK and MELAS (mitochondrial encephalopathy with lactic acidosis and stroke-like episodes) due to mutations in MT-TL1), and other CNS manifestations (seizures in MERRF, encephalopathy in MNGIE and MELAS) [44, 45]. Thus, peripheral nerves appear to be particularly vulnerable to defects in mitochondrial dynamics and mitochondrial energetics.
The study of defects due to pathogenic MFN2 mutations provides a window into the myriad mechanisms by which mitochondrial dynamics can contribute to axonal pathology. The majority of pathogenic MFN2 mutations affect amino acid residues within or near the N-terminal GTPase domain, with few mutations affecting the C-terminal region of the protein [46]. In vitro, most mutations lead to dominant-negative effects that impair mitochondrial fusion, thereby leading to mitochondrial clustering [47] and ultimately, defective mitochondrial quality control [48]. Disease mutations also impair long-range axonal transport and distribution of mitochondria [49,50,51], which could explain the particular vulnerability of the most distal portions of peripheral axons to degeneration in CMT2A. In addition, MFN2 function in facilitating mitochondrial-ER contacts may also be altered by neuropathy mutations [52]. This molecular understanding of CMT2A pathogenesis presents multiple opportunities for treatment as discussed below.
Messenger RNA Processing and Protein Translation
The discovery of multiple aminoacyl-tRNA synthetase (ARS) genes as causes of dominantly inherited axonal neuropathies was completely unexpected. The original identification of dominant mutations in glycyl-ARS (GARS) as the cause of CMT2D [53] was followed by reports of dominant mutations in AARS, HARS, KARS, MARS, WARS, and YARS [54,55,56,57,58]. ARS genes encode enzymes that catalyze the charging of tRNAs with the appropriate amino acid, thus playing a critical role in protein translation in all tissues [59]. Inherited neuropathies caused by mutations in ARS genes are classified as HMN or CMT2 and generally present with typical length-dependent sensory and/or motor axonal neuropathy.
The pathogenesis of ARS-related CMT2 remains unresolved, with multiple potential pathological mechanisms. Initial suppositions placed impairment of enzymatic activity as the most likely causative disruption in ARS mutations. Indeed, many disease mutations have been shown to disrupt tRNA charging function in vitro and in yeast complementation experiments [56, 60, 61]. However, impairment of enzymatic function is not universal among disease mutations, suggesting that other mutation-dependent effects contribute to pathology [54, 59, 60, 62, 63]. There is also evidence for disrupted dimerization and neomorphic binding activities of disease-causing ARS mutant proteins [62, 64, 65]. An alternative mechanism of pathogenesis was suggested by elegant studies in a Drosophila model of disease-causing ARS mutations [66]. Expression of mutant GARS or YARS caused motor and sensory degeneration that was associated with global reductions in protein translation rates. The impairment of global translation by mutant GARS could not be rescued by overexpression of wild-type GARS, indicating that the mutant protein had a toxic gain of function. Furthermore, genetic inhibition of translation could recapitulate some of the degenerative phenotypes due to expression of GARS or YARS mutants, demonstrating that impaired protein translation is sufficient to cause neuronal degeneration in flies [66]. Together, these studies hint at the possibility that disruption of translation and activation of downstream cellular stress pathways may provide a unifying explanation for how various ARS mutations lead to axonal degeneration. As discussed below, impairment of intra-axonal protein synthesis may also be relevant in other forms of acquired and inherited neuropathy.
Organelle-Organelle Contacts
With advancements in high-resolution microscopy has come an increasing recognition of the highly dynamic and interconnected nature of intracellular organelles. Rather than existing as discrete and disconnected biological compartments, organelles undergo extensive physical and functional interactions [67, 68]. Organelle-organelle contacts have been described to occur between mitochondria, lysosomes/late endosomes, and ER, among others. These contacts occur both within non-neuronal cells and within peripheral nerve axons. Furthermore, new evidence continues to demonstrate important functional consequences for these highly fluid and often transient interactions [69]. CMT2 disease genes have emerged as critical regulators of organelle-organelle contacts, potentially providing a previously unrecognized pathophysiologic connection among disease-associated pathways thought to be distinct and unrelated.
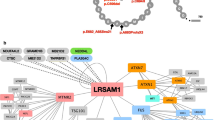
Studies of organelle-organelle contacts have revealed important roles for both MFN2 and the small GTPase Rab7 in regulating these dynamic processes. MFN2 plays a critical role in mediating contacts between ER and mitochondria, which occur at mitochondria-associated membrane regions and may facilitate transfer of Ca2+ from ER to mitochondria [70]. The formation and dissolution of ER-mitochondria contacts are regulated by MFN2 homotypic and heterotypic binding events, and CMT2A mutations alter the dynamics of these contacts with resultant ER stress [71]. Interestingly, the small GTPase Rab7, which is mutated in CMT2B, has also been implicated in regulating organelle-organelle contacts between mitochondria and lysosomes [72]. Dominant RAB7 mutations cause a severe sensory-predominant neuropathy with ulcero-mutilating features that are phenotypically similar to patients with congenital insensitivity to pain [16]. Rab7 localizes to late endosomes, lysosomes, and autophagosomes, where it recruits various effector proteins, including the retrograde microtubule motor cytoplasmic dynein [73,74,75]. Rab7 has been shown to support long-range axonal trafficking of its target organelles, and disease mutations appear to confer dysregulation of Rab7 activity and resultant gain of function [76, 77]. Recent work has shown that Rab7 plays a critical role in lysosome-mitochondria contacts, with Rab7 GTPase activity being particularly important for promoting untethering of lysosomes from mitochondria [72]. Surprisingly, lysosomes can serve as important platforms for the transport of RNA granules along axons, a process that is required to support delivery of mRNA for local axonal protein translation [78]. In addition, lysosome-mitochondria tethers are sites of local protein translation, and disruption of these contacts impairs local translation as well as mitochondrial morphology. Strikingly, Rab7 disease mutations impair axonal protein translation on late endosomes and lysosomes and disrupt mitochondrial morphology and trafficking [78]. Thus, Rab7 and MFN2 have been unexpectedly linked to common pathways involving axonal transport of RNA granules and intra-axonal protein translation.
In addition to CMT, the underlying genetics in HSP provide further evidence for the importance of organelle-organelle contacts in axonal biology. HSP is primarily a distal axonopathy of CNS spinal cord projecting neurons that results in progressive lower extremity spasticity and weakness [79]. Variable peripheral sensory and motor involvement can also occur, resulting in phenotypic overlap with CMT2. Much like CMT, HSP is genetically diverse, with over 80 genetic loci resulting in either dominant, recessive, or X-linked disease. However, a substantial fraction of HSP genes are directly implicated in various aspects of ER membrane dynamics, including ER-organelle contacts and ER membrane bending and tubulation [80, 81]. Mutations in ER-shaping proteins are particularly common, with mutations in spastin (SPAST), atlastin-1 (ATL1), reticulon 2 (RTN2), and REEP1, accounting for around half of all HSP cases [82]. The mechanisms by which HSP-causing mutations alter ER membrane dynamics and function are diverse, but serve to further highlight the vulnerability of long axons to disruption of organelle membrane formation and maintenance [81].
With the finding that CMT2-causing mutations in various amino-acyl-tRNA synthetases lead to suppression of global translation that is particularly detrimental to neuronal homeostasis, a pattern emerges suggesting that many distinct forms of CMT may share the common pathological insult of impaired axonal protein translation. Disruption of local translation within axons can result from a range of primary pathological insults in various forms of CMT2, such as impaired axonal trafficking, disrupted organelle tethering, or failure of RNA granule delivery (Fig. 1). Further highlighting these pathways, there is a growing body of evidence linking impaired local protein translation to the pathogenesis of spinal muscular atrophy and ALS [83]. Furthermore, the microtubule-disrupting chemotherapeutic agent paclitaxel may lead to axonal neuropathy in part through impaired delivery of mRNA to distal axonal compartments [84]. This confluence of neuropathy-related genes onto pathways involved in microtubule transport, mitochondrial energetics, organelle-organelle interactions, and local protein translation underscores the delicate nature of axonal physiology, particularly in the longest peripheral nerves, and suggests that multiple perturbations of metabolic processes can expose the unique vulnerability of this highly specialized portion of the nervous system. However, from an optimistic perspective, these pathophysiological connections may provide the potential to uncover therapeutic targets that could be relevant for multiple forms of CMT, and perhaps other forms of neuromuscular degenerative diseases.

The confluence of axonal neuropathy genes in the regulation of axonal transport, organelle-organelle contacts, and local protein translation. Schematic representation of multiple gene products and associated pathways implicated in inherited axonal neuropathy. (1) Mitofusin 2 (MFN2) regulates mitochondrial axonal transport through recruitment of adaptor proteins and microtubule transport machinery such as kinesin motors. (2) MFN2 regulates mitochondrial-ER contacts. (3) MFN2 regulates mitochondrial tethering and fusion. (4) Rab7 regulates late endosome and lysosome axonal transport and contacts with mitochondria. (5) Rab7-positive late endosomes and lysosomes promote long-range axonal transport of RNA granules and protein translation machinery to support local axonal protein translation. (6) Aminoacyl-tRNA synthetases charge tRNAs to support axonal protein synthesis
Treatment
General Considerations
Treatment strategies fall broadly into two categories — “specific” therapies that target a unique aspect in the pathogenesis of a subtype of neuropathy and “general” therapies that utilize an approach that can be readily applied and/or adapted to multiple neuropathy subtypes. In either case, treatment efforts rely on the underlying assumption that the pathogenic mechanisms of axonal CMT of varied causes are related to cell-autonomous dysfunction conferred by a specific genetic defect in peripheral motor and/or sensory neurons. The nature of the genetic defect largely dictates the therapeutic possibilities for a given form of axonal CMT. For instance, dominant mutations that cause disease either through a gain-of-function or dominant-negative effect can be targeted by therapeutic strategies that silence the mutant allele, inhibit the mutant protein function, or suppress downstream consequences of the mutant protein activity. For dominant-negative mutations, an additional possibility is “overwhelming” the mutant protein through supplementation of expression of the wild-type protein. On the other hand, recessive/loss-of-function forms of CMT should be amenable to gene replacement strategies.
Some specific CMT subtypes have received particular attention with respect to clinical and translational efforts for several reasons. First, CMT subtypes that are more common and thereby affect a larger fraction of the patient population, such as CMT2A, have received more consideration. Second, increased disease prevalence also impacts an often overlooked but critical facet of the therapeutic development pipeline: the identification of sufficient numbers of patients for detailed natural history studies. These studies provide longitudinal clinical information that is fundamental to multiple aspects of clinical trial design, including the identification of relevant endpoints and biomarkers, calculation of statistical power, and choice of patient subgroups most likely to demonstrate therapeutic efficacy. Third, both basic and translational researches have been dramatically aided in CMT types for which validated and robust animal models have been developed. While generation of an animal model that recapitulates important features of dominant, monogenic human disease may seem straightforward, the realities of modeling length-dependent, slowly progressive axonal neuropathies in small rodents have frequently proved challenging. Still, many robust CMT2 mouse models have now become available [85], and rat models are increasingly being generated, sometimes with improved recapitulation of the human disease phenotypic features [86, 87]. Fourth, with the gradual progression of most forms of CMT, the identification of biomarkers of disease activity and therapeutic target engagement can greatly enhance the power to detect a biological effect at an early stage. In the sections below, we discuss specific examples of genetic axonal neuropathies that are likely to be on the leading edge of therapeutic development and clinical trial initiation.
Specific Approaches for Individual Neuropathies
For many forms of axonal CMT, understanding of the specific genetic defects and molecular pathogenesis make treatments directed at the underlying pathology at least theoretically possible. Likely the most straightforward pathway for translation from molecular pathogenesis to therapeutics relates to inherited neuropathies due to metabolic defects. In these forms of neuropathy, knowledge of the affected metabolic pathway suggests therapeutic opportunities to reduce accumulation of toxic intermediates or to otherwise address enzymatic deficiencies. HSAN1 and SORD-related neuropathy are illustrative cases of metabolic neuropathies that are potentially treatable. In other specific CMT2 forms, understanding of the distinct mechanisms of pathogenesis has led to identification of small molecules that may have therapeutic benefit; CMT2A and TRPV4 neuropathy are examples. In addition to these specific examples, there are several genetic axonal neuropathies, many of which are not classically defined as CMT, which can be viewed as potentially treatable. These inherited neuropathies, which have either some experimental evidence for potential efficacy of correcting a metabolic defect, or an animal model demonstrating proof of principle for a specific therapeutic intervention, are presented briefly in Table 1.
HSAN1
HSAN1 is caused by dominant missense mutations in either SPTLC1 or SPTLC2, which encode subunits of the serine palmitoyl transferase (SPT) enzyme that functions in production of sphingolipids [110, 111]. Patients develop a sensory and motor neuropathy with predominantly axonal features, and also develop autonomic dysfunction. Sensory loss can be profound, leading to recurrent painless injuries that can be complicated by ulceration and osteomyelitis [112]. Disease-causing mutations decrease the affinity of SPT for its normal substrate, l-serine, and increased affinity for alanine and glycine, leading to production and accumulation of atypical, toxic sphingolipids [113]. Administration of l-serine in a mouse model of HSAN1 led to reduction in toxic sphingolipids as well as behavioral and histological improvements [114], and a small human clinical trial also showed reduction in toxic sphingolipid accumulation and a hint of clinical benefit [115]. While the trial size (18 patients) and short follow-up (2 years) limited the power to detect significant clinical impact, these results demonstrate successful targeting of sphingolipid accumulation and warrant further study.
SORD Neuropathy
The recent identification of recessive mutations in SORD represents a promising therapeutic opportunity [116]. SORD neuropathy is a motor-predominant, axonal neuropathy, [116], and may be the most common cause of recessively inherited peripheral neuropathy. SORD encodes sorbitol dehydrogenase, the second step in the polyol pathway that enzymatically converts glucose to fructose. In this pathway, aldose reductase first converts glucose to the poorly metabolized sugar sorbitol, and then SORD catalyzes the conversion of sorbitol to fructose. Loss of SORD activity results in accumulation of sorbitol, which has previously been implicated as a potentially neurotoxic species in diabetic neuropathy [117]. SORD mutations that cause neuropathy produce loss of function, either by introducing premature stop codons or by disrupting amino acid residues critical for enzymatic activity. Accumulation of sorbitol was demonstrated in patient serum and patient-derived cells as well as in a Drosophila model. Deletion of the fly SORD ortholog resulted in degenerative phenotypes that could be reversed by FDA-approved medications that inhibit aldose reductase (epalrestat and ranirestat) and thereby reduce sorbitol accumulation. These results have led to ongoing efforts to identify additional patients and better define the natural history of SORD neuropathy in preparation for a human clinical trial.
CMT2A
In contrast to SORD neuropathy, in which gene discovery suggested an obvious therapeutic approach, the path toward a rational therapeutic for CMT2A required hard-earned insights into molecular pathogenesis of disease-causing mutations. As described above, MFN2 has several functions — mitochondrial transport, tethering to organelles, fusion, and homeostasis. Pathogenic mutations likely act in a dominant-negative fashion, thereby impairing one or more of these normal functions [47, 50, 118]. Importantly, MFN2 is highly homologous to MFN1, and studies from various experimental systems have demonstrated that MFN1 and MFN2 can directly interact, can support mitochondrial fusion in trans, and that MFN1 can compensate for MFN2 deficiency [47, 50]. The relative paucity of expression of MFN1 within axons may explain the selective vulnerability of motor and sensory neurons to MFN2 mutations [47]. In non-neuronal tissues, there may be enough MFN1 expression to counteract the effects of mutant MFN2, whereas low levels of MFN1 in neurons make them almost solely reliant on MFN2.
Recent studies have highlighted the functional importance of conformational changes within the heptad repeat regions of MFN2 for its function in facilitating intermolecular MFN2 interactions and mitochondrial tethering [119]. Dimerization of mitofusin molecules present on adjacent mitochondria is facilitated by switching of the HR2 domain from a “closed” (HR2 binding to HR1 of the same MFN2 molecule) to an “open” conformation (HR2 binding to the HR2 of an opposing MFN2). Small molecules targeting HR1/HR2 of MFN2 can function as “mitofusin agonists” by shifting the equilibrium of mitofusins toward the open/active state and can restore mitochondrial axon trafficking defects in cultured motor neurons from MFN2 T105M mutant mice [51]. Subsequently, an orally available “mitofusin agonist” (MiM111) was shown to ameliorate disease phenotypes in the T105M mutant mice [120, 121]. As there are both mouse and rat animal models of CMT2A that recapitulate core features of the human disease, these models could be readily utilized for further pre-clinical trials of mitofusin conformation-altering drugs.
TRPV4-Related Neuropathies
Neuropathy due to mutations in TRPV4 (transient potential vanilloid 4), which encodes a calcium permeable ion channel, represents another form of axonal neuropathy that is potentially treatable. While manifestations of TRPV4 mutations result in heterogeneous forms of motor predominant or motor exclusive neuropathy, studies in cultured cells and in a Drosophila model suggest that gain of ion channel function, leading to increased Ca2+ influx, is likely the most proximal event in the pathogenic cascade [122, 123]. Mutations that cause neuropathy largely localize to a single intracellular domain, the ankyrin repeat domain, and predominantly affect conserved arginine residues [124]. Neuropathogenic mutations also disrupt protein–protein interactions, including with the small GTPase RhoA [122]. Expression of human TRPV4 with neuropathy-causing mutations results in multiple neurodegenerative phenotypes in flies, which can be rescued with genetic or pharmacologic inhibition of ion channel activity [123]. Importantly, many highly specific TRPV4 antagonists have been generated, including an orally available drug (GSK2798745) that showed good tolerability in a clinical trial for cardiogenic pulmonary edema [125]. This TRPV4 antagonist, or similar drugs, could potentially be repurposed for use in patients with TRPV4 neuropathy. Current barriers to a clinical trial include the rarity of patients with TRPV4-related neuropathy, their heterogeneous clinical manifestations, and an incomplete understanding of the natural history.
General Pharmacologic Approaches
Given the convergence of pathological pathways among multiple forms of CMT, there is great interest in identifying therapeutic targets that might be broadly applicable. The landmark identification of Sarm1 as a critical gene in axonal degeneration currently represents the most promising discovery that has emerged from these endeavors [126]. Sarm1 is an NADase, which depletes NAD+, thereby activating metabolic signaling events that culminate in axonal degeneration [127]. Inhibiting Sarm1 activation is an attractive therapeutic target, and is reviewed in another article in this issue (Volume 18, issue 4).
Abnormal tubulin acetylation has also emerged as a potentially unifying downstream pathological abnormality in multiple forms of CMT2. Acetylated tubulin is highly abundant within axons, and acetylation increases the stability of microtubules and may also facilitate transport of cargoes along microtubules, particularly in the anterograde direction, through increased affinity for kinesin motors [128]. The first description of altered tubulin acetylation in CMT came from work on a mouse model of CMT2F (related to HSPB1 mutations) [129]. Acetylated tubulin was reduced in primary neurons and sciatic nerve from these mice, but not in the spinal cord. Similar deficiencies of acetylated tubulin have subsequently been demonstrated in mutant CMT2A/Mfn2 and CMT2D/Gars mouse models, suggesting that dysregulation of tubulin acetylation within axons might be a common feature across genetically diverse forms of CMT2 [130].
Tubulin acetylation is principally regulated by the opposing activities of the deacetylase HDAC6 and the acetylase α-tubulin acetyltransferase 1. HDAC6 is a unique member of the HDAC family — its expression is restricted to the cytosol, its primary enzymatic targets are not histones, it contains a ubiquitin binding domain, and it interacts with microtubule transport machinery [131, 132]. In the same mouse models mentioned above, inhibition of HDAC6, either pharmacologically or through genetic deletion, restored levels of acetylated tubulin within the peripheral nerve and partially restored sensory and motor function. In the HSPB1 mutant mouse model, HDAC6 inhibition improved mitochondrial trafficking, rotarod performance, and the amplitudes of motor and sensory nerve responses [129]. Similar but modest improvements in sensory and motor function were seen with pharmacologic HDAC6 inhibition in a mouse model of CMT2A, but the effects were more pronounced with genetic deletion of HDAC6 in these mice [133]. Finally, in a mutant mouse model of CMT2D, pharmacological inhibition of HDAC6 also led to modest improvements in several motor and sensory parameters [130].
In addition to its function in regulating tubulin acetylation, HDAC6 serves other important roles, including facilitating protein turnover by linking proteasome-dependent and autophagy-dependent protein degradation pathways [134]. In this latter context, HDAC6 activity plays a protective role through promoting clearance of misfolded and potentially proteotoxic species. Thus, clinical use of HDAC6 inhibition to correct defects in peripheral nerve tubulin acetylation could potentially lead to important off-target effects that could be detrimental to the nervous system in other ways.
Genetic Approaches
Gene Replacement
The success of adeno-associated virus (AAV)-mediated gene delivery for spinal muscular atrophy demonstrates its feasibility for inherited axonal neuropathies [135], as current vectors can deliver the missing gene to both sensory and motor neurons [136, 137]. In addition to recessive axonal neuropathies caused by loss-of-function mutations, some dominant mutations could also be treated by overexpression of wild-type protein [47, 138], or by incorporating a small interfering RNA (siRNA) against the endogenous gene into the viral vector. In the future, gene-editing strategies, such as deleting dominant alleles, could become feasible.
The first foray into gene therapy for inherited axonal neuropathy is underway. Giant axonal neuropathy (GAN) is a recessive, multisystem neurodegenerative disease caused by loss of function mutations in gigaxonin. GAN is extremely rare and presents in early childhood with a range of signs and symptoms, including sensory and motor neuropathy, ataxia, and gait difficulty, as well as characteristic, kinky hair [139]. The disease is relentlessly progressive, with brainstem pathology leading to dysphagia, dysarthria, and ultimately death due to respiratory failure. Loss of gigaxonin, an E3 ubiquitin ligase involved in intermediate filament organization and turnover, leads to massively enlarged axons that are filled with disorganized intermediate filaments. Gan knockout mice have abnormal accumulation of intermediate filaments within cells, with mild axonal degeneration and functional motor impairment. Intrathecal delivery of GAN with AAV9 into these mice reduced intermediate filament accumulations in sensory neurons and peripheral nerve, and modestly improved rotarod performance [140]. In light of the fatal nature of GAN, these promising preclinical proof-of-principle results helped launch the first intrathecal AAV gene transfer clinical trial in humans (NCT02362438), which is currently underway. While the primary goal of the study is to evaluate the safety of this treatment approach, additional clinical endpoints will be concurrently assessed.
The overexpression of MFN1 or MFN2 is a promising approach for CMT2A. Transgenic mice that express the disease-causing R94Q mutation (a common mutation that causes severe neuropathy) of MFN2 in PNS and CNS neurons develop axonal degeneration, reduced strength, poor motor performance, vision loss, and modestly reduced survival [138]. In line with prior studies, the R94Q MFN2 functions as a dominant negative, leading to impaired mitochondrial fusion. Importantly, over-expression of human MFN1 or MFN2 within the nervous system using the prion promotor reversed mitochondrial clustering, axonal degeneration, and gliosis. Together, these results provide a proof-of-concept that gene therapy to increase expression of functional, fusion-competent mitofusins may at least partially overcome the effects of dominant negative MFN2 alleles and provide therapeutic benefit in CMT2A patients. With the recent description of the natural history of CMT2A in a large patient cohort, these pre-clinical animal studies could help pave the way for future gene therapy human trials in CMT2A [35].
Gene Knockdown
The successful employment of antisense oligonucleotides (ASOs) and siRNA in familial amyloid polyneuropathy is truly a remarkable achievement in neuromuscular medicine, and is reviewed in another article in this issue (Volume 18, issue 4). However, these strategies are not currently viewed as generally applicable for treatment in inherited axonal neuropathies. Most of the genes that cause inherited axonal neuropathies are probably essential for neuronal health and are thus poor candidates for the global knockdown strategy that was used for treating familial amyloid polyneuropathy.
Selective knockdown or removal of the disease-causing allele, sparing the wild-type allele, is not yet feasible, but some pioneering work underscores the potential of mutant allele–specific genetic silencing. Two distinct GARS mutations that cause CMT2D have been successfully targeted by allele-specific siRNA in mouse models [141]. Both alleles had multiple nucleotide substitutions, increasing the ability of ASOs to specifically target the mutant allele. In the mouse models, the mutant GARS alleles were successfully knocked down using AAV9-mediated delivery of siRNA, and the degree of phenotypic rescue was dramatic. Whether disease mutations arising from a single nucleotide change, which are by far the most common variants responsible for disease, could be efficiently and specifically targeted while sparing the wild-type allele remains to be determined. In addition, employing allele-specific knockdown strategies in human patients poses a substantial regulatory challenge in that each ASO or siRNA sequence for an individual patient would be unique and might have to undergo a separate safety and efficacy evaluation prior to approval. If these technical and regulatory hurdles can be overcome, strategies to specifically silence (or correct with gene editing) dominant missense mutant alleles would revolutionize the treatment of diseases caused by dominant alleles.
Conclusions
Research on inherited neuropathies is entering an exciting new phase — clinical and translational directions into the pathogenesis and treatment of CMT. Recent work has added granularity to the understanding of long-held notions of how disrupted axonal biology leads to neuronal degeneration and has begun to provide some unexpected pathophysiological connections that may ultimately reveal therapeutic targets shared across unrelated forms of CMT. Available drugs could very likely impact underlying pathological mechanisms of HSAN1, SORD neuropathy, and TRPV4 neuropathy. CMT2A has emerged at the leading edge of pre-clinical therapeutic development, and rational clinical trials for this form of axonal CMT can be envisioned. In addition, the field of gene therapy has made tremendous advances, with identification of viral vector-based methods capable of robust transduction of sensory motor and sensory neurons. Much work remains to be done to facilitate clinical trials for various forms of CMT, including natural history studies and biomarker identification and validation. However, with the confluence of efforts across these varied disciplines and approaches, we foresee a day when clinicians can tell some patients that their form of CMT is a treatable condition.
References
Rossor, A. M., Carr, A. S., Devine, H., Chandrashekar, H., Pelayo-Negro, A. L., Pareyson, D., Shy, M. E., Scherer, S. S. & Reilly, M. M. Peripheral neuropathy in complex inherited diseases: an approach to diagnosis. J Neurol Neurosurg Psychiatry 88, 846-863 (2017).
Scherer, S. S., Kleopa, K. A. & Benson, M. D. Rosenberg's molecular and genetic basis of neurological and psychiatric disease. 6th edn, Vol. 2 345–76 (Academic Press, 2020).
Harding, A. E. & Thomas, P. K. Genetic aspects of hereditary motor and sensory neuropathy (types I and II). J Med Genet 17, 329-336 (1980).
Skre, H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet 6, 98-118 (1974).
Barreto, L. C., Oliveira, F. S., Nunes, P. S., de Franca Costa, I. M., Garcez, C. A., Goes, G. M., Neves, E. L., de Souza Siqueira Quintans, J. & de Souza Araujo, A. A. Epidemiologic Study of Charcot-Marie-Tooth Disease: A Systematic Review. Neuroepidemiology 46, 157–165 (2016).
Pareyson, D., Marchesi, C. & Salsano, E. Dominant Charcot-Marie-Tooth syndrome and cognate disorders. Handb Clin Neurol 115, 817-845 (2013).
Bis-Brewer, D. M., Fazal, S. & Zuchner, S. Genetic modifiers and non-Mendelian aspects of CMT. Brain Res 1726, 146459 (2020).
Laura, M., Pipis, M., Rossor, A. M. & Reilly, M. M. Charcot-Marie-Tooth disease and related disorders: an evolving landscape. Curr Opin Neurol 32, 641-650 (2019).
Houlden, H. & Reilly, M. M. Molecular genetics of autosomal-dominant demyelinating Charcot-Marie-Tooth disease. Neuromolecular Med 8, 43-62 (2006).
Beijer, D., Sisto, A., Van Lent, J., Baets, J. & Timmerman, V. Defects in Axonal Transport in Inherited Neuropathies. J Neuromuscul Dis 6, 401-419 (2019).
Sleigh, J. N., Rossor, A. M., Fellows, A. D., Tosolini, A. P. & Schiavo, G. Axonal transport and neurological disease. Nat Rev Neurol 15, 691-703 (2019).
Conde, C. & Caceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci 10, 319-332 (2009).
Neveling, K., Martinez-Carrera, L. A., Holker, I., Heister, A., Verrips, A., Hosseini-Barkooie, S. M., Gilissen, C., Vermeer, S., Pennings, M., Meijer, R., te Riele, M., Frijns, C. J., Suchowersky, O., MacLaren, L., Rudnik-Schoneborn, S., Sinke, R. J., Zerres, K., Lowry, R. B., Lemmink, H. H., Garbes, L., Veltman, J. A., Schelhaas, H. J., Scheffer, H. & Wirth, B. Mutations in BICD2, which encodes a golgin and important motor adaptor, cause congenital autosomal-dominant spinal muscular atrophy. Am J Hum Genet 92, 946-954 (2013).
Oates, E. C., Rossor, A. M., Hafezparast, M., Gonzalez, M., Speziani, F., MacArthur, D. G., Lek, M., Cottenie, E., Scoto, M., Foley, A. R., Hurles, M., Houlden, H., Greensmith, L., Auer-Grumbach, M., Pieber, T. R., Strom, T. M., Schule, R., Herrmann, D. N., Sowden, J. E., Acsadi, G., Menezes, M. P., Clarke, N. F., Zuchner, S., Uk10K, Muntoni, F., North, K. N. & Reilly, M. M. Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia. Am J Hum Genet 92, 965–973 (2013).
Peeters, K., Litvinenko, I., Asselbergh, B., Almeida-Souza, L., Chamova, T., Geuens, T., Ydens, E., Zimon, M., Irobi, J., De Vriendt, E., De Winter, V., Ooms, T., Timmerman, V., Tournev, I. & Jordanova, A. Molecular defects in the motor adaptor BICD2 cause proximal spinal muscular atrophy with autosomal-dominant inheritance. Am J Hum Genet 92, 955-964 (2013).
Verhoeven, K., De Jonghe, P., Coen, K., Verpoorten, N., Auer-Grumbach, M., Kwon, J. M., FitzPatrick, D., Schmedding, E., De Vriendt, E., Jacobs, A., Van Gerwen, V., Wagner, K., Hartung, H. P. & Timmerman, V. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet 72, 722-727 (2003).
Puls, I., Jonnakuty, C., LaMonte, B. H., Holzbaur, E. L., Tokito, M., Mann, E., Floeter, M. K., Bidus, K., Drayna, D., Oh, S. J., Brown, R. H., Jr., Ludlow, C. L. & Fischbeck, K. H. Mutant dynactin in motor neuron disease. Nat Genet 33, 455-456 (2003).
Hafezparast, M., Klocke, R., Ruhrberg, C., Marquardt, A., Ahmad-Annuar, A., Bowen, S., Lalli, G., Witherden, A. S., Hummerich, H., Nicholson, S., Morgan, P. J., Oozageer, R., Priestley, J. V., Averill, S., King, V. R., Ball, S., Peters, J., Toda, T., Yamamoto, A., Hiraoka, Y., Augustin, M., Korthaus, D., Wattler, S., Wabnitz, P., Dickneite, C., Lampel, S., Boehme, F., Peraus, G., Popp, A., Rudelius, M., Schlegel, J., Fuchs, H., Hrabe de Angelis, M., Schiavo, G., Shima, D. T., Russ, A. P., Stumm, G., Martin, J. E. & Fisher, E. M. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science 300, 808–812 (2003).
Weedon, M. N., Hastings, R., Caswell, R., Xie, W., Paszkiewicz, K., Antoniadi, T., Williams, M., King, C., Greenhalgh, L., Newbury-Ecob, R. & Ellard, S. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet 89, 308-312 (2011).
Riviere, J. B., Ramalingam, S., Lavastre, V., Shekarabi, M., Holbert, S., Lafontaine, J., Srour, M., Merner, N., Rochefort, D., Hince, P., Gaudet, R., Mes-Masson, A. M., Baets, J., Houlden, H., Brais, B., Nicholson, G. A., Van Esch, H., Nafissi, S., De Jonghe, P., Reilly, M. M., Timmerman, V., Dion, P. A. & Rouleau, G. A. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am J Hum Genet 89, 219-230 (2011).
Nicolas, A., Kenna, K. P., Renton, A. E., Ticozzi, N., Faghri, F., Chia, R., Dominov, J. A., Kenna, B. J., Nalls, M. A., Keagle, P., Rivera, A. M., van Rheenen, W., Murphy, N. A., van Vugt, J., Geiger, J. T., Van der Spek, R. A., Pliner, H. A., Shankaracharya, Smith, B. N., Marangi, G., Topp, S. D., Abramzon, Y., Gkazi, A. S., Eicher, J. D., Kenna, A., Consortium, I., Mora, G., Calvo, A., Mazzini, L., Riva, N., Mandrioli, J., Caponnetto, C., Battistini, S., Volanti, P., La Bella, V., Conforti, F. L., Borghero, G., Messina, S., Simone, I. L., Trojsi, F., Salvi, F., Logullo, F. O., D'Alfonso, S., Corrado, L., Capasso, M., Ferrucci, L., Genomic Translation for, A. L. S. C. C., Moreno, C. A. M., Kamalakaran, S., Goldstein, D. B., Consortium, A. L. S. S., Gitler, A. D., Harris, T., Myers, R. M., Consortium, N. A., Phatnani, H., Musunuri, R. L., Evani, U. S., Abhyankar, A., Zody, M. C., Answer, A. L. S. F., Kaye, J., Finkbeiner, S., Wyman, S. K., LeNail, A., Lima, L., Fraenkel, E., Svendsen, C. N., Thompson, L. M., Van Eyk, J. E., Berry, J. D., Miller, T. M., Kolb, S. J., Cudkowicz, M., Baxi, E., Clinical Research in, A. L. S., Related Disorders for Therapeutic Development, C., Benatar, M., Taylor, J. P., Rampersaud, E., Wu, G., Wuu, J., Consortium, S., Lauria, G., Verde, F., Fogh, I., Tiloca, C., Comi, G. P., Soraru, G., Cereda, C., French, A. L. S. C., Corcia, P., Laaksovirta, H., Myllykangas, L., Jansson, L., Valori, M., Ealing, J., Hamdalla, H., Rollinson, S., Pickering-Brown, S., Orrell, R. W., Sidle, K. C., Malaspina, A., Hardy, J., Singleton, A. B., Johnson, J. O., Arepalli, S., Sapp, P. C., McKenna-Yasek, D., Polak, M., Asress, S., Al-Sarraj, S., King, A., Troakes, C., Vance, C., de Belleroche, J., Baas, F., Ten Asbroek, A., Munoz-Blanco, J. L., Hernandez, D. G., Ding, J., Gibbs, J. R., Scholz, S. W., Floeter, M. K., Campbell, R. H., Landi, F., Bowser, R., Pulst, S. M., Ravits, J. M., MacGowan, D. J. L., Kirby, J., Pioro, E. P., Pamphlett, R., Broach, J., Gerhard, G., Dunckley, T. L., Brady, C. B., Kowall, N. W., Troncoso, J. C., Le Ber, I., Mouzat, K., Lumbroso, S., Heiman-Patterson, T. D., Kamel, F., Van Den Bosch, L., Baloh, R. H., Strom, T. M., Meitinger, T., Shatunov, A., Van Eijk, K. R., de Carvalho, M., Kooyman, M., Middelkoop, B., Moisse, M., McLaughlin, R. L., Van Es, M. A., Weber, M., Boylan, K. B., Van Blitterswijk, M., Rademakers, R., Morrison, K. E., Basak, A. N., Mora, J. S., Drory, V. E., Shaw, P. J., Turner, M. R., Talbot, K., Hardiman, O., Williams, K. L., Fifita, J. A., Nicholson, G. A., Blair, I. P., Rouleau, G. A., Esteban-Perez, J., Garcia-Redondo, A., Al-Chalabi, A., Project Min, E. A. L. S. S. C., Rogaeva, E., Zinman, L., Ostrow, L. W., Maragakis, N. J., Rothstein, J. D., Simmons, Z., Cooper-Knock, J., Brice, A., Goutman, S. A., Feldman, E. L., Gibson, S. B., Taroni, F., Ratti, A., Gellera, C., Van Damme, P., Robberecht, W., Fratta, P., Sabatelli, M., Lunetta, C., Ludolph, A. C., Andersen, P. M., Weishaupt, J. H., Camu, W., Trojanowski, J. Q., Van Deerlin, V. M., Brown, R. H., Jr., van den Berg, L. H., Veldink, J. H., Harms, M. B., Glass, J. D., Stone, D. J., Tienari, P., Silani, V., Chio, A., Shaw, C. E., Traynor, B. J. & Landers, J. E. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 97, 1268–1283 e1266 (2018).
Liu, Y. T., Laura, M., Hersheson, J., Horga, A., Jaunmuktane, Z., Brandner, S., Pittman, A., Hughes, D., Polke, J. M., Sweeney, M. G., Proukakis, C., Janssen, J. C., Auer-Grumbach, M., Zuchner, S., Shields, K. G., Reilly, M. M. & Houlden, H. Extended phenotypic spectrum of KIF5A mutations: From spastic paraplegia to axonal neuropathy. Neurology 83, 612-619 (2014).
Crimella, C., Baschirotto, C., Arnoldi, A., Tonelli, A., Tenderini, E., Airoldi, G., Martinuzzi, A., Trabacca, A., Losito, L., Scarlato, M., Benedetti, S., Scarpini, E., Spinicci, G., Bresolin, N. & Bassi, M. T. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin Genet 82, 157-164 (2012).
Ori-McKenney, K. M., Xu, J., Gross, S. P. & Vallee, R. B. A cytoplasmic dynein tail mutation impairs motor processivity. Nat Cell Biol 12, 1228-1234 (2010).
Staff, N. P., Fehrenbacher, J. C., Caillaud, M., Damaj, M. I., Segal, R. A. & Rieger, S. Pathogenesis of paclitaxel-induced peripheral neuropathy: A current review of in vitro and in vivo findings using rodent and human model systems. Exp Neurol 324, 113121 (2020).
Shah, A., Hoffman, E. M., Mauermann, M. L., Loprinzi, C. L., Windebank, A. J., Klein, C. J. & Staff, N. P. Incidence and disease burden of chemotherapy-induced peripheral neuropathy in a population-based cohort. J Neurol Neurosurg Psychiatry 89, 636-641 (2018).
LaPointe, N. E., Morfini, G., Brady, S. T., Feinstein, S. C., Wilson, L. & Jordan, M. A. Effects of eribulin, vincristine, paclitaxel and ixabepilone on fast axonal transport and kinesin-1 driven microtubule gliding: implications for chemotherapy-induced peripheral neuropathy. Neurotoxicology 37, 231-239 (2013).
Shemesh, O. A. & Spira, M. E. Paclitaxel induces axonal microtubules polar reconfiguration and impaired organelle transport: implications for the pathogenesis of paclitaxel-induced polyneuropathy. Acta Neuropathol 119, 235-248 (2010).
Chauvenet, A. R., Shashi, V., Selsky, C., Morgan, E., Kurtzberg, J., Bell, B. & Pediatric Oncology Group, S. Vincristine-induced neuropathy as the initial presentation of charcot-marie-tooth disease in acute lymphoblastic leukemia: a Pediatric Oncology Group study. J Pediatr Hematol Oncol 25, 316–320 (2003).
Ibanez-Julia, M. J., Berzero, G., Reyes-Botero, G., Maisonobe, T., Lenglet, T., Slim, M., Louis, S., Balaguer, A., Sanson, M., Le Guern, E., Latour, P., Ricard, D., Stojkovic, T. & Psimaras, D. Antineoplastic agents exacerbating Charcot Marie Tooth disease: red flags to avoid permanent disability. Acta Oncol 57, 403-411 (2018).
Pareyson, D., Saveri, P., Sagnelli, A. & Piscosquito, G. Mitochondrial dynamics and inherited peripheral nerve diseases. Neurosci Lett 596, 66-77 (2015).
Zuchner, S., De Jonghe, P., Jordanova, A., Claeys, K. G., Guergueltcheva, V., Cherninkova, S., Hamilton, S. R., Van Stavern, G., Krajewski, K. M., Stajich, J., Tournev, I., Verhoeven, K., Langerhorst, C. T., de Visser, M., Baas, F., Bird, T., Timmerman, V., Shy, M. & Vance, J. M. Axonal neuropathy with optic atrophy is caused by mutations in mitofusin 2. Ann Neurol 59, 276-281 (2006).
Feely, S. M., Laura, M., Siskind, C. E., Sottile, S., Davis, M., Gibbons, V. S., Reilly, M. M. & Shy, M. E. MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology 76, 1690-1696 (2011).
McCorquodale, D. S., 3rd, Montenegro, G., Peguero, A., Carlson, N., Speziani, F., Price, J., Taylor, S. W., Melanson, M., Vance, J. M. & Zuchner, S. Mutation screening of mitofusin 2 in Charcot-Marie-Tooth disease type 2. J Neurol 258, 1234-1239 (2011).
Pipis, M., Feely, S. M. E., Polke, J. M., Skorupinska, M., Perez, L., Shy, R. R., Laura, M., Morrow, J. M., Moroni, I., Pisciotta, C., Taroni, F., Vujovic, D., Lloyd, T. E., Acsadi, G., Yum, S. W., Lewis, R. A., Finkel, R. S., Herrmann, D. N., Day, J. W., Li, J., Saporta, M., Sadjadi, R., Walk, D., Burns, J., Muntoni, F., Ramchandren, S., Horvath, R., Johnson, N. E., Zuchner, S., Pareyson, D., Scherer, S. S., Rossor, A. M., Shy, M. E., Reilly, M. M. & Inherited Neuropathies Consortium - Rare Disease Clinical Research, N. Natural history of Charcot-Marie-Tooth disease type 2A: a large international multicentre study. Brain 143, 3589–3602 (2020).
Koshiba, T., Detmer, S. A., Kaiser, J. T., Chen, H., McCaffery, J. M. & Chan, D. C. Structural basis of mitochondrial tethering by mitofusin complexes. Science 305, 858-862 (2004).
Misko, A., Jiang, S., Wegorzewska, I., Milbrandt, J. & Baloh, R. H. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci 30, 4232-4240 (2010).
Zimon, M., Baets, J., Fabrizi, G. M., Jaakkola, E., Kabzinska, D., Pilch, J., Schindler, A. B., Cornblath, D. R., Fischbeck, K. H., Auer-Grumbach, M., Guelly, C., Huber, N., De Vriendt, E., Timmerman, V., Suter, U., Hausmanowa-Petrusewicz, I., Niemann, A., Kochanski, A., De Jonghe, P. & Jordanova, A. Dominant GDAP1 mutations cause predominantly mild CMT phenotypes. Neurology 77, 540-548 (2011).
Birouk, N., Azzedine, H., Dubourg, O., Muriel, M. P., Benomar, A., Hamadouche, T., Maisonobe, T., Ouazzani, R., Brice, A., Yahyaoui, M., Chkili, T. & Le Guern, E. Phenotypical features of a Moroccan family with autosomal recessive Charcot-Marie-Tooth disease associated with the S194X mutation in the GDAP1 gene. Arch Neurol 60, 598-604 (2003).
Chung, K. W., Kim, S. M., Sunwoo, I. N., Cho, S. Y., Hwang, S. J., Kim, J., Kang, S. H., Park, K. D., Choi, K. G., Choi, I. S. & Choi, B. O. A novel GDAP1 Q218E mutation in autosomal dominant Charcot-Marie-Tooth disease. J Hum Genet 53, 360-364 (2008).
Alexander, C., Votruba, M., Pesch, U. E., Thiselton, D. L., Mayer, S., Moore, A., Rodriguez, M., Kellner, U., Leo-Kottler, B., Auburger, G., Bhattacharya, S. S. & Wissinger, B. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26, 211-215 (2000).
Yu-Wai-Man, P., Griffiths, P. G., Gorman, G. S., Lourenco, C. M., Wright, A. F., Auer-Grumbach, M., Toscano, A., Musumeci, O., Valentino, M. L., Caporali, L., Lamperti, C., Tallaksen, C. M., Duffey, P., Miller, J., Whittaker, R. G., Baker, M. R., Jackson, M. J., Clarke, M. P., Dhillon, B., Czermin, B., Stewart, J. D., Hudson, G., Reynier, P., Bonneau, D., Marques, W., Jr., Lenaers, G., McFarland, R., Taylor, R. W., Turnbull, D. M., Votruba, M., Zeviani, M., Carelli, V., Bindoff, L. A., Horvath, R., Amati-Bonneau, P. & Chinnery, P. F. Multi-system neurological disease is common in patients with OPA1 mutations. Brain 133, 771-786 (2010).
Horga, A., Bugiardini, E., Manole, A., Bremner, F., Jaunmuktane, Z., Dankwa, L., Rebelo, A. P., Woodward, C. E., Hargreaves, I. P., Cortese, A., Pittman, A. M., Brandner, S., Polke, J. M., Pitceathly, R. D. S., Zuchner, S., Hanna, M. G., Scherer, S. S., Houlden, H. & Reilly, M. M. Autosomal dominant optic atrophy and cataract "plus" phenotype including axonal neuropathy. Neurol Genet 5, e322 (2019).
Pareyson, D., Piscosquito, G., Moroni, I., Salsano, E. & Zeviani, M. Peripheral neuropathy in mitochondrial disorders. Lancet Neurol 12, 1011-1024 (2013).
Cassereau, J., Codron, P. & Funalot, B. Inherited peripheral neuropathies due to mitochondrial disorders. Rev Neurol (Paris) 170, 366-374 (2014).
Stuppia, G., Rizzo, F., Riboldi, G., Del Bo, R., Nizzardo, M., Simone, C., Comi, G. P., Bresolin, N. & Corti, S. MFN2-related neuropathies: Clinical features, molecular pathogenesis and therapeutic perspectives. J Neurol Sci 356, 7-18 (2015).
Detmer, S. A. & Chan, D. C. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol 176, 405-414 (2007).
Dorn, G. W., 2nd. Mitofusin 2 Dysfunction and Disease in Mice and Men. Front Physiol 11, 782 (2020).
Baloh, R. H., Schmidt, R. E., Pestronk, A. & Milbrandt, J. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J Neurosci 27, 422-430 (2007).
Misko, A. L., Sasaki, Y., Tuck, E., Milbrandt, J. & Baloh, R. H. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J Neurosci 32, 4145-4155 (2012).
Rocha, A. G., Franco, A., Krezel, A. M., Rumsey, J. M., Alberti, J. M., Knight, W. C., Biris, N., Zacharioudakis, E., Janetka, J. W., Baloh, R. H., Kitsis, R. N., Mochly-Rosen, D., Townsend, R. R., Gavathiotis, E. & Dorn, G. W., 2nd. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 360, 336–341 (2018).
Larrea, D., Pera, M., Gonnelli, A., Quintana-Cabrera, R., Akman, H. O., Guardia-Laguarta, C., Velasco, K. R., Area-Gomez, E., Dal Bello, F., De Stefani, D., Horvath, R., Shy, M. E., Schon, E. A. & Giacomello, M. MFN2 mutations in Charcot-Marie-Tooth disease alter mitochondria-associated ER membrane function but do not impair bioenergetics. Hum Mol Genet 28, 1782-1800 (2019).
Antonellis, A., Ellsworth, R. E., Sambuughin, N., Puls, I., Abel, A., Lee-Lin, S. Q., Jordanova, A., Kremensky, I., Christodoulou, K., Middleton, L. T., Sivakumar, K., Ionasescu, V., Funalot, B., Vance, J. M., Goldfarb, L. G., Fischbeck, K. H. & Green, E. D. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet 72, 1293-1299 (2003).
Jordanova, A., Irobi, J., Thomas, F. P., Van Dijck, P., Meerschaert, K., Dewil, M., Dierick, I., Jacobs, A., De Vriendt, E., Guergueltcheva, V., Rao, C. V., Tournev, I., Gondim, F. A., D'Hooghe, M., Van Gerwen, V., Callaerts, P., Van Den Bosch, L., Timmermans, J. P., Robberecht, W., Gettemans, J., Thevelein, J. M., De Jonghe, P., Kremensky, I. & Timmerman, V. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat Genet 38, 197-202 (2006).
Safka Brozkova, D., Deconinck, T., Griffin, L. B., Ferbert, A., Haberlova, J., Mazanec, R., Lassuthova, P., Roth, C., Pilunthanakul, T., Rautenstrauss, B., Janecke, A. R., Zavadakova, P., Chrast, R., Rivolta, C., Zuchner, S., Antonellis, A., Beg, A. A., De Jonghe, P., Senderek, J., Seeman, P. & Baets, J. Loss of function mutations in HARS cause a spectrum of inherited peripheral neuropathies. Brain 138, 2161–2172 (2015).
Tsai, P. C., Soong, B. W., Mademan, I., Huang, Y. H., Liu, C. R., Hsiao, C. T., Wu, H. T., Liu, T. T., Liu, Y. T., Tseng, Y. T., Lin, K. P., Yang, U. C., Chung, K. W., Choi, B. O., Nicholson, G. A., Kennerson, M. L., Chan, C. C., De Jonghe, P., Cheng, T. H., Liao, Y. C., Zuchner, S., Baets, J. & Lee, Y. C. A recurrent WARS mutation is a novel cause of autosomal dominant distal hereditary motor neuropathy. Brain 140, 1252-1266 (2017).
McLaughlin, H. M., Sakaguchi, R., Liu, C., Igarashi, T., Pehlivan, D., Chu, K., Iyer, R., Cruz, P., Cherukuri, P. F., Hansen, N. F., Mullikin, J. C., Program, N. C. S., Biesecker, L. G., Wilson, T. E., Ionasescu, V., Nicholson, G., Searby, C., Talbot, K., Vance, J. M., Zuchner, S., Szigeti, K., Lupski, J. R., Hou, Y. M., Green, E. D. & Antonellis, A. Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am J Hum Genet 87, 560-566 (2010).
Gonzalez, M., McLaughlin, H., Houlden, H., Guo, M., Yo-Tsen, L., Hadjivassilious, M., Speziani, F., Yang, X. L., Antonellis, A., Reilly, M. M., Zuchner, S. & Inherited Neuropathy, C. Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J Neurol Neurosurg Psychiatry 84, 1247–1249 (2013).
Boczonadi, V., Jennings, M. J. & Horvath, R. The role of tRNA synthetases in neurological and neuromuscular disorders. FEBS Lett 592, 703-717 (2018).
McLaughlin, H. M., Sakaguchi, R., Giblin, W., Program, N. C. S., Wilson, T. E., Biesecker, L., Lupski, J. R., Talbot, K., Vance, J. M., Zuchner, S., Lee, Y. C., Kennerson, M., Hou, Y. M., Nicholson, G. & Antonellis, A. A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with Charcot-Marie-Tooth disease type 2N (CMT2N). Hum Mutat 33, 244-253 (2012).
Stum, M., McLaughlin, H. M., Kleinbrink, E. L., Miers, K. E., Ackerman, S. L., Seburn, K. L., Antonellis, A. & Burgess, R. W. An assessment of mechanisms underlying peripheral axonal degeneration caused by aminoacyl-tRNA synthetase mutations. Mol Cell Neurosci 46, 432-443 (2011).
Nangle, L. A., Zhang, W., Xie, W., Yang, X. L. & Schimmel, P. Charcot-Marie-Tooth disease-associated mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect. Proc Natl Acad Sci U S A 104, 11239-11244 (2007).
Froelich, C. A. & First, E. A. Dominant Intermediate Charcot-Marie-Tooth disorder is not due to a catalytic defect in tyrosyl-tRNA synthetase. Biochemistry 50, 7132-7145 (2011).
Xie, W., Nangle, L. A., Zhang, W., Schimmel, P. & Yang, X. L. Long-range structural effects of a Charcot-Marie-Tooth disease-causing mutation in human glycyl-tRNA synthetase. Proc Natl Acad Sci U S A 104, 9976-9981 (2007).
He, W., Bai, G., Zhou, H., Wei, N., White, N. M., Lauer, J., Liu, H., Shi, Y., Dumitru, C. D., Lettieri, K., Shubayev, V., Jordanova, A., Guergueltcheva, V., Griffin, P. R., Burgess, R. W., Pfaff, S. L. & Yang, X. L. CMT2D neuropathy is linked to the neomorphic binding activity of glycyl-tRNA synthetase. Nature 526, 710-714 (2015).
Niehues, S., Bussmann, J., Steffes, G., Erdmann, I., Kohrer, C., Sun, L., Wagner, M., Schafer, K., Wang, G., Koerdt, S. N., Stum, M., Jaiswal, S., RajBhandary, U. L., Thomas, U., Aberle, H., Burgess, R. W., Yang, X. L., Dieterich, D. & Storkebaum, E. Impaired protein translation in Drosophila models for Charcot-Marie-Tooth neuropathy caused by mutant tRNA synthetases. Nat Commun 6, 7520 (2015).
Wu, Y., Whiteus, C., Xu, C. S., Hayworth, K. J., Weinberg, R. J., Hess, H. F. & De Camilli, P. Contacts between the endoplasmic reticulum and other membranes in neurons. Proc Natl Acad Sci U S A 114, E4859-E4867 (2017).
Valm, A. M., Cohen, S., Legant, W. R., Melunis, J., Hershberg, U., Wait, E., Cohen, A. R., Davidson, M. W., Betzig, E. & Lippincott-Schwartz, J. Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546, 162-167 (2017).
Phillips, M. J. & Voeltz, G. K. Structure and function of ER membrane contact sites with other organelles. Nat Rev Mol Cell Biol 17, 69-82 (2016).
de Brito, O. M. & Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605-610 (2008).
Bernard-Marissal, N., van Hameren, G., Juneja, M., Pellegrino, C., Louhivuori, L., Bartesaghi, L., Rochat, C., El Mansour, O., Medard, J. J., Croisier, M., Maclachlan, C., Poirot, O., Uhlen, P., Timmerman, V., Tricaud, N., Schneider, B. L. & Chrast, R. Altered interplay between endoplasmic reticulum and mitochondria in Charcot-Marie-Tooth type 2A neuropathy. Proc Natl Acad Sci U S A 116, 2328-2337 (2019).
Wong, Y. C., Ysselstein, D. & Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554, 382-386 (2018).
Bucci, C., Thomsen, P., Nicoziani, P., McCarthy, J. & van Deurs, B. Rab7: a key to lysosome biogenesis. Mol Biol Cell 11, 467-480 (2000).
Vitelli, R., Santillo, M., Lattero, D., Chiariello, M., Bifulco, M., Bruni, C. B. & Bucci, C. Role of the small GTPase Rab7 in the late endocytic pathway. J Biol Chem 272, 4391-4397 (1997).
Jordens, I., Fernandez-Borja, M., Marsman, M., Dusseljee, S., Janssen, L., Calafat, J., Janssen, H., Wubbolts, R. & Neefjes, J. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr Biol 11, 1680-1685 (2001).
Deinhardt, K., Salinas, S., Verastegui, C., Watson, R., Worth, D., Hanrahan, S., Bucci, C. & Schiavo, G. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron 52, 293-305 (2006).
McCray, B. A., Skordalakes, E. & Taylor, J. P. Disease mutations in Rab7 result in unregulated nucleotide exchange and inappropriate activation. Hum Mol Genet 19, 1033-1047 (2010).
Cioni, J. M., Lin, J. Q., Holtermann, A. V., Koppers, M., Jakobs, M. A. H., Azizi, A., Turner-Bridger, B., Shigeoka, T., Franze, K., Harris, W. A. & Holt, C. E. Late Endosomes Act as mRNA Translation Platforms and Sustain Mitochondria in Axons. Cell 176, 56–72 e15 (2019).
Timmerman, V., Clowes, V. E. & Reid, E. Overlapping molecular pathological themes link Charcot-Marie-Tooth neuropathies and hereditary spastic paraplegias. Exp Neurol 246, 14-25 (2013).
Hubner, C. A. & Kurth, I. Membrane-shaping disorders: a common pathway in axon degeneration. Brain 137, 3109-3121 (2014).
Ozturk, Z., O'Kane, C. J. & Perez-Moreno, J. J. Axonal Endoplasmic Reticulum Dynamics and Its Roles in Neurodegeneration. Front Neurosci 14, 48 (2020).
Boutry, M., Morais, S. & Stevanin, G. Update on the Genetics of Spastic Paraplegias. Curr Neurol Neurosci Rep 19, 18 (2019).
Thelen, M. P. & Kye, M. J. The Role of RNA Binding Proteins for Local mRNA Translation: Implications in Neurological Disorders. Front Mol Biosci 6, 161 (2019).
Bobylev, I., Joshi, A. R., Barham, M., Ritter, C., Neiss, W. F., Hoke, A. & Lehmann, H. C. Paclitaxel inhibits mRNA transport in axons. Neurobiol Dis 82, 321-331 (2015).
Juneja, M., Burns, J., Saporta, M. A. & Timmerman, V. Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development. J Neurol Neurosurg Psychiatry 90, 58-67 (2019).
Sereda, M., Griffiths, I., Puhlhofer, A., Stewart, H., Rossner, M. J., Zimmerman, F., Magyar, J. P., Schneider, A., Hund, E., Meinck, H. M., Suter, U. & Nave, K. A. A transgenic rat model of Charcot-Marie-Tooth disease. Neuron 16, 1049-1060 (1996).
De Gioia, R., Citterio, G., Abati, E., Nizzardo, M., Bresolin, N., Comi, G. P., Corti, S. & Rizzo, F. Animal Models of CMT2A: State-of-art and Therapeutic Implications. Mol Neurobiol 57, 5121-5129 (2020).
Stelten, B. M. L. et al. Long-term treatment effect in cerebrotendinous xanthomatosis depends on age at treatment start. Neurology 92, e83-e95 (2019).
Kuriyama, M., Tokimura, Y., Fujiyama, J., Utatsu, Y. & Osame, M. Treatment of cerebrotendinous xanthomatosis: effects of chenodeoxycholic acid, pravastatin, and combined use. J Neurol Sci 125, 22-28 (1994).
Andersson, H. C., Marble, M. & Shapira, E. Long-term outcome in treated combined methylmalonic acidemia and homocystinemia. Genet Med 1, 146-150 (1999).
Enns, G. M. et al. Progressive neurological deterioration and MRI changes in cblC methylmalonic acidaemia treated with hydroxocobalamin. J Inherit Metab Dis 22, 599-607 (1999).
Bartholomew, D. W. et al. Therapeutic approaches to cobalamin-C methylmalonic acidemia and homocystinuria. J Pediatr 112, 32-39 (1988).
Uziel, G., Garavaglia, B., Ciceri, E., Moroni, I. & Rimoldi, M. Riboflavin-responsive glutaric aciduria type II presenting as a leukodystrophy. Pediatr Neurol 13, 333-335 (1995).
Plantone, D., Pardini, M. & Rinaldi, G. Riboflavin in Neurological Diseases: A Narrative Review. Clin Drug Investig 41, 513-527 (2021).
De Biase, I. et al. Diagnosis, Treatment, and Clinical Outcome of Patients with Mitochondrial Trifunctional Protein/Long-Chain 3-Hydroxy Acyl-CoA Dehydrogenase Deficiency. JIMD Rep 31, 63-71 (2017).
Chelban, V. et al. PDXK mutations cause polyneuropathy responsive to pyridoxal 5'-phosphate supplementation. Ann Neurol 86, 225-240 (2019).
Ortiz, A. et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol Genet Metab 123, 416-427 (2018).
Germain, D. P. et al. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin Genet 96, 107-117 (2019).
Gibberd, F. B., Billimoria, J. D., Page, N. G. & Retsas, S. Heredopathia atactica polyneuritiformis (refsum's disease) treated by diet and plasma-exchange. Lancet 1, 575-578 (1979).
Moser, H. W. et al. Therapeutic trial of plasmapheresis in Refsum disease and in Fabry disease. Birth Defects Orig Artic Ser 16, 491-497 (1980).
Van Calcar, S. C. et al. Nutrition management guideline for very-long chain acyl-CoA dehydrogenase deficiency (VLCAD): An evidence- and consensus-based approach. Mol Genet Metab 131, 23-37 (2020).
Marce-Grau, A., Marti-Sanchez, L., Baide-Mairena, H., Ortigoza-Escobar, J. D. & Perez-Duenas, B. Genetic defects of thiamine transport and metabolism: A review of clinical phenotypes, genetics, and functional studies. J Inherit Metab Dis 42, 581-597 (2019).
Will, E. J. & Bijvoet, O. L. Primary oxalosis: clinical and biochemical response to high-dose pyridoxine therapy. Metabolism 28, 542-548 (1979).17Albers, J. W. & Fink, J. K. Porphyric neuropathy. Muscle Nerve 30, 410-422 (2004).
Albers, J. W. & Fink, J. K. Porphyric neuropathy. Muscle Nerve 30, 410-422 (2004).
Balwani, M. et al. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N Engl J Med 382, 2289-2301 (2020).
Lindstedt, S., Holme, E., Lock, E. A., Hjalmarson, O. & Strandvik, B. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet 340, 813-817 (1992).
Burnett, J. R., Hooper, A. J. & Hegele, R. A. in GeneReviews((R)) (eds M. P. Adam et al.) (1993).
Martinelli, D. et al. MEDNIK syndrome: a novel defect of copper metabolism treatable by zinc acetate therapy. Brain 136, 872-881 (2013).
Hashizume, A., Fischbeck, K. H., Pennuto, M., Fratta, P. & Katsuno, M. Disease mechanism, biomarker and therapeutics for spinal and bulbar muscular atrophy (SBMA). J Neurol Neurosurg Psychiatry 91, 1085-1091 (2020).
Dawkins, J. L., Hulme, D. J., Brahmbhatt, S. B., Auer-Grumbach, M. & Nicholson, G. A. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet 27, 309-312 (2001).
Bejaoui, K., Wu, C., Scheffler, M. D., Haan, G., Ashby, P., Wu, L., de Jong, P. & Brown, R. H., Jr. SPTLC1 is mutated in hereditary sensory neuropathy, type 1. Nat Genet 27, 261-262 (2001).
Houlden, H., King, R., Blake, J., Groves, M., Love, S., Woodward, C., Hammans, S., Nicoll, J., Lennox, G., O'Donovan, D. G., Gabriel, C., Thomas, P. K. & Reilly, M. M. Clinical, pathological and genetic characterization of hereditary sensory and autonomic neuropathy type 1 (HSAN I). Brain 129, 411-425 (2006).
Penno, A., Reilly, M. M., Houlden, H., Laura, M., Rentsch, K., Niederkofler, V., Stoeckli, E. T., Nicholson, G., Eichler, F., Brown, R. H., Jr., von Eckardstein, A. & Hornemann, T. Hereditary sensory neuropathy type 1 is caused by the accumulation of two neurotoxic sphingolipids. J Biol Chem 285, 11178-11187 (2010).
Garofalo, K., Penno, A., Schmidt, B. P., Lee, H. J., Frosch, M. P., von Eckardstein, A., Brown, R. H., Hornemann, T. & Eichler, F. S. Oral L-serine supplementation reduces production of neurotoxic deoxysphingolipids in mice and humans with hereditary sensory autonomic neuropathy type 1. J Clin Invest 121, 4735-4745 (2011).
Fridman, V., Suriyanarayanan, S., Novak, P., David, W., Macklin, E. A., McKenna-Yasek, D., Walsh, K., Aziz-Bose, R., Oaklander, A. L., Brown, R., Hornemann, T. & Eichler, F. Randomized trial of l-serine in patients with hereditary sensory and autonomic neuropathy type 1. Neurology 92, e359-e370 (2019).
Cortese, A., Zhu, Y., Rebelo, A. P., Negri, S., Courel, S., Abreu, L., Bacon, C. J., Bai, Y., Bis-Brewer, D. M., Bugiardini, E., Buglo, E., Danzi, M. C., Feely, S. M. E., Athanasiou-Fragkouli, A., Haridy, N. A., Inherited Neuropathy, C., Isasi, R., Khan, A., Laura, M., Magri, S., Pipis, M., Pisciotta, C., Powell, E., Rossor, A. M., Saveri, P., Sowden, J. E., Tozza, S., Vandrovcova, J., Dallman, J., Grignani, E., Marchioni, E., Scherer, S. S., Tang, B., Lin, Z., Al-Ajmi, A., Schule, R., Synofzik, M., Maisonobe, T., Stojkovic, T., Auer-Grumbach, M., Abdelhamed, M. A., Hamed, S. A., Zhang, R., Manganelli, F., Santoro, L., Taroni, F., Pareyson, D., Houlden, H., Herrmann, D. N., Reilly, M. M., Shy, M. E., Zhai, R. G. & Zuchner, S. Biallelic mutations in SORD cause a common and potentially treatable hereditary neuropathy with implications for diabetes. Nat Genet 52, 473–481 (2020).
Chalk, C., Benstead, T. J. & Moore, F. Aldose reductase inhibitors for the treatment of diabetic polyneuropathy. Cochrane Database Syst Rev, CD004572 (2007).
El Fissi, N., Rojo, M., Aouane, A., Karatas, E., Poliacikova, G., David, C., Royet, J. & Rival, T. Mitofusin gain and loss of function drive pathogenesis in Drosophila models of CMT2A neuropathy. EMBO Rep 19 (2018).
Franco, A., Kitsis, R. N., Fleischer, J. A., Gavathiotis, E., Kornfeld, O. S., Gong, G., Biris, N., Benz, A., Qvit, N., Donnelly, S. K., Chen, Y., Mennerick, S., Hodgson, L., Mochly-Rosen, D. & Dorn, G. W., II. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 540, 74-79 (2016).
Dang, X., Zhang, L., Franco, A., Li, J., Rocha, A. G., Devanathan, S., Dolle, R. E., Bernstein, P. R. & Dorn, G. W., 2nd. Discovery of 6-Phenylhexanamide Derivatives as Potent Stereoselective Mitofusin Activators for the Treatment of Mitochondrial Diseases. J Med Chem 63, 7033–7051 (2020).
Franco, A., Dang, X., Walton, E. K., Ho, J. N., Zablocka, B., Ly, C., Miller, T. M., Baloh, R. H., Shy, M. E., Yoo, A. S. & Dorn, G. W., 2nd. Burst mitofusin activation reverses neuromuscular dysfunction in murine CMT2A. Elife 9 (2020).
McCray, B. A., Diehl, E., Sullivan, J. M., Aisenberg, W. H., Zaccor, N. W., Lau, A. R., Rich, D. J., Goretzki, B., Hellmich, U. A., Lloyd, T. E. & Sumner, C. J. Neuropathy-causing TRPV4 mutations disrupt TRPV4-RhoA interactions and impair neurite extension. Nat Commun 12, 1444 (2021).
Woolums, B. M., McCray, B. A., Sung, H., Tabuchi, M., Sullivan, J. M., Ruppell, K. T., Yang, Y., Mamah, C., Aisenberg, W. H., Saavedra-Rivera, P. C., Larin, B. S., Lau, A. R., Robinson, D. N., Xiang, Y., Wu, M. N., Sumner, C. J. & Lloyd, T. E. TRPV4 disrupts mitochondrial transport and causes axonal degeneration via a CaMKII-dependent elevation of intracellular Ca(2). Nat Commun 11, 2679 (2020).
McCray, B. A., Schindler, A., Hoover-Fong, J. E. & Sumner, C. J. in GeneReviews((R)) (eds M. P. Adam et al.) (1993).
Goyal, N., Skrdla, P., Schroyer, R., Kumar, S., Fernando, D., Oughton, A., Norton, N., Sprecher, D. L. & Cheriyan, J. Clinical Pharmacokinetics, Safety, and Tolerability of a Novel, First-in-Class TRPV4 Ion Channel Inhibitor, GSK2798745, in Healthy and Heart Failure Subjects. Am J Cardiovasc Drugs 19, 335-342 (2019).
Osterloh, J. M., Yang, J., Rooney, T. M., Fox, A. N., Adalbert, R., Powell, E. H., Sheehan, A. E., Avery, M. A., Hackett, R., Logan, M. A., MacDonald, J. M., Ziegenfuss, J. S., Milde, S., Hou, Y. J., Nathan, C., Ding, A., Brown, R. H., Jr., Conforti, L., Coleman, M., Tessier-Lavigne, M., Zuchner, S. & Freeman, M. R. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481-484 (2012).
Gerdts, J., Brace, E. J., Sasaki, Y., DiAntonio, A. & Milbrandt, J. SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science 348, 453-457 (2015).
Reed, N. A., Cai, D., Blasius, T. L., Jih, G. T., Meyhofer, E., Gaertig, J. & Verhey, K. J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr Biol 16, 2166-2172 (2006).
d'Ydewalle, C., Krishnan, J., Chiheb, D. M., Van Damme, P., Irobi, J., Kozikowski, A. P., Vanden Berghe, P., Timmerman, V., Robberecht, W. & Van Den Bosch, L. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat Med 17, 968-974 (2011).
Benoy, V., Van Helleputte, L., Prior, R., d'Ydewalle, C., Haeck, W., Geens, N., Scheveneels, W., Schevenels, B., Cader, M. Z., Talbot, K., Kozikowski, A. P., Vanden Berghe, P., Van Damme, P., Robberecht, W. & Van Den Bosch, L. HDAC6 is a therapeutic target in mutant GARS-induced Charcot-Marie-Tooth disease. Brain 141, 673-687 (2018).
Kawaguchi, Y., Kovacs, J. J., McLaurin, A., Vance, J. M., Ito, A. & Yao, T. P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727-738 (2003).
Rossaert, E. & Van Den Bosch, L. HDAC6 inhibitors: Translating genetic and molecular insights into a therapy for axonal CMT. Brain Res 1733, 146692 (2020).
Picci, C., Wong, V. S. C., Costa, C. J., McKinnon, M. C., Goldberg, D. C., Swift, M., Alam, N. M., Prusky, G. T., Shen, S., Kozikowski, A. P., Willis, D. E. & Langley, B. HDAC6 inhibition promotes alpha-tubulin acetylation and ameliorates CMT2A peripheral neuropathy in mice. Exp Neurol 328, 113281 (2020).
Pandey, U. B., Nie, Z., Batlevi, Y., McCray, B. A., Ritson, G. P., Nedelsky, N. B., Schwartz, S. L., DiProspero, N. A., Knight, M. A., Schuldiner, O., Padmanabhan, R., Hild, M., Berry, D. L., Garza, D., Hubbert, C. C., Yao, T. P., Baehrecke, E. H. & Taylor, J. P. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859-863 (2007).
Mendell, J. R., Al-Zaidy, S., Shell, R., Arnold, W. D., Rodino-Klapac, L. R., Prior, T. W., Lowes, L., Alfano, L., Berry, K., Church, K., Kissel, J. T., Nagendran, S., L'Italien, J., Sproule, D. M., Wells, C., Cardenas, J. A., Heitzer, M. D., Kaspar, A., Corcoran, S., Braun, L., Likhite, S., Miranda, C., Meyer, K., Foust, K. D., Burghes, A. H. M. & Kaspar, B. K. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N Engl J Med 377, 1713-1722 (2017).
Hardcastle, N., Boulis, N. M. & Federici, T. AAV gene delivery to the spinal cord: serotypes, methods, candidate diseases, and clinical trials. Expert Opin Biol Ther 18, 293-307 (2018).
Morelli, K. H., Hatton, C. L., Harper, S. Q. & Burgess, R. W. Gene therapies for axonal neuropathies: Available strategies, successes to date, and what to target next. Brain Res 1732, 146683 (2020).
Zhou, Y., Carmona, S., Muhammad, A., Bell, S., Landeros, J., Vazquez, M., Ho, R., Franco, A., Lu, B., Dorn, G. W., 2nd, Wang, S., Lutz, C. M. & Baloh, R. H. Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J Clin Invest 129, 1756-1771 (2019).
Bomont, P., Cavalier, L., Blondeau, F., Ben Hamida, C., Belal, S., Tazir, M., Demir, E., Topaloglu, H., Korinthenberg, R., Tuysuz, B., Landrieu, P., Hentati, F. & Koenig, M. The gene encoding gigaxonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet 26, 370–374 (2000).
Bailey, R. M., Armao, D., Nagabhushan Kalburgi, S. & Gray, S. J. Development of Intrathecal AAV9 Gene Therapy for Giant Axonal Neuropathy. Mol Ther Methods Clin Dev 9, 160–171 (2018).
Morelli, K. H., Griffin, L. B., Pyne, N. K., Wallace, L. M., Fowler, A. M., Oprescu, S. N., Takase, R., Wei, N., Meyer-Schuman, R., Mellacheruvu, D., Kitzman, J. O., Kocen, S. G., Hines, T. J., Spaulding, E. L., Lupski, J. R., Nesvizhskii, A., Mancias, P., Butler, I. J., Yang, X. L., Hou, Y. M., Antonellis, A., Harper, S. Q. & Burgess, R. W. Allele-specific RNA interference prevents neuropathy in Charcot-Marie-Tooth disease type 2D mouse models. J Clin Invest 129, 5568-5583 (2019).
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Funding
This work was supported by NIH/NINDS K08 NS102509 (B.A.M.), the American Academy of Neurology Neuroscience Research Training Fellowship 0078966/129168 (B.A.M.), the Judy Seltzer Levenson Memorial Fund for CMT Research, the INC (U54NS065712), which is a part of the NCATS Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), NCATS, funded through a collaboration between NCATS and the NINDS (S.S.S). We thank Natalia Nedelsky for graphical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing Interests
S.S.S. has been compensated for serving on the scientific advisory boards of Mitochondria in Motion, which is developing drugs to treat CMT2A, and Disarm Therapeutics, which is developing drugs to block SARM1 as a treatment of peripheral neuropathies.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
McCray, B.A., Scherer, S.S. Axonal Charcot-Marie-Tooth Disease: from Common Pathogenic Mechanisms to Emerging Treatment Opportunities. Neurotherapeutics 18, 2269–2285 (2021). https://doi.org/10.1007/s13311-021-01099-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-021-01099-2