
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disorder worldwide and is characterized in part by the degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNc). The main pathological hallmark of PD is the intraneuronal accumulation of misfolded α-synuclein (α-syn) aggregates. Mutations in the SNCA gene (encoding α-syn) and variations in its copy number are associated with some forms of familial PD. In the present study, T-006, a new tetramethylpyrazine (TMP) derivative with recently reported anti-Alzheimer activity, is shown to significantly promote α-syn degradation in a cellular PD model. Moreover, we illustrate that T-006 inhibits the accumulation of both Triton-soluble and -insoluble forms of α-syn and protects against α-syn-induced neurotoxicity in A53T-α-syn transgenic mice. The mechanism of action of T-006 was verified by evaluation of a potential protein degradation pathway. We found that T-006 promotes α-syn degradation in a proteasome-dependent and autophagy-independent manner. We further confirmed that T-006 enhances proteasome activity by upregulating 20S proteasome subunit β5i (LMP7) protein expression. A functional study revealed that T-006 activates the PKA/Akt/mTOR/p70S6K pathway to trigger LMP7 expression and enhance chymotrypsin-like proteasomal activity. These findings indicate that T-006 is a potent proteasome activator and a potential therapeutic agent for the prevention and treatment of PD and related diseases.
Similar content being viewed by others

Introduction
Parkinson’s disease (PD) is the second most prevalent neurodegenerative movement disorder, after Alzheimer’s disease, and affects up to 1% of the global population aged over 60 years [1, 2]. The pathology of PD is characterized by the loss of dopaminergic neurons in the substantia nigra pars compacta (SNc), resulting in dopamine deficiency in the striatum and by the presence of intracellular insoluble inclusions termed Lewy bodies [3,4,5].
Protein accumulation is one of the two well-recognized pathogenic processes behind the development of neurodegenerative diseases such as PD [6]. Protein aggregation can be detrimental to cellular function and tissue homeostasis. Some neurodegenerative disease proteins, such as α-synuclein (α-syn), accumulate as insoluble protein aggregates, causing neurotoxicity [7]. Notably, missense mutations, variation in copy number, and increased expression of α-syn are the main factors associated with PD [8, 9]. Missense mutations in SNCA, the gene encoding α-syn, resulting in A53T, A30P, and E46K substitutions alter the physiological and pathogenic properties of α-syn by promoting its aggregation, leading to the development of Lewy body pathology [8, 10]. Therefore, strategies to degrade α-syn could be effective approaches for treating PD.
Cells employ several mechanisms to defend against misfolded, aggregated and damaged proteins [11]. The two main cellular defensive mechanisms of the proteostasis network are proteasomal degradation and autophagy/lysosome degradation [12, 13]. When misfolded or damaged proteins accumulate (e.g., due to a shortage of chaperones) and impair cellular function, they can be scavenged and degraded through the ubiquitin-proteasome system (UPS) or the autophagy-lysosome pathway (ALP) [14, 15]. A loss of function in the two types of protein degradation machinery causes aggregation-mediated proteotoxicity diseases, such as PD and age-related diseases [16]. Recently, many findings have suggested that the levels of wild-type (WT) and mutated α-syn are modulated by the UPS and ALP. For instance, previous reports have shown that the UPS decreases α-syn accumulation under conditions of an increased endogenous protein burden, while the ALP was responsible for the clearance of excessive α-syn under pathologic conditions in SNCA Tg mouse models [17]. Several small molecules have been documented in the literature to promote the clearance of pathological α-syn through the UPS or ALP [18,19,20].
T-006, a new derivative of tetramethylpyrazine (TMP), a Chinese medicinal component, was previously identified as a promising anti-Alzheimer's compound [21]. It has been reported that T-006 prevents glutamate-induced excitotoxicity in CGNs by antagonizing NMDA receptors and regulating the PI3K/AKT pathway [22]. However, the potential effect of T-006 on other neurodegenerative diseases such as PD has not yet been investigated. Herein, further functional and pathological studies are carried out.
In the present study, T-006 is shown to protect against A53T-α-syn-driven neurotoxicity. T-006 selectively degrades WT and mutated α-syn through increasing LMP7 gene expression and correspondingly enhancing its chymotrypsin-like proteasomal activity. Importantly, T-006 is shown to induce A53T-α-syn degradation by stimulating the PKA/AKT/mTOR/p70S6K pathway. We envisage that T-006 could be employed as a potent proteasome-enhancing molecule to promote A53T-α-syn degradation for the treatment of PD.
Materials and methods
Reagents
F-12K medium, fetal bovine serum (FBS), horse serum (HS), penicillin-streptomycin (PS), phosphate-buffered saline (PBS) and trypsin-EDTA were purchased from Life Technologies (Grand Island, NY, USA). A lactate dehydrogenase (LDH) kit and cocktail were purchased from Roche Applied Science (Indianapolis, IN, USA). Anti-FLAG® M2 (F1804) antibody, chloroquine (CQ), dimethyl sulfoxide (DMSO), doxycycline (Dox), paraformaldehyde (PFA), rapamycin (Rap) and thiazolyl bluet tetrazolium bromide were purchased from Sigma-Aldrich (St. Louis, MO, USA). H-89 and MG132 were purchased from Selleck Chemicals (Shanghai, China). Enhanced chemiluminescence (ECL) solution was obtained from Thermo Scientific (Rockford, IL, USA). Antibodies against α-syn, p-PKA, PKA, p-Akt, Akt, p-mTOR, mTOR, p-p70S6K, p70S6K, LC3A/B, SQSTM1/p62, LMP7, β-actin and HRP-conjugated anti-rabbit IgG were purchased from Cell Signaling Technology (Boston, MA, USA). TRIzol reagent and Dynabeads® Protein G Immunoprecipitation Kit were purchased from life technologies (Eugene, OR, USA). SYBR® Premix Ex Taq™ II kit was purchased from TaKaRa (Dalian, China). Proteasome-Glo™ 3-Substrate Cell-Based Assay System was purchased from Promega (Madison, WI, USA). All other chemicals of analytical grade were purchased from local sources.
Cell culture and treatments
To investigate the potential ability of T-006 in PD, the previously characterized inducible PC12 stable cell lines overexpressing α-syn (WT, A30P, A53T) [23] were maintained in 150 μg/ml hygromycin B in DMEM supplemented with 10% HS, 5% FBS, 100 units/mL penicillin/streptomycin, and 50 μg/ml G418 in a humidified atmosphere of 5% CO2 and 95% air at 37°C. The inducible PC12 cells were induced the expression of α-syn by treating with 1 μg/ml Dox for 24 h. Then cells were washed twice with medium to remove Dox and subsequently incubated in media containing either T-006 (30 μM), CQ (30 μM), Rap (0.2 μM), MG132 (0.1 μΜ) or DMSO for 24 h. T-006 was stocked in DMSO at 30 mM and added directly into the culture media to a final working concentration of 0.1% (v/v) DMSO.
Cell viability and cytotoxicity analysis
8 × 103 inducible PC12/A53T-α-syn cells were seeded in 96-well plates overnight and treated with different concentrations of T-006 ranging from 3 μM to 100 μM for another 24 h. The supernatant was removed and detected by LDH cytotoxicity analysis, meanwhile the remainder was analyzed by MTT cell viability assays. Absorbance was measured at 490 nm for LDH experiments and 570 nm for MTT assays using a SpectraMax M5 (Wallace, Netherlands).
Proteasomal activity assay
Proteasome-Glo™ 3-Substrate Cell-Based Assay System (Promega) was used to detect the enzymatic activities of the chymotrypsin-, trypsin- and caspase-like proteasome. In brief, aforementioned treated inducible PC12/A53T-α-syn cells were collected and cell lysates prepared by Proteasome-Glo™ Cell-Based Reagent and detected by the corresponding substrates provided in the system. The luminescence readout of each proteasomal activity in three independent experiments was recorded using the SpectraMax M5 equipment.
Extraction of Triton-soluble and -insoluble fractions of α-syn protein
The Triton-soluble and -insoluble fractions of α-syn protein from cells were sequentially extracted according to a previous report [24]. After washing twice with ice-cold PBS, the cells were lysed in Triton lysis buffer (1% Triton X-100, 0.1 mM PMSF, 1% (v/v) protease inhibitor cocktail in PBS) and incubated on ice for 20 min. Similarly, 50 mg tissue samples from mice brain were homogenized in five volumes of the same lysis buffer and incubated on ice for 20 min. After centrifugation at 500 g for 10 min at 4 °C, the supernatants from cell and tissue samples were collected and centrifuged at 18,000 g for 30 min at 4 °C. These supernatants were taken as Triton-soluble fractions. The pellets in cell samples and tissue samples were rinsed with Triton X-100 lysis buffer and re-centrifuged in order to remove any trace of the soluble fraction. The pellet was resuspended in 2% SDS-containing lysis buffer and subsequent sonication for 10 sec. The resultant solution represented the Triton-insoluble fractions.
Western blotting analysis
Protein levels were examined using western blotting analysis as previously described [25]. The same amounts of protein from cell or mice brain samples were electrophoresed on SDS-polyacrylamide gel, transferred to PVDF membrane, and incubated overnight at 4°C with various primary antibodies in 5% fat-free dry milk-TBST [each antibody was diluted 1:1000: α-syn, phospho-PKA (Thr197), PKA, phospho-Akt (Ser473), Akt, phospho-mTOR (Ser2448), mTOR, phospho-p70S6K (Thr389), p70S6K, LC3A/B, and LMP7, except β-actin (1:5000)]. The blots were then incubated with HRP-conjugated secondary antibody in TBST at a 1:5000 dilution for 1 h at room temperature. Protein bands were visualized with an enhanced chemiluminescence (ECL) kit. Blots were repeated at least three times for every condition.
Quantitative real-time reverse transcription PCR
Total RNA was extracted from T-006-treated PC12/A53T-α-syn cells using the TRIzol reagent. Each cDNA was synthesized using SuperScript II Reverse Transcriptase (Invitrogen) with random primers according to the manufacturer’s protocol. qRT-PCR was performed using the SYBR® Premix Ex Taq™ II kit and Mx3005P qPCR system (Agilent Technologies, Santa Clara, CA, USA). PCR reaction conditions were as follows: 5 min at 95 °C followed by 40 cycles of 95°C for 5 s, and 60 °C for 30 s, with fluorescence measured at the end of each cycle. The gene expression was calculated by the 2−ΔΔCT method and the relative mRNA level of experimental gene was normalized to GAPDH, which was used as an endogenous control [26]. Primers used for the qRT-PCR analysis are listed in Supplementary Material, Table S1.
Construction of lentiviral vectors and lentivirus production
The full-length rat LMP7 cDNA was obtained by RT-PCR using total RNA extracted from PC12 cells and cloned into the pCDH-CMV-MCS-EF1-copGFP lentiviral vector at NotI and XbaI sites. The primers here used were 5′-GCTCTAGAGCCACCATGGCGTTACTGGATCTG-3′ (sense) and 5′-TATGCGGCCGCTCACAGAGTGGCCTCTCGGT-3′ (antisense). A short-hairpin RNA (shRNA) designed against the rat LMP7 was cloned into the pSIH1-H1-DsRed vector at BamH I and EcoRI sites. The sequence of shRNA was GATCCGACCAGGAAAGGAAGGTTCAGCTCGAGCTGAACCTTCCTTTCCTGGTCTTTTTG; the mission non-target shRNA vector was used as a control. The identity of each construct was confirmed by sequencing. Plasmids were purified with the QiaPrep miniprep kit (QIAGEN IZASA Portugal) and transfected into HEK293T lentiviral packaging cells with a three-plasmid system according to the manufacturer's protocols, and lentiviral particles were harvested from the transfected cells after 48 h and 72 h. The lentiviruses were transfected into inducible PC12/A53T-α-syn cells, and the transfection efficiency verified by Western blotting was over 80%.
Transgenic α-syn mouse model and treatment
Ten-month-old homozygous transgenic mice B6 C3-Tg (Prnp-SNCA*A53T) 83Vle/J expressing A53T human α-syn [27, 28] were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). Mice were housed in standard cages with free access to food and water at 25°C following a 12-h:12-h light:dark cycle. A53T human α-syn Tg mice received T-006 (3 mg/kg) (10 mice per group) or olive oil as vehicle (0.2 ml) daily by gavage for 4 weeks. All animal experiments were approved by the Research Ethics Committee of the Institute of Chinese Medical Sciences, University of Macau.
Rotarod testing
Mice were first trained to stay on the rod of the rotarod, which was maintained at a constant speed (5 rpm) for at least 5 min. After training for 3 days, mice were tested for a total of three trials with a constant rotation speed of 22 rpm [29, 30]; the trial was started and sustained for 10 min. The trial stopped when the mouse fell down (activating a switch that automatically stopped the timer) or when 10 min had elapsed.
Immunohistochemistry
Mice were deeply anesthetized by 1% pentobarbital sodium (50 mg/kg) and transcardially perfused with PFA in PBS (pH 7.4). Brains were dissected, post-fixed in PFA overnight and then dehydrated in 30% sucrose in PBS at 4°C. Sections were incubated in a monoclonal anti-TH primary antibody (1:400, Millipore; MAB318) for 48 hours at 4°C, washed in PBS, and incubated with secondary anti-rabbit-HRP antibody. Immunostaining was visualized after 3-3’diaminobenzidine (DAB) staining (Vector Laboratories, Burlingame, CA, USA) using bright field microscopy (Leica, Wetzlar, Germany). Five sections through the SNc areas (-5.3 mm AP from Bregma) were randomly selected from each animal. The unbiased stereological counting of TH-positive neurons was performed bilaterally in the SNc areas. A detailed description of the stereological counting procedure was provided in supplementary materials (S2). The number of TH-positive neurons in the ipsilateral side and contralateral control side were calculated. Values was then expressed as % of the contralateral control side.
Statistical analysis
One-way ANOVA followed by Tukey’s multiple comparison test (two or more groups) and Student's t-test (two groups) were used for statistical analyses by GraphPad Prism 6.0 software (GraphPad Software Inc., San Diego, CA, USA). Data are presented as means ± SD. Statistical significance was defined as P-value that was less than 0.05.
Results
T-006 displays a promising ability to downregulate WT and mutated α-syn in PC12 cells
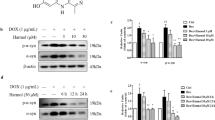
Previous studies showed that T-006 (Fig. 1A) prevented oxidative stress-induced neurotoxicity and reduced glutamate-induced excitotoxicity in cerebellar granule neurons, which implied a potential neuroprotective activity [21]. In the present study, an inducible PC12/A53T-α-syn cell model was constructed as previously addressed and used for screening assays [31]. Cells were treated with several concentrations of T-006 for 24 h. As indicated in Fig. 1B and C, at concentrations of 3-30 μM, T-006 had no impact on cell viability and toxicity. Before we assessed the effect of T-006 on α-syn, a pre-experiment was performed to evaluate the impact of Dox on the expression of α-syn. Inducible PC12/A53T-α-syn cells were treated with or without 1 μg/ml Dox for 24 h. Then cells were washed twice to remove Dox and subsequently incubated with media for different time periods. The degradation levels of A53T-α-syn in PC12 cells was increasing along with the increased treatment time without Dox (Fig. S1). To examine its effect on α-syn, we assessed the effects of various concentrations of T-006 on the protein levels of different types of α-syn, i.e., WT, A30P, and A53T. As shown in Fig. 1D and F, upon Dox induction, an increase was observed in the levels of different types of α-syn, and upon T-006 treatment, a dose-dependent decrease was observed in the levels of different types of total content of α-syn, including Triton-soluble and Triton-insoluble α-syn. Notably, the intracellular levels of WT, A30P, and A53T-α-syn decreased nearly 1.5-fold, 2-fold, and 3.5-fold at a T-006 concentration of 30 μM, respectively, compared to the Dox-induced control. The A53T mutation in α-syn has been recognized to stimulate the formation of mature fibrils, accelerating the development of PD [32, 33]. We studied the effects of T-006 at different time intervals. The results clearly show that T-006 decreases the levels of A53T-α-syn in a time-dependent fashion (Fig. 1G). Furthermore, upon T-006 treatment, the levels of both Triton-soluble and -insoluble A53T-α-syn were significantly reduced in a dose-dependent manner in Dox-induced PC12 cells (Fig. 1H and I). Altogether, these findings reveal that T-006 has the ability to promote α-syn clearance in PC12 cells.

T-006 promotes the degradation of WT and mutated α-syn in inducible PC12 cells. (a) A chemical structure of T-006. Inducible PC12/A53T-α-syn cells were treated with different concentrations of T-006 or 0.1% (v/v) DMSO for 24 h, and then analyzed by MTT assays (b) and LDH assays (c). PC12/WT-α-syn (d), A30P-α-syn (e) and A53T-α-syn (f) cells were treated with 1 μg/ml Dox for 24 h to induce the expression of WT or mutated α-syn, and then treated with different concentrations of T-006 or 0.1 % (v/v) DMSO for another 24 h. (g) PC12/A53T-α-syn cells were induced with 1 μg/ml Dox for 24 h, and then treated with T-006 (30 μM) for different time periods. After treating with T-006 (30 μM), cells were lysed in 1% Triton X-100. The Triton-soluble fractions (h) and Triton-insoluble fractions (i) were subjected to Western blotting analysis. Representative Western blotting data and analysis of three independent experiments are shown. Data are represented as the mean ± SD. ###P < 0.001 as compared to control group; *P < 0.05, **P < 0.01, ***P < 0.001, as compared to Dox group
T-006 promotes A53T-α-syn degradation by proteasomal activation
In PD, the accumulation of toxic intermediate species of oligomers disrupts the proteostasis systems, ultimately leading to cell death [34]. Hence, great efforts have been made to promote α-syn clearance. Intracellular degradation of misfolded proteins mainly depends on the proteasome and autophagy pathways. Therefore, to explore the potential pathway mediating T-006-induced α-syn degradation, we investigated A53T-α-syn protein levels by combining T-006 with the autophagy inhibitor CQ or the proteasome inhibitor MG132 (Fig. 2A). Upon T-006 treatment, the intracellular protein level of A53T-α-syn was reduced, and upon CQ treatment, it was increased. However, combining T-006 and CQ treatment, α-syn levels were reduced, suggesting that, despite the CQ-mediated inhibition of the ALP, T-006 stimulated A53T-α-syn degradation. On the other hand, A53T-α-syn levels were remarkably increased upon MG132 treatment, and pretreatment with T-006 had no significant effect on A53T-α-syn protein levels. To further verify the pathway by which T-006 promotes α-syn degradation, we detected the protein levels of the autophagy markers LC3 and p62. As shown in Fig. 2B, T-006 had no effect on the levels of LC3 and p62. These findings reveal that the UPS is the main pathway for α-syn degradation in this cell line, and T-006-induced α-syn degradation is selectively mediated by the proteasome. Protein degradation by the UPS requires activity of the 20S core particle, which contains the proteolytically active β-subunits caspase-like β1, trypsin-like β2, and chymotrypsin-like β5 [12]. We measured the chymotrypsin-, trypsin-, and caspase-like proteasomal activities after T-006 treatment using a luminescent assay. We found that T-006 significantly increased the chymotrypsin-like, but not the caspase- and trypsin-like proteasomal activities (Fig. 2C-E) in Dox-induced PC12/A53T-α-syn cells. Treatment with MG132 dramatically decreased trypsin- and chymotrypsin-like, but not caspase-like proteasomal activity. Moreover, the caspase-, trypsin-, and chymotrypsin-like proteasomal activities observed upon MG132 treatment could not be reversed by treatment with T-006, suggesting that T-006-mediated protein degradation is attenuated by MG132-mediated proteasomal inhibition. In summary, T-006 selectively promoted intracellular proteasome-dependent A53T-α-syn degradation by increasing the activity of the chymotrypsin-like in the 20S core particle, without affecting the ALP.

T-006 promotes α-syn degradationviathe proteasome. (a) Inducible PC12/A53T-α-syn cells were treated with or without 1 μg/ml Dox for 24 h, and then treated with 30 μΜ T-006, 30 μΜ CQ, 0.1 μΜ MG132 or 0.1% (v/v) DMSO for another 24 h. (b) Inducible PC12/A53T-α-syn cells were treated with 1 μg/ml Dox for 24 h, and then treated with 30 μΜ T-006 for different time periods. Representative Western blotting and data analysis of three independent experiments are shown. The proteasomal enzyme activities were measured using specific bioluminogenic enzyme substrates for chymotrypsin-like (c), trypsin-like (d), and caspase-like (e) activity. Data are represented as the mean ± SD. ###P < 0.001 as compared to control group; *P < 0.05, **P < 0.01, ***P < 0.001, as compared to Dox group; $P <0.05, as Dox/CQ compared to Dox/CQ/T-006 group
T-006 prevents A53T-α-syn from accumulation by increasing gene and protein expression of LMP7
Furthermore, we assessed the effects of T-006 on proteasome-related genes. Among the 43 tested genes, intracellular mRNA levels of low molecular weight polypeptide 7 (LMP7) were obviously increased upon T-006 treatment in Dox-inducible PC12/A53T-α-syn cells (Fig. 3A and S2), while the mRNA levels of other proteasome subunit genes were not significantly elevated (data not shown). It was previously reported that LMP7 encodes the 20S immunoproteasome subunit β5i, which contains chymotrypsin-like enzymatic activity [35].

T-006 promotes α-syn degradation through upregulating the activity of LMP7. PC12/A53T-α-syn cells were treated with 1 μg/ml Dox for 24 h and subsequently with 30 μΜ T-006 for another 24 h. (a) The mRNA levels of LMP7 were determined by qRT-PCR. (b) Protein levels of LMP7 were detected by Western blotting analysis. (c) PC12/A53T-α-syn cells were transfected with LMP7-shRNA or control-shRNA, then treated with Dox for 24 h before incubating with 30 μΜ T-006 for another 24 h. (d) PC12/A53T-α-syn cells were transfected with or without LMP7 cDNA plasmid for 24 h and were induced with Dox for another 24 h. Cell lysates were subjected to Western blotting analysis. Data are represented as mean ± SD of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, as compared to control group
Expression levels of LMP7 were increased upon T-006 treatment (Fig. 3B), in agreement with the observed increase in chymotrypsin-like proteasomal activity (Fig. 2C). By performing an LMP7 shRNA knock-down assay, we further explored the effects of T-006 on the protein levels of A53T-α-syn. Upon LMP7 knock-down, A53T-α-syn protein levels were increased (Fig. 3C). When cells were transfected with control-shRNA, T-006 significantly reduced A53T-α-syn accumulation, but T-006 treatment could not exert the same effect in LMP7 knock-down cells. Upon LMP7 overexpression, intracellular protein levels of A53T-α-syn were notably decreased (Fig. 3D). Collectively, these findings indicated that T-006 is able to upregulate the gene and protein expression levels of LMP7, which is associated with the increased activity of the chymotrypsin-like proteasome subunit, leading to a reduction in A53T-α-syn accumulation.
T-006 regulates LMP7 levels and LMP7-mediated proteasomal activity through the activation of the PKA/Akt/mTOR/p70S6K pathway
The PKA and Akt/mTOR pathways are well recognized as critical modulators in regulation of proteasomal activities [36, 37]. Given the effects of T-006 treatment on LMP7 gene and protein expression, we further investigated its effect on the PKA/Akt/mTOR pathway. T-006 significantly promoted PKA, Akt, and mTOR phosphorylation, but these three proteins exhibited different time-dependent phosphorylation patterns (Fig. 4A-C). The phosphorylation level of PKA increased more than six-fold within 5-15 min, the phosphorylation level of Akt increased 8-fold within 15 min, and the phosphorylation level of mTOR increased two-fold within 30 min. The phosphorylation level of p70S6K, which is stimulated by mTOR and controls the activation of ribosomal S6k, modulating protein synthesis, steadily increased over time, reaching a six-fold increase after 60 min (Fig. 4D). To further investigate the effects of T-006 on the PKA/Akt/mTOR/p70S6K pathway, LMP7 protein expression, and chymotrypsin-like proteasomal activity, the PKA inhibitor H-89 and the mTOR inhibitor Rap were used. Upon treatment with H89 at 10 μΜ in cells, the phosphorylation levels of p70S6K were greatly decreased. However, after incubating with T-006 in H-89-treated cells, the levels of phosphorylated p70S6K showed no visible changes (Fig. 4E). In terms of regulating p70S6K phosphorylation, similar results were obtained when the cells were pretreated with the mTOR inhibitor Rap (Fig. 4F). Similar observations were made for LMP7 protein expression and chymotrypsin-like proteasomal activity (Fig. 4G and H). These findings suggested that T-006 increases LMP7 protein expression and chymotrypsin-like proteasomal activity via activation of the PKA/Akt/mTOR/p70S6K pathway.

T-006 promotes LMP7 expression and enhances proteasomal activity through the PKA/Akt/mTOR//p70S6K pathway. Inducible PC12/A53T-α-syn cells were treated with 1 μg/ml Dox for 24 h, and then with T-006 (30 μM) for the indicated time periods; the fold change of expression levels of phosphorylated PKA/total PKA (a), phosphorylated Akt/total Akt (b), phosphorylated p-mTOR/total mTOR (c) and phosphorylated p-p70s6k/total p70s6k (d) were detected by Western blotting analysis. (e) PC12/A53T-α-syn cells were pretreated with or without the PKA inhibitor H-89 (10 μM) for 1 h and then treated with T-006 (30 μM) for 30 min. (f) PC12/A53T-α-syn cells were pretreated with or without the mTOR inhibitor Rap (0.2 μM) for 1 h and then treated with T-006 (30 μM) for 30 min. The fold change of expression levels of phosphorylated p70S6K/total p70S6K were detected by Western blotting. PC12/A53T-α-syn cells were pretreated with or without H-89 (10 μM) for 1 h and then treated with T-006 (30 μM) for 24 h. The protein levels of LMP7 were determined by Western blotting (g). Cellular chymotrypsin-like activity was determined by a commercialized kit (h). Representative Western blotting and data analysis of three independent experiments are shown. Data are represented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
T-006 exerts a protective effect against α-syn-induced neurotoxicity in A53T-α-syn transgenic mice by promoting the degradation of Triton-soluble and -insoluble α-syn
Final assessments of the neuroprotective effect and potential application of T-006 were carried out in a Tg mouse PD model. Tg mice were given a daily treatment of T-006 or olive oil as negative control by gavage for four weeks (Fig. 5A). A53T-α-syn accumulation in different areas of the mouse brains was detected (Fig. 5B–D). The results show that T-006 treatment could effectively and dramatically reverse the accumulation of Triton-soluble A53T-α-syn in the mouse cortex, midbrain, and striatum compared to controls. Additionally, T-006 also decreased the accumulation of Triton-insoluble A53T-α-syn in whole brains (Fig. 5E). Moreover, A53T-α-syn mouse behavior was evaluated by rotarod experiments. A53T-α-syn Tg mice exhibited reduced locomotor activity, and the amount of time mice stayed on the rotarod was reduced compared to WT mice (Fig. 6B). However, upon T-006 treatment, the reduction in locomotor activity in Tg mice was obviously reversed. Finally, the number of tyrosine hydroxylase-positive (TH+) neurons, which convert tyrosine to L-dopa, was examined by immunohistochemical analysis. The number of TH+ cells was decreased in Tg mice compared to WT mice. However, increased numbers of TH+ cells were detected in Tg mice when the mice were administrated with T-006 (Fig. 6C and D). Collectively, our results show that T-006 exhibits a promising protective effect against α-syn-induced neurotoxicity by promoting the degradation of Triton-soluble and -insoluble α-syn in A53T-α-syn Tg mice.

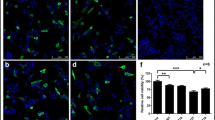
T-006 promotes the degradation of Triton-soluble and -insoluble α-syn in the A53T-α-syn Tg mouse brain. Ten-month-old homozygous A53T human α-syn Tg mice (n = 5) were gavaged daily with T-006 (3 mg/kg) or olive oil (0.2 ml) for 4 weeks. (a) An experimental scheme. The cortex (b), midbrain (c) and striatum (d) fractions were homogenized and the protein level of Triton-soluble A53T-α-syn was analyzed by Western blotting. (e) Whole-brain levels of Triton-insoluble A53T-α-syn were analyzed by Western blotting. Representative Western blotting and data analysis of three independent experiments are shown. Data are represented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001

T-006 protects against A53T-α-syn-induced neurotoxicity in Tg mice. (a) Experimental scheme. Ten-month-old WT or homozygous A53T human α-syn Tg mice were gavaged daily with T-006 (3 mg/kg) or olive oil (0.2 ml) for 4 weeks. (b) In a rotarod test, the time spent on the rotating drum before falling was recorded and analyzed (n = 10). (c) Representative images of TH+ neurons in the SNc (n = 5). (D) Quantification of TH-positive cells in the SNc (n = 5). Data are represented as means ± SD. ##P < 0.01, compared to the WT group; *P < 0.05, compared to the Tg group
Discussion
In the present study, we followed up on our previous study and continued our investigation of the neuroprotective effects of T-006. T-006 significantly decreased intracellular α-syn accumulation by activating the proteasome rather than the ALP. In addition, proteasomal activity of the chymotrypsin-like β5i subunit was more significantly upregulated upon T-006 treatment than the activity of the other two proteasomes involved in proteasomal degradation. A further assessment of gene expression revealed that expression of the LMP7 gene, which encodes the chymotrypsin-like β5i protein, was dramatically increased upon T-006 treatment. Moreover, the inhibitory effects of T-006 on A53T-α-syn accumulation can be explained by the activation of the PKA/Akt/mTOR/p70S6K signaling pathway. Finally, by using an A53T-α-syn Tg mouse PD model, we demonstrated the potential therapeutic utility of T-006 for PD treatment. T-006 reversed the accumulation of Triton-soluble and -insoluble A53T-α-syn and exerted a promising neuroprotective effect against α-syn-induced toxicity in Tg mice. These findings suggest that T-006 reduces α-syn accumulation by activating the proteasome and is therefore a promising drug candidate for the prevention and treatment of PD.
The accumulation of α-syn in dopaminergic neurons is the key indicator of the pathological progression of PD [38,39,40], even though the mechanisms causing α-syn accumulation and Lewy body formation remain unclear [41]. Abnormal α-syn accumulates in β-sheet-rich fibrils, and such structures induce cellular dysfunction and cell death. With a majority of point mutations (A30P, E46K, H50Q, G51D, A53T) cluster in gene of α-syn, an enhanced accumulation of α-syn and correlation of toxicity are identified [42,43,44]. In this study, T-006 treatment was shown to decrease the intracellular accumulation of WT, A30P, and A53T-α-syn; degradation of A53T-α-syn was most strongly influenced by T-006. In a Tg mouse model, T-006 also markedly promoted A53T-α-syn accumulation, suggestive of a promising neuroprotective characteristic. Soluble α-syn has been shown to be degraded by both the proteasome and chaperone-mediated autophagy, but once aggregated, it is scavenged by the autophagy-lysosome pathway [23].
Furthermore, we explored the functional mechanism of T-006. We have shown that the effects of T-006 on A53T-α-syn accumulation are lost when the cells are pretreated with MG132, a proteasome inhibitor, whereas T-006 was able to reverse the accumulation of A53T-α-syn in cells pretreated with CQ, an autophagy inhibitor, suggesting the UPS rather than the ALP is modulated by T-006. It is well documented that changes in autophagy and proteasome function when the α-syn expression is increased have been reported; however, in our model, we did not see obvious changes on the autophagy and proteasome function at 24 hours. But longer time (48h or even a few days) induction of α-syn will affect both autophagy and proteasome function in our model. Further examinations of the activities of three subunits of the 20S core particle, which is responsible for proteasomal degradation, demonstrated that the chymotrypsin-like proteasomal activity was influenced by T-006.
Recently, several small molecules have been reported to exhibit similar mechanistic neuroprotective effects. Salidroside promotes WT/A30P-α-syn clearance and preserves cell viability by recovering 20S proteasomal activity in SH-SY5Y cells [18]. Isorhynchophylline, a natural alkaloid, promotes the degradation of α-syn monomers, oligomers, and α-syn/synphilin-1 aggresomes in a panel of neuronal cells via activating the ALP [20]. Our study of the T-006-induced reduction in pathogenic A53T-α-syn accumulation highlighted that natural products could serve as a means to stimulate proteasomal activity, thereby exerting preventive and therapeutic effects against PD by promoting pathogenic protein clearance. A further assessment of target genes demonstrated that T-006 increases LMP7 gene expression, resulting in an enhancement of chymotrypsin-like proteasomal activity. LMP genes have been reported to mediate α-syn clearance with more genetic susceptibility to PD by modulating the immunoproteasome in dopaminergic neurons [45]. To date, few small molecular inhibitors of α-syn accumulation have been found that target the LMP7 gene or protein expression, but T-006 is one of them. Finally, T-006 was confirmed to promote α-syn degradation by activating the PKA/Akt/mTOR/p70S6K signaling pathway. Collectively, the evidence pointing toward the proteasome being a critical player in α-syn clearance has indicated proteasome-activating approaches can be promising therapies to prevent and reduce α-syn accumulation in PD.
Conclusions
In the present study, we demonstrated the neuroprotective effects of T-006 by the mediation of the proteasome via the activation of the PKA/Akt/mTOR/p70S6K signaling pathway. T-006 enhances proteasome activity, demonstrating its potential therapeutic application in PD treatment. More insights into the pathological aspects and molecular mechanisms of T-006 can additionally aid in the identification of other potent small molecules and the development of proteasome-enhancing or -activating therapies for PD treatment.
Abbreviations
- A30P:
-
mutant A30P α-synuclein
- A53T:
-
mutant A53T-α-synuclein
- ALP:
-
autophagy lysosomal pathway
- α-syn:
-
α-synuclein
- CQ:
-
chloroquine
- Dox:
-
doxycycline
- LDH:
-
Lactate dehydrogenase
- mTOR:
-
mammalian target of rapamycin
- PD:
-
Parkinson’s disease
- PFA:
-
paraformaldehyde
- PKA:
-
protein kinase A
- PMSF:
-
Phenylmethanesulfonyl fluoride
- qRT-PCR:
-
quantitative real-time reverse transcription PCR
- shRNA:
-
small hairpin RNA
- SNc:
-
Substantia nigra pars compacta
- Tg:
-
transgenic
- TH:
-
tyrosine hydrogenase
- UPS:
-
ubiquitin-proteasome system
- WT:
-
wild type
References
Pires AO, Teixeira FG, Mendes-Pinheiro B, et al. Old and new challenges in Parkinson's disease therapeutics. Prog Neurobiol 2017;156:69-89.
Fathy YY, Jonker AJ, Oudejans E, et al. Differential insular cortex subregional vulnerability to alpha-synuclein pathology in Parkinson's disease and dementia with Lewy bodies. Neuropathol Appl Neurobiol 2019;45:262-277.
Spillantini MG, Crowther RA, Jakes R, et al. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson's disease and dementia with Lewy bodies. Neurosci Lett 1998;251:205-208.
Sala G, Marinig D, Arosio A, Ferrarese C. Role of chaperone-mediated autophagy dysfunctions in the pathogenesis of Parkinson's disease. Front Mol Neurosci 2016;9:157.
Jan A, Jansonius B, Delaidelli A, et al. Activity of translation regulator eukaryotic elongation factor-2 kinase is increased in Parkinson disease brain and its inhibition reduces alpha synuclein toxicity. Acta Neuropathologica Communications 2018;6:54.
Ludtmann MHR, Angelova PR, Horrocks MH, et al. alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson's disease. Nature Communications 2018;9:2293.
Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nature Medicine 2004;10 Suppl:S10-17.
Zarranz JJ, Alegre J, Gomez-Esteban JC, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Annals of Neurology 2004;55:164-173.
Fuchs J, Tichopad A, Golub Y, et al. Genetic variability in the SNCA gene influences alpha-synuclein levels in the blood and brain. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology 2008;22:1327-1334.
Li J, Uversky VN, Fink AL. Effect of familial Parkinson's disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human alpha-synuclein. Biochemistry 2001;40:11604-11613.
Tanaka K, Matsuda N. Proteostasis and neurodegeneration: the roles of proteasomal degradation and autophagy. Biochimica et Biophysica Acta 2014;1843:197-204.
Vilchez D, Saez I, Dillin A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nature Communications 2014;5:5659.
Kim H, Calatayud C, Guha S, et al. The small GTPase RAC1/CED-10 is essential in maintaining dopaminergic neuron function and survival against alpha-synuclein-induced toxicity. Mol Neurobiol 2018;55:7533-7552.
Wilkaniec A, Lenkiewicz AM, Czapski GA, et al. Extracellular alpha-synuclein oligomers induce Parkin S-nitrosylation: relevance to sporadic Parkinson's disease etiopathology. Mol Neurobiol 2019;56:125-140.
Zhang M, Deng YN, Zhang JY, et al. SIRT3 protects rotenone-induced injury in SH-SY5Y cells by promoting autophagy through the LKB1-AMPK-mTOR pathway. Aging Dis 2018;9:273-286.
Schmidt M, Finley D. Regulation of proteasome activity in health and disease. Biochimica et Biophysica acta 2014;1843:13-25.
Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, et al. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. The Journal of Neuroscience : the official journal of the Society for Neuroscience 2011;31:14508-14520.
Li T, Feng Y, Yang R, et al. Salidroside promotes the pathological alpha-synuclein clearance through ubiquitin-proteasome system in SH-SY5Y cells. Frontiers in Pharmacology 2018;9:377.
Lan D, Wang W, Zhuang J, Zhao Z. Proteasome inhibitor-induced autophagy in PC12 cells overexpressing A53T mutant alpha-synuclein. Molecular Medicine Reports 2015;11:1655-1660.
Lu JH, Tan JQ, Durairajan SS, et al. Isorhynchophylline, a natural alkaloid, promotes the degradation of alpha-synuclein in neuronal cells via inducing autophagy. Autophagy 2012;8:98-108.
Chen H-Y, Xu D-P, Tan G-L, et al. A potent multi-functional neuroprotective derivative of Tetramethylpyrazine. J Mol Neurosci 2015;56:977-987.
Xu D, Chen H, Mak S, et al. Neuroprotection against glutamate-induced excitotoxicity and induction of neurite outgrowth by T-006, a novel multifunctional derivative of tetramethylpyrazine in neuronal cell models. Neurochem Int 2016;99:194-205.
Webb JL, Ravikumar B, Atkins J, et al. Alpha-Synuclein is degraded by both autophagy and the proteasome. The Journal of Biological Chemistry 2003;278:25009-25013.
Gan L, Vargas MR, Johnson DA, Johnson JA. Astrocyte-specific overexpression of Nrf2 delays motor pathology and synuclein aggregation throughout the CNS in the alpha-synuclein mutant (A53T) mouse model. J Neurosci 2012;32:17775-17787.
Yang X, Xu Y, Wang T, et al. Inhibition of cancer migration and invasion by knocking down delta-5-desaturase in COX-2 overexpressed cancer cells. Redox Biol 2017;11:653-662.
Ye M, Chung HS, Lee C, et al. Bee venom phospholipase A2 ameliorates motor dysfunction and modulates microglia activation in Parkinson's disease alpha-synuclein transgenic mice. Exp Mol Med 2016;48:e244.
Du RH, Zhou Y, Xia ML, et al. Alpha-Synuclein disrupts the anti-inflammatory role of Drd2 via interfering beta-arrestin2-TAB1 interaction in astrocytes. J Neuroinflammation 2018;15:258.
Zhou Y, Lu M, Du RH, et al. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson's disease. Molecular Neurodegeneration 2016;11:28.
Tatenhorst L, Tonges L, Saal KA, et al. Rho kinase inhibition by fasudil in the striatal 6-hydroxydopamine lesion mouse model of Parkinson disease. Journal of Neuropathology and Experimental Neurology 2014;73:770-779.
Morroni F, Tarozzi A, Sita G, et al. Neuroprotective effect of sulforaphane in 6-hydroxydopamine-lesioned mouse model of Parkinson's disease. Neurotoxicology 2013;36:63-71.
Song JX, Lu JH, Liu LF, et al. HMGB1 is involved in autophagy inhibition caused by SNCA/alpha-synuclein overexpression: a process modulated by the natural autophagy inducer corynoxine B. Autophagy 2014;10:144-154.
Stefanis L. Alpha-Synuclein in Parkinson's disease. Cold Spring Harb Perspect Med 2012;2:a009399.
Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nature Medicine 1998;4:1318-1320.
Lim J, Yue Z. Neuronal aggregates: formation, clearance, and spreading. Developmental Cell 2015;32:491-501.
Ferrington DA, Gregerson DS. Immunoproteasomes: structure, function, and antigen presentation. Prog Mol Biol Transl Sci 2012;109:75-112.
Collins GA, Goldberg AL. The logic of the 26S proteasome. Cell 2017;169:792-806.
Zhang F, Hu Y, Huang P, et al. Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. The Journal of Biological Chemistry 2007;282:22460-22471.
Rodriguez-Perez AI, Sucunza D, Pedrosa MA, et al. Angiotensin type 1 receptor antagonists protect against alpha-synuclein-induced neuroinflammation and dopaminergic neuron death. Neurotherapeutics 2018;15:1063-1081.
Nelson MP, Boutin M, Tse TE, et al. The lysosomal enzyme alpha-Galactosidase A is deficient in Parkinson's disease brain in association with the pathologic accumulation of alpha-synuclein. Neurobiol Dis 2018;110:68-81.
Zhang S, Feng R, Li Y, et al. Degradation of alpha-synuclein by dendritic cell factor 1 delays neurodegeneration and extends lifespan in Drosophila. Neurobiology of Aging 2018;67:67-74.
Dettmer U, Newman AJ, Soldner F, et al. Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nature Communications 2015;6:7314.
Mason JM, Fairlie DP. Toward peptide-based inhibitors as therapies for Parkinson's disease. Future Medicinal Chemistry 2015;7:2103-2105.
Horvath I, Iashchishyn IA, Moskalenko RA, et al. Co-aggregation of pro-inflammatory S100A9 with alpha-synuclein in Parkinson's disease: ex vivo and in vitro studies. J Neuroinflammation 2018;15:172.
Maguire-Zeiss KA. Alpha-Synuclein: a therapeutic target for Parkinson's disease? Pharmacol Res 2008;58:271-280.
Mo MS, Huang W, Sun CC, et al. Association analysis of proteasome subunits and transporter associated with antigen processing on Chinese patients with Parkinson's disease. Chinese Medical Journal 2016;129:1053-1058.
Acknowledgements
This study was supported by grants from the Science and Technology Development Fund (FDCT) of Macao SAR (Ref. No. 069/2015/A2, No. 134/2014/A3 and No. 062-2017-AIR), Research Committee, University of Macau (MYRG2015-00182-ICMS-QRCM, MYRG2015-00214-ICMS-QRCM, MYRG139(Y1-L4)-ICMS12-LMY and MYRG2016-00129-ICMS-QRCM) awarded to Simon M.Y. Lee, and the Natural Science Foundation of Guangdong Province awarded to Zaijun Zhang (2015A030313317).
Required Author FormsDisclosure forms provided by the authors are available with the online version of this article
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhou, H., Shao, M., Guo, B. et al. Tetramethylpyrazine Analogue T-006 Promotes the Clearance of Alpha-synuclein by Enhancing Proteasome Activity in Parkinson’s Disease Models. Neurotherapeutics 16, 1225–1236 (2019). https://doi.org/10.1007/s13311-019-00759-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-019-00759-8