
Abstract
We have previously shown that roflupram (ROF) protects against MPP+-induced neuronal damage in models of Parkinson’s disease (PD). Since impaired degradation of α-synuclein (α-syn) is one of the key factors that lead to PD, here we investigated whether and how ROF affects the degradation of α-syn in rotenone (ROT)-induced PD models in vivo and in vitro. We showed that pretreatment with ROF (10 μM) significantly attenuated cell apoptosis and reduced the level of α-syn in ROT-treated SH-SY5Y cells. Furthermore, ROF significantly enhanced the lysosomal function, as evidenced by the increased levels of mature cathepsin D (CTSD) and lysosomal-associated membrane protein 1 (LAMP1) through increasing NAD+/NADH and the expression of sirtuin 1 (SIRT1). Pretreatment with an SIRT1 inhibitor selisistat (SELI, 10 μM) attenuated the neuroprotection of ROF, ROF-reduced expression of α-syn, and ROF-increased expression levels of LAMP1 and mature CTSD. Moreover, inhibition of CTSD by pepstatin A (20 μM) attenuated ROF-reduced expression of α-syn. In vivo study was conducted in mice exposed to ROT (10 mg·kg−1·d−1, i.g.) for 6 weeks; then, ROT-treated mice received ROF (0.5, 1, or 2 mg·kg−1·d−1; i.g.) for four weeks. ROF significantly ameliorated motor deficits, which was accompanied by increased expression levels of tyrosine hydroxylase, SIRT1, mature CTSD, and LAMP1, and a reduced level of α-syn in the substantia nigra pars compacta. Taken together, these results demonstrate that ROF exerts a neuroprotective action and reduces the α-syn level in PD models. The mechanisms underlying ROF neuroprotective effects appear to be associated with NAD+/SIRT1-dependent activation of lysosomal function.
Similar content being viewed by others
Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder that is characterized by resting tremor, muscle rigidity, postural instability, and bradykinesia. It can also cause non-motor symptoms, such as sleep disorders, hyposmia, constipation, depression, and anxiety [1]. It is considered that the degeneration of dopaminergic neurons in the substantia nigra (SN), with the subsequent loss of dopamine in the brain, is a pathological substrate of PD. Thus far, great advances have been made in understanding the pathology leading to motor and non-motor symptoms of PD, and emerging therapies have been used for modulating brain circuitry and human behavior in patients suffering from PD [2]. For example, deep brain stimulation is now commonly indicated for the treatment of PD in patients without surgical contraindications [3]. Nevertheless, medications are still the most effective therapy for PD. Dopaminergic medications and anticholinergics are recommended to ameliorate symptoms, especially in the early phase of the disease [4]. However, the currently available therapies used for PD have no effects on slowing or halting disease progression. Thus, there is a need for a comprehensive understanding of the pathogenesis of PD and the identification of novel drug targets.
The precise cause of PD and the mechanism underlying the loss of dopaminergic neurons remain largely unclear. Misfolding and abnormal aggregation of α-synuclein (α-syn) are one of the key factors promoting the apoptosis and even necrosis of dopaminergic neurons [5]. The abnormal aggregation or impaired degradation of α-syn leads to the development of Lewy bodies, which is one of the typical pathological features of PD [6]. In eukaryotic cells, α-syn is mainly degraded through two pathways: the ubiquitination-protease system (UPS) and autophagy-lysosome pathway (ALP) [7]. Both the UPS and ALP decline with aging, leading to the accumulation of α-syn due to impaired clearance [8]. In addition, during the pathogenesis of PD, the excessive accumulation of reactive oxygen species in cells triggers an increase in lysosomal membrane permeabilization [9], and lysosomal depletion and dysfunction cause delayed clearance of α-syn. In summary, current findings support that α-syn accumulation and impaired intracellular proteolytic systems interact with each other, thereby generating a bidirectional positive feedback loop that leads to neurodegeneration [8]. Given the link between α-syn accumulation and intracellular proteolytic systems, therapeutic strategies that reduce the level of α-syn in the brain should be promising for counteracting α-syn pathology.
Phosphodiesterase 4 (PDE4) is one of 11 PDE enzymes; it specially hydrolyzes cAMP in cells [10]. Considering that PDE4 is highly expressed in neurons, the role of PDE4 in neurodegenerative diseases has received increasing attention [11, 12]. Rolipram (ROLI), a PDE4 inhibitor, enhances neuronal survival in primary cultured neurons [13]. Selective inhibition of PDE4 reduces the loss of neurons in the SN and increases the concentration of dopamine in the striatum of mice subjected to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [14]. Our previous studies have shown that inhibition of PDE4 by FCPR16 protects SH-SY5Y cells and primary neurons against 1-methyl-4-phenylpyridine (MPP+)-induced neuronal toxicity by enhancing autophagy [15]. In mice treated with MPTP, we have also verified that PDE4 inhibition by roflupram (ROF) ameliorates the apoptosis of dopaminergic neurons in the SN, restores mitochondrial functions, and reduces oxidative stress [16]. Collectively, these data suggest that PDE4 inhibition may be a promising strategy for the treatment of PD.
Although PDE4 inhibition has shown neuroprotective effects in multiple experimental PD models, the underlying mechanisms are largely unknown. The interaction between PDE4 and α-syn has not been illustrated. The ALP is involved in the degradation of α-syn, and our previous study has shown that PDE4 inhibition promotes autophagy by activating AMP-activated protein kinase (AMPK) in SH-SY5Y cells and neurons treated with MPP+ [15]. Those findings have set the basis to study the role of PDE4 inhibition in lysosomes. For the development of novel drugs for the treatment of PD, it is of great significance to explore how to enhance the function of lysosomes and thereby promote the clearance of α-syn. ROF is a selective PDE4 inhibitor, and its chemical structure is shown in Fig. 1a. The design and synthetic route of this compound were reported by Zheng et al. [17]. Compared with the first-generation PDE4 inhibitor ROLI, ROF has a stronger inhibitory effect on PDE4 [18]. Additionally, ROF has an excellent blood–brain barrier penetration ability [18], which makes it more suitable for the treatment of central nervous system diseases compared with the second-generation PDE4 inhibitor, roflumilast. Most importantly, ROF has little emetic potential [18], which has garnered particular interest regarding its neuroprotective effects. Our previous study has verified that ROF exerts neuroprotective effects in APP/PS1 mice and MPTP-treated mice [16, 18]. Our study has also demonstrated that ROF inhibits the proinflammatory response in both cultured microglial cells and animals injected with lipopolysaccharides [19, 20]. These results indicate that ROF is a promising candidate for the treatment of neurological and neurodegenerative diseases.

a Chemical structure of ROF. b SH-SY5Y cells were treated with different concentrations of ROT (0.5–4 μM) for 48 h, and cell viability was determined by the MTT assay. c Cells were treated with different concentrations of ROT (0.5–4 μM) for 24 h, and cell viability was determined by the MTT assay. d SH-SY5Y cells were pretreated with ROF (10 μM) for 1 h and then treated with ROT (2 μM) for 24 h. The level of phosphorylated PKA was determined by the Western blotting. e Relative level of p-PKA versus total PKA was determined by densitometry of the blots. f SH-SY5Y cells were pretreated with ROF at concentrations of 2.5, 5, and 10 μM for 1 h and then treated with ROT (2 μM) for 24 h. Cell viability was measured with the MTT assay. g, h SH-SY5Y cells pretreated with ROF (10 μM) were treated with ROT (2 μM) for 48 h. The level of cleaved caspase-3 was determined by Western blotting and the immunofluorescent assay. Data are shown as mean ± SD, n = 3. #P < 0.05, ##P < 0.01, ###P < 0.001 versus the control (CTL) group. *P < 0.05, **P < 0.01 versus the ROT-treated group.
Considering that PDE4 is a promising target for the treatment of PD, and ROF is a selective PDE4 inhibitor with the advantages of little emetic potential and excellent blood–brain barrier penetration, the treatment with ROF has shown neuroprotective effects in models of PD. However, the effects of ROF on α-syn clearance and lysosomes remain unclear. This study evaluated the effects of the PDE4 inhibitor ROF on the clearance of α-syn as well as the underlying mechanisms in experimental models induced by rotenone (ROT). We hypothesized that inhibition of PDE4 by ROF would protect against neuronal apoptosis and promote α-syn clearance by maintaining lysosomal activity.
Materials and methods
Chemicals and reagents
ROF (#SML2106) was purchased from Sigma-Aldrich (St. Louis, MO, USA). ROT (#S2348), ROLI (#S1430), selisistat (SELI) (#S1541) and pepstatin A (#S7381) were obtained from Selleck (Houston, TX, USA). Antibodies against GAPDH (#2118; 1:5000), cleaved caspase-3 (#9662; 1:1000), protein kinase A (PKA) (#4782; 1:1000), phosphorylated PKA (p-PKA) (#5661; Thr197; 1:1000), and sirtuin-1 (SIRT1) (#9475; 1:1000) were purchased from Cell Signaling Technology (Danvers, MA, USA). The antibody against lysosomal-associated membrane protein 1 (LAMP1) (#SAB3500285; 1:1000) was obtained from Sigma-Aldrich. The antibody against tyrosine hydroxylase (TH) (#ab152; 1:1000) was purchased from Merck-Calbiochem (St. Louis, MO, USA). Antibodies against α-syn (#10842-1-AP; 1:1000) and lysosomal protease cathepsin D (CTSD) (#21327-1-AP; 1:1000) were purchased from Proteintech Group, Inc. (Chicago, IL, USA). The anti-phospho-α-syn antibody (p-α-syn; Ser129; #ab51253; 1:1000), the anti-PDE4B antibody (#ab112014; 1:1000), and the Universal SIRT Activity Assay Kit (#ab156915) were purchased from Abcam (Cambridge, MA, USA). The Annexin V-FITC/PI Cell Apoptosis Detection Kit (#KGA108) was obtained from KeyGen Biotech (Nanjing, China). The nicotinamide adenine dinucleotide (NAD+)/NADH Assay Kit (#S0175), Lyso-Tracker (LYT) Red (#C1046), and the Nuclear and Cytoplasmic Protein Extraction Kit (#P0027) were from the Beyotime Institute of Biotechnology (Shanghai, China). Hoechst 33342 (#H1399), Lipofectamine 2000 (#11668019), and Opti-MEM (#31985062) were bought from Thermo Fisher Scientific. 3-(4,5-Dimethyl-2- thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) (#M2128) was obtained from Sigma-Aldrich. Fetal bovine serum (FBS) (#10099141) and Dulbecco’s Modified Eagle’s Medium (DMEM)/F-12 basic (1:1) (#C11330500BT) were purchased from Gibco (Carlsbad, CA, USA). The hydroxypropyl-β-cyclodextrin (#H108813) was obtained from Aladdin Biological Technology Co., Ltd. (Shanghai China), and Solutol HS-15 was bought from Melone Biological Technology Co., Ltd. (Dalian, Liaoning Province, China). Acridine Orange (AO) hydrochloride solution (#A8097) was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
The SH-SY5Y cell line was obtained from the American Type Culture Collection (#CRL-2266). The cells were cultured in the medium of DMEM/F-12 supplemented with 1% penicillin/streptomycin and 10% (v/v) FBS. The cells were cultivated at 37 °C in humidified air with 5% CO2. The culture medium was replaced with fresh medium every 48 h. After 3–5 passages, when growth density reached 75%–85%, the cells were used in the experiments.
MTT assay
SH-SY5Y cells were seeded in a 96-well plate (1.2 × 104 cells/well) and cultured overnight. After different pharmacological treatments for 48 h, the culture medium was replaced by MTT solution diluted in DMEM/F-12 to obtain a concentration of 0.5 mg/mL and incubated for 4 h. The MTT solution was discarded, and 150 μL DMSO was added to dissolve the formazan crystals by shaking for 10 min. Absorbance was measured at 570 nm using a microplate reader (Synergy HT; BioTek, Broadview, IL, USA).
Flow cytometry
Cellular apoptosis was detected using the Annexin V-FITC/propidium iodide (PI) Cell Apoptosis Detection Kit. SH-SY5Y cells were seeded in a 6-well plate (1.5 × 105 cells/well) and cultured overnight. After different pharmacological treatments for 24 h, the cells were washed with phosphate-buffered saline (PBS) and resuspended in 500 μL binding buffer. Then, 5 μL Annexin and PI was added to the cell suspension. Finally, the cells were detected by flow cytometry (BD FACS-VerseTM Flow Cytometer, BD Biosciences, San Jose, CA, USA).
Transfection of small interfering RNA
Small interfering RNA (siRNA) for PDE4B was synthesized by GenePharma Co. (Shanghai, China). The sequences were as follows: sense, 5′-CCUGCAAGAAGAAUCAUAUTT-3′, and antisense, 5′-AUAUGAUUCUUCUUGCAGGTT-3′. Scrambled siRNA sequences served as the negative control. SH-SY5Y cells were seeded in a six-well plate (1.5 × 105 cells/well) and cultured overnight. Negative control siRNA or PDE4B siRNA and Lipofectamine 2000 were mixed with Opti-MEM for 20 min. The mixture was added to the cell plates for 6 h. Then the culture medium was replaced with DMEM/F-12 supplemented with 10% FBS. The cells were treated with ROT 24 h after transfection.
NAD+/NADH measurement
The relative ratio of NAD+/NADH was detected by an NAD+/NADH assay kit, following the manufacturer’s instructions. SH-SY5Y cells were seeded in a 6-well plate (1.5 × 105 cells/well) and cultured overnight. After different pharmacological treatments for 24 h, the cells were lysed in NAD+/NADH lysis buffer (200 μL/well). To measure total NAD+ and NADH (NADtotal), 20 μL of cell lysates followed by 90 μL of alcohol dehydrogenase working solution was added into a new 96-well plate. To measure NADH, 80 μL of cell lysates was heated at 60 °C for 30 min to decompose NAD+ and 20 μL was added to the 96-well-plate. After 10 min of incubation at 37 °C, 10 μL of chromogenic solution was added to the plate and incubated for 40 min. A standard curve was determined on the same plate on which the sample was tested. Absorbance was measured at 450 nm by a microplate reader (Synergy HT; BioTek). The relative NAD+/NADH was calculated as follows: NAD+/NADH = (NADtotal − NADH)/NADH.
Immunofluorescence analysis
SH-SY5Y cells were seeded in a confocal dish (0.5 × 105 cells/well) and cultured overnight. After different pharmacological treatments for 24 h, the cells were washed three times with PBS and fixed in 4% paraformaldehyde for 20 min. Then, the cells were washed again three times with PBS and permeabilized with 0.2% Triton X-100 for 15 min at room temperature. After 1 h of blocking in 5% bovine serum albumin (BSA), the cells were incubated with primary antibodies against cleaved caspase-3 (1:50) or LAMP1 (1:200) at 4 °C for 12 h followed by incubation with secondary antibody (1:200, A23320, Abbkine) for 2 h at room temperature. Subsequently, the cells were washed three times with PBS and incubated with Hoechst 33342 (2 μg/mL) for 10 min at 37 °C. Finally, the cells were observed and captured with a confocal microscope (Nikon, Tokyo, Japan).
LYT Red stain and AO stain
The lysosome and the acidic compartment of the cells were stained with LYT red stain and AO stain, respectively. After different pharmacological treatments for 24 h, SH-SY5Y cells were incubated with LYT Red (50 nM) or AO (1 μg/mL) for 15 min at 37 °C in the dark. The cells were then washed three times with PBS. Subsequently, the samples were observed and captured with a confocal microscope (Nikon, Tokyo, Japan).
Reverse-transcription PCR for α-syn mRNA
Total RNA from cultured SH-SY5Y cells was extracted using the Total RNA Kit (#R6834; OMEGA, BioTek, Norcross, GA, USA) in accordance with the manufacturer’s instructions. The cDNA was obtained by reverse transcription with the PrimeScriptTM RT Master Mix Kit (#RR036A; Takara Biotechnology Co., Ltd.). PCR products were amplified with 2 × Taq Plus Master Mix (#KT201, TIANGEN BIOTECH, Beijing, China). α-Syn primer sequences were as follows:[21] forward; 5′-ATGTTGGAGGAGCAGTGGTG-3′; reverse; 5′-AATTCCTTCCTGTGGGGCTC-3′. GAPDH was used as the internal control, and GAPDH primer sequences were as follows: forwards, 5′-GCACCACCAACTGCTTAGCA-3′; reverse; 5′-TCTTCTGGGTGGCAGTGATG-3′. Finally, the amplified fragments were resolved by electrophoresis on 2% agarose gels with ethidium bromide. The image of the band of the amplified product was captured using an ultraviolet (UV) transilluminator (BioDoc-It 220 Imaging System, Upland, CA, USA).
SIRT1 activity assay
SIRT1 activity assay was carried out as described previously with minor modification [22]. In brief, SH-SY5Y cells were treated with ROF (10 μM) for 1 h and then treated with ROT (2 μM) for 24 h. After the treatments, the nuclear extracts were collected with a Nuclear and Cytoplasmic Protein Extraction Kit, and the SIRT1 deacetylase activity was measured with a SIRT Activity Assay Kit in line with the manufacturer’s instructions.
Animal treatments
C57BL/6 J male mice (8 weeks old, 20–25 g) were purchased from the Animal Center of Southern Medical University (Guangzhou, China). They have housed five mice per cage at a temperature of 24 °C ± 1 °C under a 12-h light/dark cycle. The mice had free access to food and water. Animal experiments were performed in accordance with the National Institutes of Health (Bethesda, MD, USA) guidelines for animal care and use. The study was approved by the Laboratory Animal Ethics Committee of Southern Medical University. The design of the experiments was based on the 3 R principles (replacement, refinement, and reduction) to ease the pain and reduce the number of mice to the extent possible. Mice in the vehicle group received sunflower seed oil. The other mice were orally administered (i.g.) with ROT (10 mg·kg−1·d−1). ROT was dissolved in sunflower seed oil. After six weeks, ROT-exposed mice were randomly divided into five groups and received ROF (0.5, 1, or 2 mg·kg−1·d−1; i.g.), ROLI (2.5 mg·kg−1·d−1; i.g.), or solvent (0.9% saline containing 40% [2-hydroxypropyl]-β-cyclodextrin and 6% Solutol HS-15; i.g.) for 4 weeks. After the last behavioral test, the mice were sacrificed after inhaling excessive amounts of 5% isoflurane to collect tissue samples (SN) for Western blot analysis.
Pole test
The degree of bradykinesia in mice was evaluated using the pole test as described previously [23]. The pole was a straight wooden rod with gauze to avoid slipping; its height was 60 cm and the diameter was 0.8 cm. The top of the rod was a wooden ball with a diameter of 2.5 cm. Before the test, the mice were trained for 2 days to learn how to climb down the rod. In the test, the mice were placed on the top of the rod, and the time needed to reach the bottom was recorded.
Rotarod test
The motor function in mice was analyzed by the rotarod test as described previously [23, 24]. The mice were trained for 3 days to walk on the rotarod with 20 r/min for 5 min. In the test, the mice were placed on the rotarod with a speed of 15 r/min, and the time required to drop from the rotarod was recorded.
Balance beam test
The balance beam test was used to observe motor coordination in mice and was carried out as previously described [25]. The balance beam was 125 cm long with a width of 6 mm; it was fixed to two poles 50 cm above the floor. The endpoint consisted of a black box placed at the end of the beam. The starting point consisted of a lamp providing stimulatory effects to force the mice to escape to the black box. The distance between the lamp and the box was 100 cm. There were three days of training before the test. The investigator made sure that each mouse could cross the balance beam successfully. In the test, each mouse was placed to the starting point with a lamp open, and the time needed to go through the balance beam to the black box was recorded. At the same time, to evaluate motor deficits in mice, the slipping count was recorded as foot fault.
Western blot analysis
Western blotting was performed as described earlier with minor modification [16]. After different treatments, cells or tissues (SN) were lysed in lysis buffer (#P0013; Beyotime Institute of Biotechnology) on ice for 40 min, and the extracts were centrifuged at 20000 × g in 4 °C for 25 min to obtain the supernatant. After quantitative determination of protein concentrations in the supernatant by the BCA Protein Assay Kit, the mixture contained 5 × loading buffer, and the supernatant was boiled at 95 °C for 10 min. Next, the mixture of protein extracts was resolved by 10% or 12% SDS-PAGE, and then transferred to PVDF membrane, followed by blocking with 5% BSA for 1.5 h at room temperature and washing with TBST three times for 10 min each. Subsequently, the membrane was incubated with primary antibody at 4 °C for 12 h and washed with TBST three times for 10 min. Finally, after 2 h of incubation with secondary antibody at room temperature, the membrane was washed four times with TBST for 5 min each. The proteins were detected by chemiluminescence and quantified using an image analysis system (Version 6.0; Image-Pro Plus).
Immunofluorescence
The mice were perfused with cold physiological saline and then with by 4% PFA. Their brains were moved into 4% PFA and then dehydrated with sucrose solutions (20% and 30%) at 4 °C. The brains were removed from 30% sucrose for frozen sectioning of the SN, and the sections were washed three times with PBS. Then, 5% BSA containing 0.3% Triton 100-X TBST was used to block the sections for 1 h at room temperature, and the sections were incubated with the solution of primary antibody against TH (1:200) at 4 °C for 12 h, followed by the secondary antibody for 2 h at room temperature. After washing with PBS for three times, the tissue sections were removed to glass slides and mounted with antifade Polyvinylpyrrolidone (PVP) Mounting Medium under the cover glass. Images were captured with a confocal microscope.
Statistical analysis
Quantitative analysis of fluorescence intensity, mRNA level, and protein expression was normalized to the mean of the control group. Data are shown as the mean ± standard deviation (SD). The analyses of differences between the means were carried out using one-way ANOVA, followed by Bonferroni’s post-hoc test for pairwise comparisons using SPSS 19.0 (SPSS Inc., Armonk, NY, USA). P < 0.05 was considered to be statistically significant. Graphs were plotted using GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA, USA).
Results
PDE4 inhibition by ROF protects against neuronal apoptosis induced by ROT in SH-SY5Y cells
The chemical structure of ROF is shown in Fig. 1a. SH-SY5Y cells treated with ROT were used as an in vitro cellular model to evaluate the neuroprotective effects of ROF. As shown in Fig. 1b, treatment with ROT (0.5–4 μM) for 48 h triggered cell death in a concentration-dependent manner, with approximately 50%–60% of the cells left viable at 2 μM (P < 0.001). Similarly, treatment with 2 μM of ROT for 24 h induced a significant decrease in the viability of SH-SY5Y cells (Fig. 1c, P < 0.01). Considering that PKA is the canonical downstream protein kinase of PDE4, and the inhibition of PDE4 leads to the activation of PKA [18], we first determined the phosphorylation level of PKA after treatment with ROF. Our results showed that ROT treatment caused a significant decrease in the phosphorylation of PKA at Thr197 (Fig. 1d, e; P < 0.05), while the inhibition of PDE4 by ROF reversed the effect of ROT on PKA phosphorylation (Fig. 1d, e; P < 0.05). Then we detected the viability of SH-SY5Y cells with or without ROF treatment via the MTT assay. As shown in Fig. 1f, ROF (2.5–10 μM) enhanced cell viability in a dose-dependent manner (Fig. 1f). To further verify the protective effects of ROF, we assessed cellular apoptosis through detecting the level of cleaved caspase-3 by Western blotting. We found that ROF (10 μM) alone had no effects on apoptosis, whereas treatment with ROT for 24 h significantly increased the apoptosis of SH-SY5Y cells, as evidenced by increased level of cleaved caspase-3 (P < 0.001). ROF antagonized the neurotoxicity of ROT and significantly decreased the level of cleaved caspase-3 (Fig. 1g, P < 0.01). These data were further confirmed by the immunostaining of cleaved caspase-3 in SH-SY5Y cells. Consistent with the results shown in Fig. 1h, ROT increased the fluorescent intensity, while ROF blocked the effect of ROT and decreased the fluorescence intensity of cleaved caspase-3 (Fig. 1h). These results showed that ROF protected against cellular apoptosis induced by ROT in SH-SY5Y cells.
PDE4 knockdown reduces cellular apoptosis induced by ROT in SH-SY5Y cells
Because inhibition of PDE4 by ROF protects against cellular apoptosis in SH-SY5Y cells, we next investigated whether PDE4 knockdown has similar effects as ROF. SH-SY5Y cells were transfected with specific siRNA to PDE4B, and the knockdown efficiency was confirmed by Western blotting (Fig. 2a, b). Flow cytometry was used to analyze the percentage of apoptotic cells. Compared with the level of apoptosis observed in cells in the negative control (NC) group, ROT-treated cells had a significantly higher percentage of apoptotic cells (Fig. 2c, d; P < 0.001). Similarly, PDE4B siRNA alone did not affect the survival of SH-SY5Y cells. Treatment with ROT decreased the percentage of surviving cells to 60% (P < 0.001). As expected, knockdown of PDE4B reversed the effects of ROT and increased the cell viability to 80% (Fig. 2e. P < 0.001). Taken together, these results suggested that inhibition or knockdown of PDE4 expression exerted neuroprotective effects on SH-SY5Y cells treated with ROT.

a, b The knockdown efficiency of PDE4B siRNA in SH-SY5Y cells was verified by Western blotting. c After transfection with PDE4B siRNA for 24 h, SH-SY5Y cells were treated with ROT (2 μM) for 24 h. The cells were then analyzed using flow cytometry. The rates of apoptosis (d) and survival (e) were detected by flow cytometry. Data are shown as the mean ± SD, n = 3. ##P<0.01, ###P < 0.001 versus the negative control (NC) group. ***P < 0.001 versus the ROT-treated group or NC + ROT group.
ROF decreases the expression of α-syn and enhances the levels of LAMP1 and mature CTSD in SH-SY5Y cells treated with ROT
A previous study has shown that ROT increases the level of α-syn in SH-SY5Y cells [26]. Here, we aimed to clarify whether ROF could attenuate the level of α-syn. The cells were pretreated with ROF (10 μM) for 1 h and then exposed to ROT (2 μM) for 24 h. The protein expression level of α-syn was determined by Western blotting. As shown in Figs. 3a and 3b, compared with cells in the control group, ROT significantly stimulated the expression of α-syn (P < 0.001), whereas ROF attenuated the effect of ROT on the expression of α-syn (P < 0.05). The level of phosphorylated α-syn in SH-SY5Y cells was also detected. We found that ROT (2 μM) treatment caused a significant increase in the phosphorylation of α-syn at Ser129 (Fig. 3c, d; P < 0.01). In contrast, ROF (10 μM) blocked the role of ROT and downregulated the level of phosphorylated α-syn (Fig. 3c, d; P < 0.05). Then, we investigated the role of ROF in the lysosome. ROT decreased the expression level of LAMP1 (P < 0.01), while ROF abolished the effect of ROT (P < 0.05; Fig. 3e, f). Simultaneously, ROF increased the level of mature CTSD in SH-SY5Y cells treated with ROT (P < 0.05; Fig. 3g, h). To further confirm the influence of ROF on lysosome, we labeled the lysosome with a deep-red fluorescent dye (LysoTracker® Deep Red). As shown in Figs. 3i and 3j, compared with the cells in the control group, ROF increased the fluorescence intensity in SH-SY5Y cells (P < 0.01). These results indicated that ROF decreased the level of α-syn, a phenomenon that was accompanied by enhanced function of the lysosome.

SH-SY5Y cells were treated with ROF (10 μM) for 1 h and then treated with ROT (2 μM) for 24 h. (a, c, e, g) The protein expression levels of α-syn, p-α-syn, LAMP1, and CTSD were detected by Western blotting. (b, d, f, h) The relative levels of α-syn/GAPDH, p-α-syn/GAPDH, LAMP1/GAPDH, and mCTSD/proCTSD were measured by densitometric quantitation. (i) LYT Red staining was used to detect the number of the lysosomes. Scale bar = 10 μm. j Quantification of lysotracker fluorescence intensity. Data are shown as the mean ± SD, n = 3. ##P < 0.01, ###P < 0.001 versus the CTL group. *P < 0.05, **P < 0.01 versus the ROT-treated group.
ROF protects SH-SY5Y cells against ROT-induced apoptosis in an NAD+/SIRT1-dependent manner
SIRT1 exerts protective effects against PD [27]. In addition, our previous study has shown that ROF activates AMPK/SIRT1 in microglial cells treated with lipopolysaccharide [20]. Thus, we investigated whether ROF produces neuroprotective effects in an SIRT1-dependent manner. As shown in Fig. 4a, treatment with ROT caused a decrease in the relative content of NAD+/NADH (P < 0.001), while ROF reversed the decrease in NAD+/NADH in the cells treated with ROT (P < 0.01), indicating that NAD+/NADH is possibly involved in the protective effects of ROF. Western blotting further demonstrated an increase in the expression of SIRT1 (P < 0.05; Figs. 4b and 4c), an evolutionarily conserved NAD+-dependent deacetylase. We also verified whether ROF treatment increases SIRT1 deacetylation activity. Our results showed that ROT induced a significant decrease in the activity of SIRT1 in SH-SY5Y cells (P < 0.05), whereas 10 μM ROF reversed the effect of ROT (Fig. 4d, P < 0.05). Next, we investigated the essential role of SIRT1 in the protective effects of ROF. Cells pretreated with SELI (10 μM) were treated with ROF (10 μM) and ROT (2 μM) for 24 h. After the treatment, cell viability and cellular apoptosis were determined with the MTT assay and flow cytometry, respectively. The SIRT1 inhibitor SELI produced no toxic effects at the concentration of 2.5–10 μM (Fig. 4e); hence, SELI at 10 μM was used for the following cell survival and apoptosis assay. As shown in Fig. 4f–h, ROT significantly reduced cell viability and increased apoptosis, while ROF protected SH-SY5Y cells against cell death induced by ROT. Interestingly, the protective effects of ROF against cell death induced by ROT were inhibited by pretreatment with SELI. These results suggested that the NAD+/SIRT1 pathway was involved in the protective effect of ROF.

SH-SY5Y cells were treated with ROF (10 μM) for 1 h and then treated with ROT (2 μM) for 24 h. a The relative level of NAD+/NADH was determined using the NAD+/NADH Assay kit. b The protein expression of SIRT1 was detected by Western blotting. c The relative level of SIRT1/GAPDH was measured by densitometric quantitation. d The activity of SIRT1 was measured by an SIRT Activity Assay Kit. e SH-SY5Y cells were treated with various concentrations of SELI for 48 h. Cell viability was determined by the MTT assay. f After 1 h pretreatment with SELI (10 μM), SH-SY5Y cells were treated with ROF (10 μM) for 1 h and then treated with ROT (2 μM) for 48 h. Cell viability was measured by the MTT assay. g, h After 1 h pretreatment with SELI (10 μM), SH-SY5Y cells were treated with ROF (10 μM) for 1 h and then exposed to ROT (2 μM) for 24 h. Cell apoptosis was analyzed by flow cytometry. Data are shown as the mean ± SD, n = 3. #P < 0.05, ##P < 0.01, ###P < 0.001 versus the CTL group. *P < 0.05, **P < 0.01, ***P < 0.001 versus the ROT-treated group. $P<0.05, $$P < 0.01 versus the ROF + ROT-treated group.
Effects of ROF on the clearance of α-syn are mediated by the SIRT1 pathway
Because ROF protects SH-SY5Y cells against apoptotic cell death induced by ROT in the SIRT1-dependent manner, we next investigated the involvement of SIRT1 in the clearance of α-syn. As shown in Figs. 5a and 5b, treatment with ROT led to a significant increase in the protein level of α-syn (P < 0.01). ROF reduced the protein level of α-syn (P < 0.05), whereas SELI reversed the effect of ROF and decreased the level of α-syn (P < 0.05). To determine whether the alteration of α-syn was due to increased protein synthesis or hindered degradation, we detected the mRNA level of α-syn. The results are shown in Figs. 5c and 5d. We found that SELI, ROT, and ROF had no effects on the mRNA level of α-syn, indicating that ROF had no effects on the transcription of α-syn. ROF may decrease the expression of α-syn by promoting the degradation of α-syn; hence, we studied the roles of ROF and SELI on the function of lysosome. ROF significantly enhanced the expression of LAMP1 in the cells treated with ROT (P < 0.05). SELI, at 10 μM, partly inhibited the expression of LAMP1 (P < 0.05; Figs. 5e and 5f). We also performed immunofluorescence assays to investigate the involvement of SIRT1 in the protective actions of ROF described above. ROF appreciably increased the level of LAMP1 in SH-SY5Y cells, while SELI strongly decreased the intensity of immunofluorescence of LAMP1 (Fig. 5g). Consistently, ROF enhanced the level of mature CTSD, while the inhibition of SIRT1 blocked the effect of ROF and reduced the level of mature CTSD (Figs. 5h and 5i). Given that inhibition of SIRT1 had no effect on the transcription of α-syn, our results support the role of SIRT1 in mediating the function of the lysosome. These results indicated that ROF could promote the clearance of α-syn by upregulating the protein expression of SIRT1 and subsequently activating the lysosome.

After 1 h treatment with SELI (10 μM), SH-SY5Y cells were treated with ROF (10 μM) for 1 h and then treated with ROT (2 μM) for 24 h. (a) The protein expression of α-syn was detected by Western blotting. (b) The relative level of α-syn/GAPDH was measured by densitometric quantitation. (c and d) The mRNA level of α-syn was detected by RT-PCR. (e and h) The protein expression levels of LAMP1 and CTSD were detected by Western blotting. (f and i) The relative levels of LAMP1/GAPDH and mCTSD/proCTSD were measured by densitometric quantitation. (g) The expression of LAMP1 was determined by immunofluorescence analysis. Scale bar, 10 μm. The protein expression level of CTSD was detected by Western blotting. Data are shown as the mean ± SD, n = 3. ##P < 0.01, ###P < 0.001 versus the CTL group. *P < 0.05 versus the ROT-treated group. $P < 0.05, $$P < 0.01 versus the ROF + ROT-treated group.
Inhibition of lysosomal protease CTSD abolishes the effect of ROF in clearing α-syn
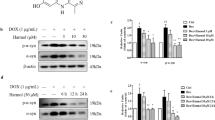
Because the degradation of α-syn is primarily mediated through the lysosomal pathway, we evaluated whether a lysosomal inhibitor would block the clearance of α-syn. Pepstatin A was used to inhibit the function of the lysosome to further explore the effects of ROF on α-syn clearance. We first confirmed that PA produced no toxic effects on SH-SY5Y cells at the concentration of 1.25–20 μM (Fig. 6a). The cells were then pretreated with pepstatin A (20 μM) in the presence of ROF (10 μM), and then stimulated with ROT (2 μM) for 24 h. Lysotracker staining and acridine orange (AO) staining were used to observe the changes in the number of lysosomes and acidic vesicles. The results are shown in Fig. 6b. ROT treatment caused a decrease in the intensity of red fluorescence compared with the control cells, indicating that the number of lysosomes in the cells was reduced. ROF pretreatment enhanced the intensity of red fluorescence, while the effect of ROF was reversed by pepstatin A. Similarly, AO staining showed that ROF reversed the decrease in AO fluorescence intensity caused by ROT, while pepstatin A blocked the effects of ROF on the number of acidic vesicles (Fig. 6c). We investigated the consequence of pepstatin A treatment on the survival of SH-SY5Y cells. As shown in Fig. 6d, pepstatin A clearly blocked the pro-survival effects of ROF in a dose-dependent manner (Fig. 6d). Then, we determined whether pepstatin A affected the protein level of α-syn. ROF reduced the protein level of α-syn (P < 0.05; Fig. 6e and f). As expected, the lysosomal inhibitor pepstatin A blocked the clearance of α-syn by ROF (P < 0.01). These results indicated that inhibiting the function of lysosome blocked the scavenging effects of ROF on α-syn.

a SH-SY5Y cells were treated with different concentrations of pepstatin A (1.25–80 μM) for 48 h. Cell viability was determined by the MTT assay. b, c After 1 h pretreatment with pepstatin A (20 μM), SH-SY5Y cells were treated with ROF (10 μM) for 1 h and then treated with ROT (2 μM) for 24 h. LYT Red and AO staining were used to detect the numbers of the lysosomes and acidic vesicles. Scale bar, 10 μm. d After 1 h pretreatment with pepstain A, SH-SY5Y cells were treated with ROF (10 μM) for 1 h and then treated with ROT (2 μM) for 48 h. Cell viability was evaluated with the MTT assay. e After 1 h pretreatment with pepstatin A (20 μM), SH-SY5Y cells were treated with ROF (10 μM) for 1 h and then exposed to ROT (2 μM) for 24 h. The protein expression of α-syn was detected by Western blotting. f The relative level of α-syn/GAPDH was measured by densitometric quantitation. Data are shown as mean ± SD, n = 3. ##P < 0.01, ###P < 0.001 versus the CTL group. *P < 0.05, ***P < 0.001 versus the ROT-treated group. $$P < 0.01, $$$P < 0.001 versus the ROF + ROT-treated group. PA, pepstatin A.
ROF ameliorates motor deficits in mice treated with ROT
Next, we determined whether ROF would protect against neuronal apoptosis and ameliorate motor deficits in mice treated with ROT. A schematic diagram of the experimental procedure is shown in Fig. 7a. ROT was administered to C57BL/6 J mice by intragastric administration at a dose of 10 mg·kg−1·d−1 for 6 weeks. ROF (0.5, 1, or 2 mg·kg−1·d−1) was administered once a day for 4 consecutive weeks from week 7. ROLI, a canonical PDE4 inhibitor, was used as a positive control. The doses of ROF and ROLI were based on our previous studies [18, 28]. The pole test, rotarod test, and balance beam test were used to evaluate the motor functions. The results are shown in Fig. 7b-e. Compared with the control mice, the time required for the model group mice to successfully climb from the top of the rod to the bottom (latency period) was significantly longer (13.60 ± 2.27 s; P < 0.001). Compared with the mice in the model group, ROF (1, or 2 mg·kg−1·d−1) significantly shortened the latency period (Fig. 7b). The rotarod test showed that the mice in the control group continued to move on the roller for about 60 s (56.50 ± 13.48 s), and treatment with ROT greatly shortened the time for the mouse to move on the roller (the latency to fall) (P < 0.001). However, ROF increased the latency to fall in a dose-dependent manner (Fig. 7c). When the dose was 1 mg·kg−1·d−1, the latency to fall was 31.90 ± 6.54 s (P < 0.01), and the latency to fall at 2 mg·kg−1·d−1 was 40.70 ± 11.46 s (P < 0.001). We set up a balance beam experiment to explore the coordination ability of the mice. The results are shown in Figs. 7d and 7e. Compared with the control mice, the continuous administration of ROT increased foot fault count when the mice crossed the balance beam (P < 0.001), and increased the length of time for the mice to cross the balance beam (17.30 ± 3.89 s, P < 0.001). ROF at doses of 1 and 2 mg·kg−1·d−1 significantly reduced the foot fault count and shortened the amount of time it took to cross the balance beam. In all of these experiments, and ROLI showed similar effects on ameliorating motor deficits. These data suggested that ROF was able to improve the motor coordination ability in mice treated with ROT.

a Experimental timeline. C57BL/6 J male mice were orally treated with ROT (10 mg·kg−1·d−1) for 6 weeks and then treated with ROF (0.5, 1, or 2 mg·kg−1·d−1) or ROLI (2.5 mg·kg−1·d−1) for 4 weeks. b, c, d, e The pole, rotarod, and balanced beam tests were used to evaluate the degree of bradykinesia, motor function, and motor coordination, respectively. Data are shown as the mean ± SD, n = 10. ###P < 0.001 versus the vehicle group. *P < 0.01, **P < 0.01, ***P < 0.001, versus the ROT-treated group.
ROF protects against dopaminergic degeneration and promotes the clearance of α-syn in the SN of mice treated with ROT
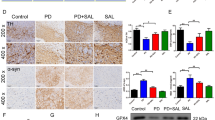
ROF is a specific PDE4 inhibitor that can cross the blood–brain barrier. The phosphorylation of PKA was determined in the SN. We found that ROT treatment decreased the level of phosphorylated PKA (Fig. 8a, b; P < 0.01). Consistent with the data obtained in SH-SY5Y cells, ROF increased the phosphorylated PKA level in the SN in a concentration-dependent manner (Fig. 8a, b). The typical pathological feature of PD is the loss of dopaminergic neurons in the SN. We detected the level of TH, a marker of dopaminergic neurons in the SN by Western blotting. The administration of ROF and ROLI significantly increased the protein expression level of TH in the SN of the mice (Fig. 8c, d). These data were further confirmed by the immunostaining, showing that ROT caused a significant loss of dopaminergic neurons, whereas ROF reversed the effects of ROT and increased the fluorescence intensity of TH (Fig. 8e, f). We found that ROF significantly reduced the content of α-syn in the SN of the mice subjected to ROT (Fig. 8g, h). Consistent with the data from SH-SY5Y cells, the levels of LAMP1 and SIRT1 were significantly decreased in the SN of mice treated with ROT. ROF treatment almost completely abolished the decrease in expression levels of LAMP1 and SIRT1 in the SN of ROT-treated mice (Fig. 8i–l). Taken together, these data suggested that ROF enhanced the function of lysosomes by upregulating SIRT1 to promote the clearance of α-syn.

a, c, g, i, k Protein expression levels of p-PKA, TH, α-syn, SIRT1, and LAMP1 in the SN of the brain in mice were detected by Western blotting. b, d, h, j, l The relative levels of p-PKA/PKA, TH/GAPDH, α-syn/GAPDH, SIRT1/GAPDH, and LAMP1/GAPDH were measured by densitometric quantitation. e Representative photographs showing TH protein expression in SN of the brain in mice were detected by immunofluorescence staining. f Fluorescence intensity of TH staining. Scale bar, 50 μm. Data are shown as the mean ± SD, n = 3. ##P < 0.01, ###P < 0.001 versus the vehicle group. *P < 0.05, **P < 0.01, ***P < 0.001 versus the ROT-treated group.
Discussion
The major finding of this study is that the inhibition of PDE4 by ROF enhanced the viability of SH-SY5Y cells treated with ROT. In addition, ROF ameliorated the motor dysfunction and increased the protein level of TH in the SN in a mouse model of PD induced by ROT. The underlying mechanism of action is that ROF enhances the function of lysosomes by activating NAD + /SIRT1 signaling, thus promoting the clearance of α-syn by the lysosome. Summary of the protective effects of ROF against ROT-induced injury and the underlying mechanism is shown in Fig. 9. To the best of our knowledge, this is the first study to investigate the role of PDE4 in the clearance of α-syn and the potential mechanisms. Our results suggest that PDE4 can be used as a potential target for the treatment of PD, and ROF is a promising candidate compound for promoting the clearance of α-syn. In the present study, we verified that the inhibition of PDE4 exerted neuroprotective effects in PD models. Although the inhibitory effect of ROF against the PDE4 enzyme has been tested previously [18], the PDE4 enzymatic activity in SH-SY5Y cells treated with ROT was not determined in the present study. Determination of PDE4 enzymatic activity would be useful to justify the use of PDE4 inhibitors in the treatment of PD.

ROT treatment leads to the impairment of the lysosome and the decreased degradation of α-syn. Whereas ROF promotes the clearance of α-syn by enhancing lysosomal function in a NAD+/SIRT1-dependent manner, thereby increasing the cell viability of SH-SY5Y cells and restoring motor functions in ROT-treated mice.
During the development of PD, as the abnormal accumulation of α-syn increases, the damage of neurons is further aggravated, eventually leading to the death of neurons. We found that ROT induced damage of SH-SY5Y cells, resulting in a decreased cell survival rate, and knockdown of PDE4B expression in the cells significantly enhanced the survival rate of the injured cells. In line with these findings, ROF also exerted neuroprotective effects in the mice subjected to ROT. These data verified that treatment with ROF counteracted the cell damage caused by ROT and exhibited excellent neuroprotective effects in PD models. Attenuating the accumulation of α-syn in the cells is a potential strategy for preventing the death of neurons [29]. The lysosomal system is one of the major pathways mediating the degradation of α-syn. Therefore, enhancing the function of lysosomes to promote the elimination of α-syn may be a therapeutic strategy for reducing neuronal damage [30]. Based on the above observations, we next explored the underlying mechanisms, and found that ROT increased the protein level of α-syn in SH-SY5Y cells, consistent with the results reported by other laboratories [26, 31]. It is encouraging that ROF reversed the increase in α-syn. We found that ROF was also able to increase the fluorescence intensity of lysosomes and increase the protein levels of LAMP1 and mature CTSD in the cells, emphasizing the ability of ROF to enhance lysosomal function. Therefore, we propose the hypothesis that the effect of ROF on the clearance of α-syn is mediated through enhancing the function of lysosomes. Our results showed that the level of α-syn decreased after treatment with ROF. Of note, α-syn is a component of the Lewy body. One of the limitations of the present study is that the effect of ROF on the formation of the Lewy body was not determined. Further studies should investigate whether PDE4 is involved in the processes that govern α-syn fibrillization and Lewy body formation. It is worth noting that α-syn can be digested by both the ALP and UPS. This study mainly focused on the ALP. Of note, promoting the degradation of α-syn by enhancing the UPS also produces neuroprotective effects [32]. Therefore, the effects of ROF on ubiquitination and protease activity need to be further studied. Additionally, ROT is an inhibitor of the respiratory chain complex I in the mitochondria [33]. Substantial studies have focused on oxidative stress and the subsequent cell death induced by ROT [34, 35]. Our present study links the neurotoxic effects of ROT to the impairment of the lysosome both in vitro and in vivo, as evidenced by the decreased levels of LAMP1 and mature CTSD. Our data are consistent with the previous findings that showed that the average number of lysosomal puncta per dopamine neuron is reduced in the brains of individuals suffering from PD compared with that of age-matched controls [36]. Taken together, these results support the role of lysosomal dysfunction in the pathogenesis of PD. Importantly, we identified that inhibition of PDE4 by ROF was effective to enhance the function of the lysosome. This study provides support for PDE4 as a therapeutic target for individuals with PD.
As a member of the SIRT family, SIRT1 is essentially a histone deacetylase regulated by NAD+ [37]. However, SIRT1 may also interact with multiple non-histone proteins, thereby participating in the regulation of protein degradation, mitochondrial dysfunction, oxidative stress, and neuroinflammation [38]. The protein expression of SIRT1 is downregulated in PD models both in vitro and in vivo;[39] similarly, clinical studies have indicated that compared with healthy individuals, the enzyme activity of SIRT1 is significantly lower in PD patients [40]. Hence, SIRT1 is viewed as a pro-survival protein that is downregulated under conditions of cellular stress, including PD [40]. The activation or increased expression of SIRT1 can combat the damage of dopaminergic neurons caused by various neurotoxins. For example, resveratrol can reduce the toxic effects of α-syn by activating SIRT1 in the SH-SY5Y cells treated with 6-hydroxydopamine [41]. Overexpression of SIRT1 can also reduce the production of α-syn oligomers in cells to resist the damage of SH-SY5Y cells induced by ROT [40]. Consistent with these findings, our study verified that the inhibition of PDE4 by ROF promoted the clearance of α-syn by activating the NAD+/SIRT1 signaling pathway. It is worth noting that ROT causes the accumulation of α-syn in neurons before dopaminergic degeneration [36]. ROF can prevent ROT-triggered α-syn accumulation and neurodegeneration. Hence, ROF is also promising for the prevention of PD. Taken together, these data indicate that downregulation of SIRT1 plays a critical role in PD development, and SIRT1 activators might be potential molecules for PD therapy. Our results support that regulating SIRT1 expression by PDE4 inhibitors is a potential therapeutic strategy in PD. The inhibition of PDE4 promotes the accumulation of cAMP, and the subsequent activation of the downstream molecules, such as PKA [42]. The effects of cAMP-mediated signaling on the activation of SIRT1 have been reported in different diseases. For example, activation of SIRT1 by cAMP/PKA signaling prevents doxorubicin-associated cardiotoxicity [43]. Activation of the cAMP/PKA pathway also protects against palmitate-induced toxicity in hepatocytes through activating NAD+/SIRT1 [44]. In this study, we propose that ROF exerts neuroprotection by upregulating NAD+/SIRT1. However, it remains to be determined whether the effects of ROF on SIRT1 expression are mediated directly or indirectly. Additionally, Our study showed that the inhibition of SIRT1 impaired the lysosome function. However, it is less understood how SIRT1 controls lysosomal activity. Previous studies have suggested that SIRT1 is a positive regulator of ATP6V1A, which plays a pivotal role in the acidification of lysosomes. In addition, SIRT1 is an upstream molecule of the transcription factor EB (TFEB), which is a master gene for lysosomal biogenesis. Further studies are needed to unravel whether PDE4 inhibition will affect the functions of ATP6V1A and TFEB.
In summary, we explored the protective effects of ROF on the clearance of α-syn as well as the underlying mechanisms. We first evaluated the neuroprotective effect of ROF both in vitro and in vivo. Then we examined the levels of α-syn, LAMP1, and the mature form of CTSD. Moreover, we detected the levels of NAD+ and SIRT1 in these experimental models. Our findings showed that ROF increased the cell viability of SH-SY5Y cells and restored motor functions in ROT-treated mice. According to our results, ROF is a positive regulator of SIRT1 in neuronal cells. The ROF-mediated lysosomal system is critical for regulating α-syn homeostasis; thus, we propose that ROF is a promising candidate for the treatment of PD.
References
Elfil M, Bahbah EI, Attia MM, Eldokmak M, Koo BB. Impact of obstructive sleep apnea on cognitive and motor functions in Parkinson’s disease. Mov Disord. 2021;36:570–80.
Kabra A, Sharma R, Kabra R, Baghel US. Emerging and alternative therapies for Parkinson disease: an updated review. Curr Pharm Des. 2018;24:2573–82.
Krauss JK, Lipsman N, Aziz T, Boutet A, Brown P, Chang JW, et al. Technology of deep brain stimulation: current status and future directions. Nat Rev Neurol. 2021;17:75–87.
Fox SH, Katzenschlager R, Lim SY, Barton B, de Bie RMA, Seppi K, et al. International Parkinson and movement disorder society evidence-based medicine review: Update on treatments for the motor symptoms of Parkinson’s disease. Mov Disord. 2018;33:1248–66.
Borghammer P. The alpha-synuclein origin and connectome model (SOC Model) of Parkinson’s disease: explaining motor asymmetry, non-motor phenotypes, and cognitive decline. J Parkinsons Dis. 2021;11:455–74.
Fares MB, Jagannath S, Lashuel HA. Reverse engineering Lewy bodies: how far have we come and how far can we go? Nat Rev Neurosci. 2021;22:111–31.
Jiang TF, Chen SD. Dysfunction of two lysosome degradation pathways of alpha-synuclein in Parkinson’s disease: potential therapeutic targets? Neurosci Bull. 2012;28:649–57.
Xilouri M, Brekk OR, Stefanis L. alpha-Synuclein and protein degradation systems: a reciprocal relationship. Mol Neurobiol. 2013;47:537–51.
Abuarab N, Munsey TS, Jiang LH, Li J, Sivaprasadarao A. High glucose-induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn2+-mediated mitochondrial fission. Sci Signal. 2017;10:eaal4161.
Wang H, Gaur U, Xiao J, Xu B, Xu J, Zheng W. Targeting phosphodiesterase 4 as a potential therapeutic strategy for enhancing neuroplasticity following ischemic stroke. Int J Biol Sci. 2018;14:1745–54.
Zhang C, Cheng Y, Wang H, Wang C, Wilson SP, Xu J, et al. RNA interference-mediated knockdown of long-form phosphodiesterase-4D (PDE4D) enzyme reverses amyloid-beta42-induced memory deficits in mice. J Alzheimers Dis. 2014;38:269–80.
Bhat A, Ray B, Mahalakshmi AM, Tuladhar S, Nandakumar DN, Srinivasan M, et al. Phosphodiesterase-4 enzyme as a therapeutic target in neurological disorders. Pharmacol Res. 2020;160:105078.
Yamashita N, Hayashi A, Baba J, Sawa A. Rolipram, a phosphodiesterase-4-selective inhibitor, promotes the survival of cultured rat dopaminergic neurons. Jpn J Pharmacol. 1997;75:155–9.
Hulley P, Hartikka J, Abdel’Al S, Engels P, Buerki HR, Wiederhold KH, et al. Inhibitors of type IV phosphodiesterases reduce the toxicity of MPTP in substantia nigra neurons in vivo. Eur J Neurosci. 1995;7:2431–40.
Zhong J, Xie J, Xiao J, Li D, Xu B, Wang X, et al. Inhibition of PDE4 by FCPR16 induces AMPK-dependent autophagy and confers neuroprotection in SH-SY5Y cells and neurons exposed to MPP+-induced oxidative insult. Free Radic Biol Med. 2019;135:87–1.
Zhong J, Dong W, Qin Y, Xie J, Xiao J, Xu J, et al. Roflupram exerts neuroprotection via activation of CREB/PGC-1alpha signalling in experimental models of Parkinson’s disease. Br J Pharmacol. 2020;177:2333–50.
Zheng S, Kaur G, Wang H, Li M, Macnaughtan M, Yang X, et al. Design, synthesis, and structure-activity relationship, molecular modeling, and NMR studies of a series of phenyl alkyl ketones as highly potent and selective phosphodiesterase-4 inhibitors. J Med Chem. 2008;51:7673–88.
Guo H, Cheng Y, Wang C, Wu J, Zou Z, Niu B, et al. FFPM, a PDE4 inhibitor, reverses learning and memory deficits in APP/PS1 transgenic mice via cAMP/PKA/CREB signaling and anti-inflammatory effects. Neuropharmacology. 2017;116:260–9.
You T, Cheng Y, Zhong J, Bi B, Zeng B, Zheng W, et al. Roflupram, a phosphodiesterase 4 inhibitior, suppresses inflammasome activation through autophagy in microglial cells. ACS Chem Neurosci. 2017;8:2381–92.
Li D, Xu J, Qin Y, Cai N, Cheng Y, Wang H. Roflupram, a novel phosphodiesterase 4 inhibitor, inhibits lipopolysaccharide-induced neuroinflammatory responses through activation of the AMPK/Sirt1 pathway. Int Immunopharmacol. 2021;90:107176.
St-Amour I, Turgeon A, Goupil C, Planel E, Hebert SS. Co-occurrence of mixed proteinopathies in late-stage Huntington’s disease. Acta Neuropathol. 2018;135:249–65.
Kurano M, Hara M, Nojiri T, Ikeda H, Tsukamoto K, Yatomi Y. Resveratrol exerts a biphasic effect on apolipoprotein M. Br J Pharmacol. 2016;173:222–33.
Guo YL, Duan WJ, Lu DH, Ma XH, Li XX, Li Z, et al. Autophagy-dependent removal of alpha-synuclein: a novel mechanism of GM1 ganglioside neuroprotection against Parkinson’s disease. Acta Pharmacol Sin. 2021;42:518–28.
Liu J, Liu W, Lu Y, Tian H, Duan C, Lu L, et al. Piperlongumine restores the balance of autophagy and apoptosis by increasing BCL2 phosphorylation in rotenone-induced Parkinson disease models. Autophagy. 2018;14:845–61.
Ren ZL, Wang CD, Wang T, Ding H, Zhou M, Yang N, et al. Ganoderma lucidum extract ameliorates MPTP-induced parkinsonism and protects dopaminergic neurons from oxidative stress via regulating mitochondrial function, autophagy, and apoptosis. Acta Pharmacol Sin. 2019;40:441–50.
Ramalingam M, Huh YJ, Lee YI. The impairments of alpha-synuclein and mechanistic target of rapamycin in rotenone-induced SH-SY5Y cells and mice model of Parkinson’s disease. Front Neurosci. 2019;13:1028.
Feng Y, Liu T, Dong SY, Guo YJ, Jankovic J, Xu H, et al. Rotenone affects p53 transcriptional activity and apoptosis via targeting SIRT1 and H3K9 acetylation in SH-SY5Y cells. J Neurochem. 2015;134:668–76.
Xie J, Bi B, Qin Y, Dong W, Zhong J, Li M, et al. Inhibition of phosphodiesterase-4 suppresses HMGB1/RAGE signaling pathway and NLRP3 inflammasome activation in mice exposed to chronic unpredictable mild stress. Brain Behav Immun. 2021;92:67–77.
Xu MM, Ryan P, Rudrawar S, Quinn RJ, Zhang HY, Mellick GD. Advances in the development of imaging probes and aggregation inhibitors for alpha-synuclein. Acta Pharmacol Sin. 2020;41:483–98.
Schmidt MF, Gan ZY, Komander D, Dewson G. Ubiquitin signalling in neurodegeneration: mechanisms and therapeutic opportunities. Cell Death Differ. 2021;28:570–90.
Li M, Hu J, Yuan X, Shen L, Zhu L, Luo Q. Hepcidin decreases rotenone-induced alpha-Synuclein accumulation via autophagy in SH-SY5Y cells. Front Mol Neurosci. 2020;13:560891.
Cai CZ, Zhou HF, Yuan NN, Wu MY, Lee SM, Ren JY, et al. Natural alkaloid harmine promotes degradation of alpha-synuclein via PKA-mediated ubiquitin-proteasome system activation. Phytomedicine. 2019;61:152842.
Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, et al. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003;278:8516–25.
Tapias V, McCoy JL, Greenamyre JT. Phenothiazine normalizes the NADH/NAD+ ratio, maintains mitochondrial integrity and protects the nigrostriatal dopamine system in a chronic rotenone model of Parkinson’s disease. Redox Biol. 2019;24:101164.
Farombi EO, Awogbindin IO, Olorunkalu PD, Ogbuewu E, Oyetunde BF, Agedah AE, et al. Kolaviron protects against nigrostriatal degeneration and gut oxidative damage in a stereotaxic rotenone model of Parkinson’s disease. Psychopharmacol (Berl). 2020;237:3225–36.
Rocha EM, De Miranda BR, Castro S, Drolet R, Hatcher NG, Yao L, et al. LRRK2 inhibition prevents endolysosomal deficits seen in human Parkinson’s disease. Neurobiol Dis. 2020;134:104626.
Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–6.
Li X, Feng Y, Wang XX, Truong D, Wu YC. The critical role of SIRT1 in Parkinson’s disease: mechanism and therapeutic considerations. Aging Dis. 2020;11:1608–22.
Pallas M, Pizarro JG, Gutierrez-Cuesta J, Crespo-Biel N, Alvira D, Tajes M, et al. Modulation of SIRT1 expression in different neurodegenerative models and human pathologies. Neuroscience. 2008;154:1388–97.
Singh P, Hanson PS, Morris CM. SIRT1 ameliorates oxidative stress induced neural cell death and is down-regulated in Parkinson’s disease. BMC Neurosci. 2017;18:46.
Albani D, Polito L, Batelli S, De Mauro S, Fracasso C, Martelli G, et al. The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by alpha-synuclein or amyloid-beta (1-42) peptide. J Neurochem. 2009;110:1445–56.
Schreiber R, Hollands R, Blokland A. A mechanistic rationale for PDE-4 inhibitors to treat residual cognitive deficits in acquired brain injury. Curr Neuropharmacol. 2020;18:188–201.
Hu C, Zhang X, Song P, Yuan YP, Kong CY, Wu HM, et al. Meteorin-like protein attenuates doxorubicin-induced cardiotoxicity via activating cAMP/PKA/SIRT1 pathway. Redox Biol. 2020;37:101747.
Li J, Dou X, Li S, Zhang X, Zeng Y, Song Z. Nicotinamide ameliorates palmitate-induced ER stress in hepatocytes via cAMP/PKA/CREB pathway-dependent Sirt1 upregulation. Biochim Biophys Acta. 2015;1853:2929–36.
Acknowledgements
This work was supported by Guangdong Basic and Applied Basic Research Foundation (2021A1515011133), Science and Technology Program of Guangzhou (No. 202002030494), National Natural Science Foundation of China (Nos. 81773698 and 81974501), Key-Area Research and Development Program of Guangdong Province (No. 2018B030334001), the Program for Changjiang Scholars and Innovative Research Team in University (IRT_16R37).
Author information
Authors and Affiliations
Contributions
WLD, JHZ, YQC, JFX, YYQ, NBC, LL and MFL performed research and analyzed data. WLD, HTW and JPX wrote the paper. HTW and JPX conceived and designed research. All authors have given their final approval for the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Dong, Wl., Zhong, Jh., Chen, Yq. et al. Roflupram protects against rotenone-induced neurotoxicity and facilitates α-synuclein degradation in Parkinson’s disease models. Acta Pharmacol Sin 42, 1991–2003 (2021). https://doi.org/10.1038/s41401-021-00768-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00768-4
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
PI3K/AKT signaling activation by roflumilast ameliorates rotenone-induced Parkinson’s disease in rats
Inflammopharmacology (2024)
-
Targeting NAD Metabolism for the Therapy of Age-Related Neurodegenerative Diseases
Neuroscience Bulletin (2024)
-
Targeting phosphodiesterase 4 as a potential therapy for Parkinson’s disease: a review
Molecular Biology Reports (2024)
-
Dopaminergic neurodegeneration in the substantia nigra is associated with olfactory dysfunction in mice models of Parkinson’s disease
Cell Death Discovery (2023)
-
Phosphodiesterase-4 Inhibition in Parkinson’s Disease: Molecular Insights and Therapeutic Potential
Cellular and Molecular Neurobiology (2023)