
Abstract
This study tested the efficacy of the phosphodiesterase type III inhibitor cilostazol in Alzheimer’s disease patients with white matter lesions treated with donepezil in comparison with donepezil monotherapy using fluorodeoxyglucose (18F) positron-emission tomography (FDG PET). A 24-week, randomized, double-blind, placebo-controlled, parallel-group study was conducted. Thirty-six Alzheimer’s disease patients with white matter lesions who received donepezil (n = 18 each in the cilostazol and placebo groups) were enrolled. Participants underwent pre and post FDG PET imaging scans and three rounds of clinical and neuropsychological tests. The cilostazol group did not show a significant decrease of regional glucose metabolism; however, regional glucose metabolism was significantly decreased in the parietal and frontal lobes of the placebo group. The repeated measures ANOVA measuring differences in uptake change revealed that regional glucose metabolism in the left inferior frontal gyrus was significantly more preserved in the cilostazol group than that in the placebo group (p < 0.005). Mean changes from baseline on the Mini-Mental State Exam, Alzheimer’s Disease Assessment Scale-cognitive subscale, Alzheimer’s Disease Cooperative Study-Activities of Daily Living Inventory, and the Clinical Dementia Rating Sum of Boxes did not differ between the two groups. In the cilostazol group, the increase of glucose metabolism correlated with the improvment of the Alzheimer’s Disease Assessment Scale-cognitive score. We conclude that cilostazol treatment added to donepezil may delay the decline in regional cerebral metabolism in Alzheimer’s disease with white matter lesions compared with donepezil monotherapy. In additon, our results verified the efficacy of cilostazol in improving or protecting cognitive function in Alzheimer’s disease through increased glucose metabolism. However, the long-term effect of cilostazol on cognitive function and Alzheimer’s disease modification must be tested in further studies with larger sample size and longer study period. Trial registration: http://clinicaltrials.gov: NCT01409564
Similar content being viewed by others


Introduction
Until recently, acetylcholinesterase inhibitors (AChEI), which increase acetylcholine levels, and memantine, a partial N-methyl-d-aspartate receptor antagonist, were the only drugs approved for the treatment of Alzheimer’s disease (AD). However, an increasing number of preclinical animal studies suggest that phosphodiesterase (PDE) inhibitors may delay AD progression.
PDEs are key enzymes in the intracellular signal transduction pathways. They hydrolyze and inactivate cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP), which act as second messengers in several biochemical processes [1]. PDE inhibitor–induced increase of cAMP activates protein kinase A and the subsequent phosphorylation of cAMP response element-binding (CREB) protein. CREB phosphorylation activates several target genes that trigger new protein synthesis, thus strengthening existing synaptic connections and forming new ones related to memory consolidation [2]. PDE inhibitors may improve long-term memory and exert a neuroprotective effect through these mechanisms.
To date, PDE 2~5 inhibitors including cilostazol have been shown to improve memory and protect brain synapses in preclinical animal models [3,4,5,6]. Cilostazol is a common PDE3 inhibitor which strongly upregulates both cAMP and cGMP. It prevents platelet aggregation and is usually used to treat peripheral arterial disease and prevent stroke [7, 8]. Additionally, cilostazol has been shown to have a protective effect against hypoperfusion-induced white matter damage [4] and to enhance generation of immature neuroblasts in the ischemic hippocampus in animal studies [9]. Furthermore, in an AD mouse model, cilostazol has been shown to decrease the deposition of neurotoxic beta amyloid protein accumulation and to increase its degradation [10]. In a study of AD patients who received combination therapy with donepezil and cilostazol, improved Mini-Mental State Exam (MMSE) scores were reported, suggesting potential for cilostazol treatment in delaying cognitive decline in patients receiving donepezil with mild AD [11]. A similar finding was also reported that combination therapy with cilostazol was a good therapeutic indicator for improving MMSE scores [12].
These findings suggest that cilostazol can delay the progression of AD and the changes of white matter related to vascular damages by enhancing neuronal protection and regeneration. Therefore, it is hypothesized that patients who have AD with white matter lesions will be especially benefited by cilostazol administration. Cilostazol also has synergistic effect on protecting and strengthening neuronal functions with donepezil, which increases synaptic acetylcholine concentration level [13]. To test this hypothesis, we conducted a 24-week, randomized, double-blind, placebo-controlled, parallel-group Cilostazol Administration Study In Dementia (CASID) for AD patients with white matter lesions who have received donepezil monotherapy. We used fluorodeoxyglucose (18F) positron-emission tomography (FDG PET) as the primary outcome measure because the changes in regional cerebral metabolism detected by FDG PET are highly sensitive to synaptic changes which are estimated to have begun before the onset of cognitive symptom changes [14], and these FDG PET metabolic changes are highly correlated with clinical disease severity changes [15].
Methods
Participants
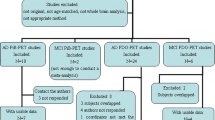
Drawing from one dementia clinic, we enrolled 46 participants with a diagnosis of AD with white matter lesions and concomitant use of donepezil (10 mg) for more than 3 months before onset of the trial. According to the previous study, 11~39 subjects are needed in each group based on power analysis study reflecting treatment responses which was planned to determine the number of patients per group needed to detect reflecting 50% percentage FDG PET regional reduction changes of decline in patients with AD with 80% power and p ≤ 0.05 (two-tailed) [16]. There was no participant referral from the pharmaceutical company. The diagnosis of probable AD was made according to the criteria of the National Institute of Neurological and Communicative Disorders and Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) [17]. Only patients with any white matter lesions according to the Fazekas criteria [18] were enrolled. The additional inclusion criteria were as follows: screening and baseline MMSE scores between 10 and 26 [19], aged 60 years or older with no history of schizophrenia, major depressive disorder, mental retardation, drug dependence, visual or auditory impairment, congestive heart failure, or clinically significant uncontrolled neurological or medical diseases that could cause cognitive impairment. Exclusion criteria included hypersensitivity to cilostazol, a tendency to bleed or use of an anticoagulation agent, abnormally low levels of vitamin B12 or folate, and a positive venereal disease research laboratory test. Concomitant medications such as anticholinergics, anticonvulsants, antidepressants, and antipsychotics were not permitted to newly start within 3 months before the baseline measurement or during the course of the study. All other medications were permitted. Those with territory infarction or strategic infarction, multiple sclerosis, brain hemorrhage, brain infection, normal-pressure hydrocephalus, or brain tumor on brain MRI were excluded.
Patients were required to have a reliable caregiver. Written informed consent was obtained from the patients and their caregivers. The review board of our institution approved the study protocol, and our research was completed in accordance with guidelines for good clinical practice and the guidelines of the Helsinki Declaration.
Study Design
The present study was a 24-week, randomized, double-blind, placebo-controlled, parallel-group single-center study. Patients were randomly assigned to receive daily cilostazol (200 mg) or placebo using a computerized randomization schedule. Placebo were supplied by the Otsuka International Asia Arab Company. The initial dose of cilostazol was 100 mg (50 mg twice daily) and was increased to 200 mg (100 mg twice daily) after 2 weeks. The clinical outcome variables were assessed at baseline and at 12-week intervals. Each patient underwent a FDG PET scan at baseline and at 24 weeks while receiving the full-dose of cilostazol. Safety was assessed at 12-week intervals using physical examinations, clinical laboratory tests, and adverse event monitoring. We used a per-protocol set to test the biological efficacy of cilostazol. The protocol-specified primary outcome measure was the voxel-based analysis of changes from baseline in the cerebral metabolic rate of glucose consumption (CMRglc) measured using FDG PET. The protocol-specified secondary outcome measures were the scores of MMSE, Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-cog) [20], Alzheimer’s Disease Cooperative Study-Activities of Daily Living (ADCS-ADL) Inventory [21], and the Sum of Boxes of the Clinical Dementia Rating (CDR-SB) [22].
Data Management
A case report form (CRF) is designed by the research team for data collection according to the protocol. The CRF exists in a paper version. The Clinical Data Management (CDM) team developed the Data Management Plan (DMP), which details how the data are to be handled according to how the study is anticipated to be run. Discrepancies should be reviewed at 1-month intervals by the CDM team to ensure that they are being resolved in a timely manner. Finally, a database is put in place after all data management activities are completed to ensure there was no manipulation of study data after unmasking of the treatment groups.
Clinical Measures
The MMSE is a neurocognitive test designed to screen for cognitive impairment that can be administered in 5 to 10 min. Scores range from 0 to 30; high scores indicate normal cognition, and scores between 10 and 26 represent mild to moderate dementia. The Korean version of the MMSE was validated by Lee et al. [23].
The ADAS-cog is a valid and reliable 11-item neuropsychological test set designed to assess the severity of cognitive impairment. Scores range from 0 to 70, with higher scores indicating greater severity. The ADAS-cog is widely used in drug studies in patients with AD. On average, untreated patients with mild to moderate AD show an increase of 7–11 points per year in the ADAS-cog score [24]. The Korean version of the ADAS-cog was validated by Suh et al. [25].
The ADCS-ADL is a valid 23-item ADL test used to measure daily life dysfunctions. Scores range from 0 to 78, with lower scores indicating greater severity.
The CDR is an observer rating scale designed to rate severity in dementia patients. It comprises six performance areas: memory, orientation, judgment and problem solving, community affairs, home and hobbies, and personal care. Each area is rated on a 5-level scale, in which 0 indicates the absence of dementia; 0.5, questionable; 1, mild; 2, moderate; and 3, severe dementia. We used the sum of CDR scores (CDR-SB) across these six areas. The Korean CDR was validated by Choi et al. [26].
PET Acquisition
FDG PET/CT was performed using a Gemini TF64 PET/CT scanner (Philips Healthcare, Andover, MA, USA). All subjects had fasted for at least 6 h before scanning. They received intravenous injection of 4.8 MBq/kg of FDG in a dimly lit, quiet waiting room and were instructed to remain lying comfortably. After a 40-min FDG equilibration period, the brain emission imaging was started and continued for 10 min with 2-mm thickness, 90 slices, and 256 × 256 matrix size. CT acquisition for attenuation map was done with the following parameters: 120 kVp, 50 mAs, 2-mm slice thickness, 512 × 512 matrix. Images were reconstructed through 3D Row-Action Maximum-Likelihood Algorithm (RAMLA) by 90 slices with 2-mm thickness in 128 × 128 matrix, and corrected for attenuation and scatter.
PET Image Analysis
Preprocessing and statistical analyses were performed using Statistical Parametric Mapping (SPM8, Wellcome Department of Imaging Neuroscience, London, UK, http://www.fil.ion.ucl.ac.uk/spm) implemented in Matlab 7.6 (The Mathworks Inc., Natick, MA). The FDG PET images of each participant were averaged and this mean image was spatially normalized onto the Montreal Neurological Institute (MNI) standard PET template, using the parameters estimated during the transformation of mean image into the MNI space. The standardized image of each subject was smoothed with a 12-mm full-width at half-maximum (FWHM) isotropic Gaussian kernel. Brain glucose metabolism at each voxel was proportionally scaled to the global mean FDG uptake value; hence, the relative regional CMRglc was calculated. To analyze the main and interaction effects of the placebo and cilostazol groups, a 2 × 2 repeated measures ANOVA including time (pre and post treatment) as within-subjects factor and treatment group (placebo and cilostazol) as between-subjects factor was performed for every voxel within the brain.
The results were mapped as t statistic value, and superimposed onto MNI template. For the statistical analysis of the images, a statistical threshold was set at the voxel level of p value < 0.005 (uncorrected) with a cluster level of < 0.05 (family-wise error corrected). Only voxels that were adjacent to at least 100 voxels with similar p values were counted (kE > 100).
To confirm the treatment effect, a comparison of metabolic changes on region of interest basis was also performed. The averaged regional glucose metabolism within the significant clusters was extracted using MarsBaR (http://marsbar.sourceforge.net). The significant differences of each cluster in each group were analyzed using repeated measures ANOVA of SPSS for Windows, Version 16 (SPSS Inc., 2007, Chicago, IL). Brain region locations were verified on a standard brain atlas of Talairach.
Clinical Data Analysis
Normally distributed continuous variables were analyzed using independent t tests, and the Mann–Whitney U test was used to analyze non-normally distributed continuous variables. Fisher’s exact test was used for categorical variables. We analyzed a per-protocol set to compare clinical outcomes in the two groups. Secondary outcome measures, such as MMSE, ADAS-cog, ADCS-ADL, and CDR-SB scores, were analyzed using repeated measures ANOVA, with treatment condition (cilostazol vs placebo) as between-subject factors and time (baseline, week 12, and week 24) as within-subject factors. Statistical tests were conducted using SPSS. All hypothesis tests were two-sided, and the threshold of significance was set at p = 0.05.
Results
Patients
A total of 46 participants were enrolled in the study, and 36 (18 in the cilostazol group and 18 in the placebo group) completed the 24-week follow-up assessment. Two participants in the cilostazol group dropped out because of adverse events. One participant in the placebo group was discontinued because of poor compliance, and seven participants withdrew consent (three in the cilostazol group and four in the placebo group). We found no group differences in the demographic, clinical characteristics, and the severity of white matter regions of the participants who completed the study (Table 1).
Cerebral Glucose Metabolism Change
Groupwise comparison of pre-medication FDG PET uptake level of the placebo and the cilostazol group showed that there were no baseline differences of metabolic rate (p > 0.1). According to FDG PET image comparison between pre- and postmedication conditions, glucose uptake was not decreased in the cilostazol group; however, the placebo group showed a significant decrease in glucose metabolism in the bilateral parietal lobes, posterior cingulate cortex, precuneus, and inferior frontal gyri (p < 0.005; Figs. 1A, B and 2). The paired t test analysis based on regions of interest also showed that there was a significant metabolic rate decrease in the bilateral parietal lobe and inferior frontal gyri of the placebo group (p < 0.005), whereas none was found in the cilostazol group (p > 0.1; Fig. 2).

Voxel-based analysis of changes from baseline in the cerebral metabolic rate of glucose consumption (CMRglc) for participants who completed the study (uncorrected p < 0.001). (A) CMRglc was preserved in the cilostazol group, whereas (B) CMRglc decreased in the parietal lobes and inferior frontal gyri (IFG) in the placebo group. (C) Between-group comparison of changes in CMRglc. CMRglc in the left IFG was higher in the cilostazol group than that in the placebo group

Changes from baseline in the cerebral metabolic rate of glucose consumption (CMRglc) in the parietal lobes and inferior frontal gyri (IFG) in the cilostazol and placebo groups. p values were obtained from paired t tests between pre- and postmedication conditions
The voxel-based repeated measures ANOVA of glucose metabolic changes from baseline revealed that cilostazol treatment most prominently protected the left inferior frontal gyrus from the decrease in glucose uptake compared to the placebo group (Fig. 1C). No brain region showing greater metabolic decreases was found in the cilostazol group compared to the placebo group. The region analyzed showed that the percent of metabolic change after treatement was 1.08 ± 3.58% CMRglc in the cilostazol group and − 2.86 ± 3.28% CMRglc in the placebo group (p = 0.001).
The severity of white matter lesions did not affect the FDG PET changes in the region of interest analysis (left parietal lobe, p = 0.872; right parietal lobe, p = 0.841; left inferior frontal gyrus, p = 0.964; right inferior frontal gyrus, p = 0.296, corrected for age, gender, years of education, baseline MMSE, and premedication uptake level). In the repeated measures ANOVA of regional glucose uptake changes, the left inferior frontal gyrus showed significant time-group effect (p < 0.01; Table 2).
Clinical Changes
The mean changes from baseline in the MMSE, ADAS-cog, ADCS-ADL, and CDR sum scores were not different between groups at any time point, and there were no significant interaction effects of the between- and within-subject factors; the ADAS-cog score showed distinguishable tendency to deteriorate over time (p < 0.001; Table 3) in both groups.
However, we found positive effects of cilostazol on ADAS-cog scores especially in the increased glucose uptake subgroup in the cilostazol group unlike the decreased glucose uptake subgroup. When we tested that the cerebral metabolic change would predict the cognitive change in the cilostazol group, left parietal CMRglc change (t = 2.23, p = 0.04), right parietal CMRglc change (t = 2.28, p = 0.04), left inferior frontal CMRglc change (t = 2.09, p = 0.05), right inferior frontal CMRglc change (t = 2.74, p = 0.01) predicted the ADAS-cog score improvement (Fig. 3).

Cerebral metabolic rate of glucose (CMRglc) changes predict the cognitive portion of Alzheimer’s Disease Assessment Scale (ADAS-cog) improvement
Adverse Events
Four participants from the cilostazol group (17% out of initially enrolled 23 subjects) and 3 from the placebo group (13% out of 23) experienced adverse events (Table 4). All of the adverse events were transient and mild. The percentage of the participants who experienced adverse events was not significantly different between groups (p = 0.70).
Discussion
We used FDG PET imaging to sensitively measure longitudinal changes in brain glucose metabolism in AD with white matter lesion patients during this study. The imaging results showed that CMRglc did not decrease significantly in patients treated with cilostazol. Regional glucose metabolism was significantly reduced in the bilateral parietal lobes of patients who received the donepezil monotherapy, and prominently preserved in the left inferior frontal gyrus of patients who received cilostazol (Fig. 1C). In the repeated measures ANOVA of regionally averaged uptake values, the change of uptake in the cilostazol-treated group by time was significantly different from that of the placebo group (p < 0.001; Table 2). Our 24-week randomized, double-blind, placebo-controlled trial found that cilostazol is a safe treatment for the maintenance of brain function in mild-to-moderate AD patients with white matter lesions.
We found that cilostazol prevented functional decline in the AD with white matter lesion brains. Hypometabolism in FDG PET scans indicates reduced synaptic activity [27], which commonly results in impaired brain function [14]; thus, cilostazol appears to prevent the decrease in general synaptic activity caused by the progression of AD. This protective effect of cilostazol may also be applied to AD patients without white matter lesions as well, because the comparison of metabolic change between the cilostazol and placebo group was not affected by the severity of white matter lesions.
The biochemical mechanism of cilostazol is related to the upregulation of CREB protein [28]. CREB is a cellular transcription factor protein that stimulates target genes for nerve cell growth and, consequently, long-term synaptic plasticity [29, 30]. Therefore, cilostazol may act to strengthen existing synaptic connections and to form new ones, which may delay or reverse the progressive degenerative synaptic changes accelerated by amyloid plaques in AD [31, 32]. It may be related to the neuroprotective effect of cilostazol. Animal studies also reported that cilostazol prevented memory impairment and protected brain oxidative stress [33, 34], and also suppressed Aβ peptide-induced neurotoxicity and inhibited Aβ oligomerization [35].
Additionally, we suggested a hypothetical mechanism which can explain and understand dynamic relationships among Aβ, p-Tau, and glucose related to AD pathology and cilostazol effects on their dynamic interactions. The action of Aβ may contribute to decreased glucose uptake and neuronal degeneration in AD [36]. The increase of glucose by vasodilation with cilostazol may protect synaptic loss and neuronal degeneration in AD patients. We found that cilostazol contributed to the increase of glucose in some specific regions in AD brain, and this increase is related to the cognitive improvement in this study [33,34,35]. Thus, the glucose uptake increase by cilostazol may prevent Aβ-induced impairment of glucose transport in neurons of mild AD and this glucose transport maintenance with cilostazol can delay cognitive deterioration.
Hypoperfusion and oxygen–glucose deprivation also induced both necrotic and apoptotic cell death of astrocytes by tau phosphorylation [37]. Glucose deprivation increases tau phosphorylation via P38 mitogen-activated protein kinase, and then finally, glucose deprivation induces cell apoptosis by tau phosphorylation [38]. Therefore, cilostazol may prevent or attenuate tauopathy-related cell apoptosis with positive effects of increased glucose uptake by cilostazol.
The regions found to be hypometabolic in our placebo group have been associated with the progression of AD and vulnerability to AD-related pathologies [16, 18]. Furthermore, Aβ deposition in these regions is reported to be significantly higher in patients with AD than that in control subjects [39]. Previous research has shown that Aβ plaques have a neurobiological influence and disrupt CMRglc [22, 40], which may explain the significant negative correlation between FDG PET uptake and the Pittsburgh Compound B PET binding potential [41]. These findings provide further evidences that cilostazol protects against AD progression by maintaining brain metabolism in the regions that are most susceptible to AD.
Functional neuroprotective effects exerted by upregulated phosphorylation of CREB with administration of cilostazol has been already reported in rat models [13, 28]. Recently, a SPECT imaging study found an increase of regional cerebral blood flow in the right anterior cingulate lobe in human patients with AD and cerebrovascular disease [42]. In the current study, the comparison of the glucose metabolic rates in the cilostazol and placebo groups revealed that cilostazol had a significant protective effect in the left inferior frontal gyrus (Table 2). A previous longitudinal study found that the left inferior frontal gyrus was not as severely degenerated as the parietal lobes in early course of AD; however, cerebral metabolic decline was similar in the two brain regions in late AD [16]. It suggests that cilostazol was more effective in delaying the degenerative synaptic changes caused by AD in the inferior frontal gyrus because this is one of the regions that are damaged in the more advanced stage of AD. It also can be inferred that cilostazol added to donopezil will be more effective if applied early, before irreversible pathological changes afflict wider regions.
We found that in both frontal and parietal lobes, the increase of glucose 24 weeks after cilostazol administration was associated with the improvements of cognition. The increase of glucose by cilostazol may protect tauopathy and cell apoptosis in AD patients.
Our clinical measures of cognitive and global functions showed no significant changes in the cilostazol group by time (Table 3). This finding is not surprising because neuropsychological tests in AD clinical studies require a large sample size and long duration to yield significant results [43]. FDG PET is a sensitive tool for detecting subtle neurobiological changes driven by the early phase of AD pathologies [17, 44]. Because abnormal CMRglc likely precedes deterioration in cognitive function [45, 46], hypometabolism in the affected regions may not be accompanied by related cognitive functional deficits [14, 47]. Although the group–time interaction was not significant in our study, it is possible that a longer-duration study with a larger sample size would show clinical improvement with cilostazol treatment.
The education years were 5.56 in the cilostazol group and 4.72 in the placebo group. The education years are normally longer in the developed countries. But, Korean elderly people have had colonial rule and Korean War when they were young, so there are few people who received formal education more than primary school. The lower education level of the elderly was common in most Asian countries such as Hong Kong and Singapore.
The recommended maintenance dosage of cilostazol is 100 mg bid. We started a dose of 50 mg bid and increased the dose to 100 mg bid to decrease side effect as headache and diarrhea. In general, Korean elderly women experience more cilostazol side effects due to their lower body weight in our clinical experiences. A similar side effect of combination drug—AchEI—was also considered in this decision.
Certain aspects of our study require further investigation. Due to the small sample size, further longitudinal investigations with delayed-start design will be necessary in larger groups over longer treatment durations, in order to test the disease-modifying effects of cilostazol added to donepezil in AD progress. In addition to the current study in AD patients with white matter lesions, efficacy of cilostazol administration on pure AD patients should be investigated. Furthermore, although FDG PET imaging is highly correlated with Aβ plaque deposition, molecular imaging should be conducted to validate a possible anti-amyloid effect of cilostazol. Aβ plaque neuroimaging can indicate whether cilostazol treatment prevents further plaque deposition and subsequent cell death in other regions of the brain.
Conclusions
Our CASID results show that cilostazol administration added to donepezil threapy preserved cerebral glucose metabolic rate of the left inferior frontal gyrus in patients with AD and white matter lesions despite the small sample size. Although our results did not verify the efficacy of cilostazol in improving or protecting cognitive function related to AD progression, FDG PET results proved that decrease in CMRglc was not found in the cilostazol group. Additionally, our results also suggested the possiblilities of cilostazol in improving or protecting cognitive functions through the increase of glucose uptake by cilostazol. The AD-modifying effects of cilostazol must be verified using Aβ PET imaging and larger clinical trials with a delayed-start long-term design.
Abbreviations
- AD:
-
Alzheimer’s disease
- ADAS-cog:
-
Alzheimer’s Disease Assessment Scale-cognitive subscale
- ADCS-ADL:
-
Alzheimer’s Disease Cooperative Study-Activities of Daily Living Inventory
- CDR:
-
Clinical Dementia Rating Scale
- CDR-SB:
-
the Sum of the Boxes of the Clinical Dementia Rating Scale
- IFG:
-
inferior frontal gyrus
- CMRglc:
-
cerebral metabolic rate of glucose consumption
References
Halene TB, Siegel SJ. PDE inhibitors in psychiatry--future options for dementia, depression and schizophrenia? Drug Discov Today. 2007;12(19–20):870–8.
Benito E, Barco A. CREB’s control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci. 2010;33(5):230–40.
Erceg S, Monfort P, Hernandez-Viadel M, Llansola M, Montoliu C, Felipo V. Restoration of learning ability in hyperammonemic rats by increasing extracellular cGMP in brain. Brain research. 2005;1036(1–2):115–21.
Lee JH, Park SY, Shin YW, Hong KW, Kim CD, Sung SM, et al. Neuroprotection by cilostazol, a phosphodiesterase type 3 inhibitor, against apoptotic white matter changes in rat after chronic cerebral hypoperfusion. Brain research. 2006;1082(1):182–91.
Patil CS, Singh VP, Kulkarni SK. Modulatory effect of sildenafil in diabetes and electroconvulsive shock-induced cognitive dysfunction in rats. Pharmacol Rep. 2006;58(3):373–80.
Sasaki T, Kitagawa K, Omura-Matsuoka E, Todo K, Terasaki Y, Sugiura S, et al. The phosphodiesterase inhibitor rolipram promotes survival of newborn hippocampal neurons after ischemia. Stroke; a journal of cerebral circulation. 2007;38(5):1597–605.
Lee JH, Park SY, Shin HK, Kim CD, Lee WS, Hong KW. Protective effects of cilostazol against transient focal cerebral ischemia and chronic cerebral hypoperfusion injury. CNS Neurosci Ther. 2008;14(2):143–52.
Gotoh F, Tohgi H, Hirai S, Terashi A, Fukuuchi Y, Otomo E, et al. Cilostazol stroke prevention study: A placebo-controlled double-blind trial for secondary prevention of cerebral infarction. Journal of Stroke and Cerebrovascular Diseases. 2000;9(4):147–57.
Lee JH, Shin HK, Park SY, Kim CD, Lee WS, Hong KW. Cilostazol preserves CA1 hippocampus and enhances generation of immature neuroblasts in dentate gyrus after transient forebrain ischemia in rats. Experimental neurology. 2009;215(1):87–94.
Park SH, Kim JH, Bae SS, Hong KW, Lee DS, Leem JY, et al. Protective effect of the phosphodiesterase III inhibitor cilostazol on amyloid beta-induced cognitive deficits associated with decreased amyloid beta accumulation. Biochem Biophys Res Commun. 2011;408(4):602–8.
Ihara M, Nishino M, Taguchi A, Yamamoto Y, Hattori Y, Saito S, et al. Cilostazol add-on therapy in patients with mild dementia receiving donepezil: a retrospective study. PLoS One. 2014;9(2):e89516.
Tai S-Y, Chien C-Y, Chang Y-H, Yang Y-H. Cilostazol use is associated with reduced risk of dementia: a nationwide cohort study. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics. 2017;14(3):784–91.
Lee JH, Park SY, Shin YW, Kim CD, Lee WS, Hong KW. Concurrent administration of cilostazol with donepezil effectively improves cognitive dysfunction with increased neuroprotection after chronic cerebral hypoperfusion in rats. Brain research. 2007;1185:246–55.
Jack CR, Jr., Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet neurology. 2010;9(1):119–28.
Reiman EM, Alzheimer’s Disease Biomarkers Working Group for the Alliance for Aging R. Fluorodeoxyglucose positron emission tomography: emerging roles in the evaluation of putative Alzheimer’s disease-modifying treatments. Neurobiol Aging. 2011;32 Suppl 1:S44–7.
Alexander GE, Chen K, Pietrini P, Rapoport SI, Reiman EM. Longitudinal PET Evaluation of Cerebral Metabolic Decline in Dementia: A Potential Outcome Measure in Alzheimer’s Disease Treatment Studies. Am J Psychiatry. 2002;159(5):738–45.
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–44.
Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA. MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. AJR Am J Roentgenol. 1987;149(2):351–6.
Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. Journal of psychiatric research. 1975;12(3):189–98.
Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry. 1984;141(11):1356–64.
Galasko D, Bennett D, Sano M, Ernesto C, Thomas R, Grundman M, et al. An inventory to assess activities of daily living for clinical trials in Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. Alzheimer Dis Assoc Disord. 1997;11 Suppl 2:S33–9.
Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412–4.
Lee JH, Lee KU, Lee DY, Kim KW, Jhoo JH, Kim JH, et al. Development of the Korean version of the Consortium to Establish a Registry for Alzheimer’s Disease Assessment Packet (CERAD-K): clinical and neuropsychological assessment batteries. The journals of gerontology Series B, Psychological sciences and social sciences. 2002;57(1):P47–53.
Kramer-Ginsberg E, Mohs RC, Aryan M, Lobel D, Silverman J, Davidson M, et al. Clinical predictors of course for Alzheimer patients in a longitudinal study: a preliminary report. Psychopharmacology bulletin. 1988;24(3):458–62.
Suh GH, Kang CJ. Validation of the Severe Impairment Battery for patients with Alzheimer’s disease in Korea. International journal of geriatric psychiatry. 2006;21(7):626–32.
Choi SH, Na DL, Lee BH, Hahm DS, Jeong JH, Yoon SJ, et al. Estimating the validity of the Korean version of expanded Clinical Dementia Rating (CDR) scale. J Korean Neurol Assoc. 2001;19(6):585–91.
Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21(10):1133–45.
Watanabe T, Zhang N, Liu M, Tanaka R, Mizuno Y, Urabe T. Cilostazol protects against brain white matter damage and cognitive impairment in a rat model of chronic cerebral hypoperfusion. Stroke; a journal of cerebral circulation. 2006;37(6):1539–45.
Walton MR, Dragunow I. Is CREB a key to neuronal survival? Trends Neurosci. 2000;23(2):48–53.
Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–48.
Yankner BA. Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron. 1996;16(5):921–32.
Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature. 1999;399(6738 Suppl):A23–31.
Oguchi T, Ono R, Tsuji M, Shozawa H, Somei M, Inagaki M, et al. Cilostazol Suppresses Aβ-induced Neurotoxicity in SH-SY5Y Cells through Inhibition of Oxidative Stress and MAPK Signaling Pathway. Frontiers in Aging Neuroscience. 2017;9:337.
Hiramatsu M, Takiguchi O, Nishiyama A, Mori H. Cilostazol prevents amyloid β peptide25-35-induced memory impairment and oxidative stress in mice. British journal of pharmacology. 2010;161(8):1899–912.
Shozawa H, Oguchi T, Tsuji M, Yano S, Kiuchi Y, Ono K. Supratherapeutic concentrations of cilostazol inhibits β-amyloid oligomerization in vitro. Neuroscience letters. 2018;677:19–25.
Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP. Amyloid β-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. Journal of Neuroscience. 1997;17(3):1046–54.
Paquet M, Ribeiro FM, Guadagno J, Esseltine JL, Ferguson SS, Cregan SP. Role of metabotropic glutamate receptor 5 signaling and homer in oxygen glucose deprivation-mediated astrocyte apoptosis. Molecular brain. 2013;6(1):9.
Lauretti E, Pratico D. Glucose deprivation increases tau phosphorylation via P 38 mitogen-activated protein kinase. Aging cell. 2015;14(6):1067–74.
Potkin SG, Anand R, Fleming K, Alva G, Keator D, Carreon D, et al. Brain metabolic and clinical effects of rivastigmine in Alzheimer’s disease. Int J Neuropsychopharmacol. 2001;4(3):223–30.
Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med. 2008;14(2):45–53.
Li Y, Rinne JO, Mosconi L, Pirraglia E, Rusinek H, DeSanti S, et al. Regional analysis of FDG and PIB-PET images in normal aging, mild cognitive impairment, and Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2008;35(12):2169–81.
Sakurai H, Hanyu H, Sato T, Kume K, Hirao K, Kanetaka H, et al. Effects of cilostazol on cognition and regional cerebral blood flow in patients with Alzheimer’s disease and cerebrovascular disease: a pilot study. Geriatr Gerontol Int. 2013;13(1):90–7.
Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Green RC, et al. The Alzheimer’s Disease Neuroimaging Initiative: a review of papers published since its inception. Alzheimer’s & dementia. 2012;8(1 Suppl):S1–68.
Jagust W, Reed B, Mungas D, Ellis W, Decarli C. What does fluorodeoxyglucose PET imaging add to a clinical diagnosis of dementia? Neurology. 2007;69(9):871–7.
de Leon MJ, Convit A, Wolf OT, Tarshish CY, DeSanti S, Rusinek H, et al. Prediction of cognitive decline in normal elderly subjects with 2-[(18)F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET). Proc Natl Acad Sci U S A. 2001;98(19):10966–71.
Silverman DH, Small GW, Chang CY, Lu CS, Kung De Aburto MA, Chen W, et al. Positron emission tomography in evaluation of dementia: Regional brain metabolism and long-term outcome. JAMA. 2001;286(17):2120–7.
Tartaglia MC, Rosen HJ, Miller BL. Neuroimaging in dementia. Neurotherapeutics. 2011;8(1):82–92.
Acknowledgments
The authors wish to thank all patients and their caregivers who participated in the study. The English in this document has been checked by at least two professional editors, both native speakers of English (http://textcheck.com).
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Availability of Data and Materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Funding
JYL, EJY, and YKK was partially supported by a grant from Ministry of Science, ICT and Future Planning (Grant No: NRF-2014M3C7A1046042). Dr. Jun-Young Lee has been financially supported by the Otsuka International Asia Arab Company, limited for this research project.
Author information
Authors and Affiliations
Contributions
JYL, HYL, and YKK designed the study and carried out the analyses presented in this article. JYL, HYL, and YKK provided the meta-database of clinical trials and contributed to the analysis of this dataset. JYL, HYL, YKK, HWL, HBY, JSC, HYJ, EJY, YHJ, and HRK all contributed to the writing of the manuscript and to valuable discussion. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
This study was performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and was approved by the Ethics Board of Seoul National University College of Medicine & SMG-SNU Boramae Medical Center. All subjects or responsible caregivers, whichever appropriate, gave their informed consent.
Consent for Publication
Not applicable.
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PDF 1718 kb)
Rights and permissions
About this article

Cite this article
Lee, JY., Lee, H., Yoo, H.B. et al. Efficacy of Cilostazol Administration in Alzheimer’s Disease Patients with White Matter Lesions: A Positron-Emission Tomography Study. Neurotherapeutics 16, 394–403 (2019). https://doi.org/10.1007/s13311-018-00708-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-018-00708-x