
Abstract
Objective
Liver fibrosis is a chronic liver disease caused by a variety of pathophysiological. However, there are no effective treatments to combat it. HSCs are a major source of fibrotic cells and exploring the mechanisms of HSC activation may provide new strategies for the treatment of liver fibrosis.
Objectives
To explore the role and underlying mechanism of SLIT3 in HSCs fibrosis.
Results
GSE163211 dataset analysis identified aberrant expression of SLIT3 in NASH F1-F4 tissues and SLIT3 expression level was positively correlated with fibrosis-related proteins. In vitro experiments showed that TGF-β induced upregulation of SLIT3 in LX-2 cells. Knockdown of SLIT3 significantly inhibited TGF-β-induced α-SMA, COL1A2, and COL1A1 expression, inhibited excessive cell proliferation and migration, and suppressed YAP activity.
Conclusion
Collectively, our findings suggest that SLIT3 deficiency alleviates TGF-β-induced HSCs activation by inhibiting YAP activity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic liver disease causes approximately 2 million deaths each year, accounting for 3.5% of all deaths worldwide (Asrani et al. 2019). Liver fibrosis is one of its most common consequences and manifests itself as a wound-healing response to chronic liver injury, which may be caused by viruses, alcohol abuse, steatosis, and autoimmune or genetic disorders (Angulo et al. 2015; Bruschi et al. 2020; Poilil Surendran et al. 2017). Now, it has become a major world health issue (Del Campo et al. 2018), and it is associated with functional organ damage (Del Campo et al. 2018). If left untreated, it will eventually develop into cirrhosis. To date, the only effective treatment for cirrhosis is liver transplantation (Zhao et al. 2019). Therefore, new therapeutic strategies are urgently needed to prevent the development of liver fibrosis.
Liver fibrosis is a dynamic process involving crosstalk between multiple cell types, and hepatic stellate cells (HSCs) have received widespread attention as a major source of fibrotic cells (Tsuchida et al. 2017). Activation-driven HSCs, which undergo phenotypic trans-differentiation following liver injury, can overproduce extracellular matrix (ECM) and accumulate in the subendothelial space, thereby causing their transition to proliferative or contractile myofibroblast-like cells that induce excessive scarring, eventually leading to end-stage liver disease, cirrhosis, and hepatocellular carcinoma (Marcellin et al. 2018; Tsuchida et al. 2017; Yanguas et al. 2016; Zhang et al. 2016; Zhao et al. 2019). This process is activated by various factors, and studies have shown that the fibrotic effect of TGF-β is closely related to changes in ECM composition (Hinz 2015).
Elevated levels and nuclear translocation of the transcriptional regulator YES-associated protein (YAP) have been reported to drive activation of HSCs in vivo/in vitro (Li et al. 2021; Xiang et al. 2020). YAP signaling is essential for liver regeneration. Connective tissue growth factor (CTGF), a target gene of YAP, is associated with the pathological process of tissue fibrosis. CTGF is overexpressed in liver fibrotic tissues and activates HSCs, and induces synthesis and secretion of ECM proteins, and it is also associated with cell proliferation, migration and differentiation (Makino et al. 2018; Ramazani et al. 2018). YAP have been reported to interact with TGF-β-induced Smad2/3 and participate in tissue fibrosis (Hiemer et al. 2014; Zhang et al. 2022).
Mammalian slit-guided ligands 1–3 (SLIT1-3) are highly conserved secreted glycoproteins (Plump et al. 2002). Analysis of GEO data (GSE163211) revealed that SLIT3 is up-regulated in liver tissue from patients with fibrosing non-alcoholic steatohepatitis. SLIT3 has been reported to be involved in stress overload-induced cardiac fibrosis and poor remodeling, reducing fibrillar collagen production and regulating YAP transcription and translation (Gong et al. 2020).The role of SLIT3 in liver fibrosis is unclear. This study aimed to explore the role and underlying mechanism of SLIT3 in HSCs fibrosis.
Materials and methods
GEO data processing
A gene expression microarray dataset GSE163211 was obtained from the GEO database, from the GPL29503 platform, representing datasets from hepatic fibrosis, inflammation and steatosis. Normal liver tissue (Normal, n = 6) and non-alcoholic steatohepatitis with fibrosis stages 1–4 (NASH F1–F4, n = 46) from diabetic patients were selected for analysis. The GEO online platform was used to explore the differentially expressed genes (DEGs) in Normal and NASH F1–F4 tissues. A p value of < 0.05 and logFC (fold change) of > 0.5 were set as thresholds to screen DEGs.
Cell culture and treatment
Human immortal HSCs cell line LX‐2 was bought from Merck Millipore (Beijing, China). LX‐2 cells were cultured in DMEM (Gibco, Gaithersburg, MD, USA) with 10% fetal bovine serum (Gibco, Gaithersburg, MD, USA). All culture media contained 100 U/ml of penicillin and 100 μg/ml of streptomycin. LX‐2 cells were treated with exogenous 5 ng/mL TGF‐β (R&D Systems, USA) and incubated for 24 h to be activated.
Transfection with siRNA and overexpression vector
The transfection of vectors into LX-2 cells was subjected to Lipofectamine 2000 (Invitrogen). Short interfering SLIT3 (siSLIT3), negative control siRNAs (siNC), pcDNA3.1-vector (vector) and SLIT3 overexpression vector (SLIT3) were purchased from RioBio (Guangzhou, China) and addgene (USA).
Quantitative real-time polymerase chain reaction
Total RNA was extracted from TGF-β-treated LX-2 cells using RNAiso Plus reagent (Takara; China). Then it was reverse transcribed into complementary DNA using Prime Script RT Kit (Takara; China). qRT-PCR was performed using the SYBR Green PCR kit (Takara, China) according to the manufacturer's protocol. The primer sequences used to amplify SLIT3 and 18S are as follows: SLIT3 (Forward 5’-GTCAGCGTCATCGAGAGAGG-3’; Reverse 5’-TTCGGCGTGCTCTGGAAAA-3’), 18S (Forward 5’- GGAATTGACGGAAGGGCACCACC-3’; Reverse 5’-GTGCAGCCCCGGACATCTAAGG-3’).
Western blot analysis
Western blot analysis of protein expression in LX-2 cells was performed as previously described (Seo et al. 2020). β-actin was used as an internal control. The Image J software was used to quantify protein expression. The primary antibodies included anti- SLIT3, anti-α-SMA, anti-COL1A1 and anti-COL1A2 (1:1000; Abcam, Cambridge, UK), anti-YAP, anti-p-YAP, anti-Smad2/3, anti-p-Smad2/3, anti-CTGF, and anti-β-actin (1:1000; Cell Signaling Technology, Danvers, MA).
CCK-8 assay
Lx-2 cells transfected with siSLIT3, siNC, vector and pcDNA3.1-SLIT3 were seeded into 96-well plates. 24 h after transfection, 5 ng/ml TGF-β was added. Subsequently, CCK-8 solution was added into the culture medium (100 μl) for 3 h. Absorbance was measured at 450 nm.
Wound‑healing assay
Transfected or untransfected L × 2 cells were inoculated into 12-well plates at a density of 3–7 × 105 cells/ml. Straight wounds were made on the fused monolayer and the cells were incubated with TGF-β for 24 h. Wounded monolayers are washed with PBS and photographed using an inverted microscope.
Statistical analysis
Statistical analysis was performed using GraphPad Prism Software®. Results are expressed as mean ± standard deviation (SD), and statistical differences between groups were compared by Student's t-test or ANOVA. All experiments were repeated done in triplicate. p < 0.05 was considered statistically significant.
Results
SLIT3 is up-regulated in TGF-β-induced HSCs
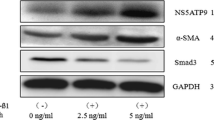
We first used the previously published RNA sequencing (RNAseq) dataset GSE163211 to assess the gene regulation associated with NASH-related liver fibrosis progression. The differential gene expression was compared between normal liver tissue and NASH F1-F4 (Fig. 1A). 45 differential genes were detected, of which 30 were up-regulated and 15 were down-regulated. Among them, SLIT3 was up-regulated in NASH F1–F4 with logFC = 0.930, p value = 0.027 and expression data were obtained from GSE163211 (Fig. 1B). In addition, it was found that SLIT3 was significantly positively correlated with the expression levels of fibrosis-related genes COL1A1 and COL1A2 (Fig. 1C). Previous reports have shown that TGF-β is capable of inducing HSCs activation. Next, LX-2 cells were incubated with 5 ng/mL TGF-β in vitro. The results showed that TGF-β induced upregulation of SLIT3 protein and mRNA levels (Fig. 1C, D). The expression level first increased with the treatment time and then decreased, and the expression level was the highest at 1 h. These results suggested that the differential expression of SLIT3 may be related to liver fibrosis.

SLIT3 is up-regulated in NASH F1-F4 patient's liver tissue and TGF-β-induced HSCs. A Volcano map of Normal vs NASH F1-F4 differentially expressed genes. B Expression level of SLIT3 in GSE163211 dataset. C In the GSE163211 dataset, SLIT3 was positively correlated with the expression levels of COL1A1 and COL1A2. D Western blot analysis showed the SLIT3 protein expression level of LX‐2 cell after 5 ng/mL TGF-β treatment for 24 h. **P < 0.01 compared with 0 h group
Knockdown of SLIT3 inhibits TGF-β-induced HSCs fibrosis
To investigate the potential role of SLIT3 in HSCs fibrosis, SLIT3 was knocked down or overexpressed in LX-2 cells. Western blot analysis verified the effect of SLIT3 knockdown and overexpression (Fig. 2). TGF-β increased the accumulation of α-SMA, COL1A2 and COL1A1 compared to control, but these increases were significantly suppressed by knockdown of SLIT3 (Fig. 2). In contrast, overexpression of SLIT3 further promoted α-SMA, COL1A2, and COL1A1 expression (Fig. 2). These results suggest that SLIT3 is involved in the induction of LX-2 cell fibrosis in response to TGF-β.

Knockdown of SLIT3 inhibits TGF-β-induced HSCs fibrosisWestern blot analysis evaluated the expression of levels of SLIT3, α-SMA, COL1A2 and COL1A1. **P < 0.01 compared with control group; + + p < 0.01 compared with TGF-β + siNC group; $p < 0.05, $$p < 0.01 compared with TGF-β + vector group
Knockdown of SLIT3 inhibits TGF-β-induced HSCs viability and migration
Another feature of HSCs activation is high proliferation and migration rates. To evaluate the effect of SLIT3 on proliferation and migration of HSCs, CCK-8 assay and wound-healing experiment were performed. As expected, TGF-β stimulation significantly enhanced the proliferation rate of LX-2 cells (Fig. 3A). Cells transfected with siNC or empty plasmid had no effect on TGF-β-induced cell proliferation. After down-regulation of SLIT3, the proliferation level of the cells was decreased and recovered to a level comparable to that of the control. Similar results were shown in wound-healing experiments. LX-2 cells transfected with SLIT3 siRNA or SLIT3 overexpression plasmids showed significant differences in their migratory responses to TGF-β (Fig. 3B, C).

Knockdown of SLIT3 inhibits TGF-β-induced HSCs viability and migration. A CCK-8 was used to examine the viability of LX‐2 cell (B, C) Wound healing assay LX-2 cell migration. The statistical analysis was shown in the bar graphs. **P < 0.01 compared with control group; + + p < 0.01 compared with TGF-β + siNC group; $p < 0.05, $$p < 0.01 compared with TGF-β + vector group
Knockdown of SLIT3 inhibits the YAP signaling pathway
Next, we investigated Fig. 4 whether SLIT3 mediates YAP signaling activation. The data showed that TGF-β significantly induced YAP expression and reduced YAP phosphorylation compared with the control. SLIT3 deficiency inhibited YAP expression and increased its phosphorylation. In addition, we also examined the protein expression of genes downstream of YAP. The results showed that SLIT3 deficiency inhibited TGF-β-induced CTGF expression and Smad2/3 phosphorylation.

Knockdown of SLIT3 inhibits the YAP signaling pathway. Western blot analysis evaluated the expression of levels of YAP, p-YAP, CTGF, SMAD2/3 and p-SMAD2/3. **P < 0.01 compared with control group; + + p < 0.01 compared with TGF-β + siNC group; $$p < 0.01 compared with TGF-β + vector group
Discussion
Non-alcoholic fatty liver disease (NAFLD) is a chronic liver disease that affects one quarter of the world's population and imposes a huge global burden on public health systems (Koyama et al. 2017, Powell et al. 2021). NAFLD initially presents as hepatic steatosis and subsequently evolves into NASH with the potential for progressive fibrosis and increased risk of cirrhosis and hepatocellular carcinoma (Bruno et al. 2020; Challa et al. 2019). The progression of NASH to liver fibrosis is a reversible stage in this process (Liu et al. 2019), but the initial triggers for the development of liver fibrosis remain largely unknown.
With the development of bioinformatics, we can obtain RNA sequencing data from the GEO public database to identify differentially expressed genes related to the progression of liver fibrosis, providing new ideas for the study of the molecular mechanism of liver fibrosis. To this end, we explored differentially expressed genes between normal liver tissue and NASH-associated fibrotic tissue in the GSE163211 dataset and found 45 genes with abnormal expression. Among them, SLIT3 caught our attention. SLIT3 was up-regulated in NASH F1-F4 tissues, and its expression level was significantly positively correlated with the levels of liver fibrosis-related factors including COL1A1. SLIT3, one of the three SLIT ligands originally discovered in the central nervous system, mediates axon guidance through a roundabout family of receptors (Long et al. 2004). Several subsequent studies revealed that SLIT3 is expressed in tissues other than the nervous system and is involved in physiological functions. For example, SLIT3 induces directional migration and proliferation of Mc3t3-e1 cells (Kim et al. 2018), and can promote the formation of H-type blood vessels and bone formation (Peng et al. 2020). In addition, SLIT3 is also involved in cardiac fibrosis and remodeling induced by pressure overload. Experiments have shown that SLIT3 is mainly present in fibrillar collagen-producing cells, and SLIT3 deficiency attenuates COL1A1 transcription in cardiac, aortic adventitia, lung, spleen and kidney tissues (Gong et al. 2020). Therefore, SLIT3 may be a potential target of fibrosis.
HSCs are a population of non-parenchymal hepatocytes (Trivedi et al. 2021). Under normal conditions, HSCs store lipids in the liver (Weiskirchen et al. 2019). During liver injury, HSCs are activated and transformed into proliferative and contractile myofibroblasts that simultaneously produce ECM, and numerous chemokines and cytokines such as COL1A1,α-SMA, fibronectin, laminin and proteoglycans, and cause tissue scaring (Mehta et al. 2019; Toosi 2015). TGF-β plays an important role in the pathogenesis of liver fibrosis and is one of the most potent fibrosis activators, which can induce quiescent HSCs differentiate into myofibroblast-like cells and induce ECM production (Massagué 2012).
To explore the potential role of SLIT3 in liver fibrosis, we constructed a TGF-β-induced LX-2 cell model in vitro. Western blotting results showed that TGF-β induced the expression of SLIT3 in LX-2 cells, and at the same time induced the activation of LX-2 cells, as well as increased the expressions of α-SMA, COL1A2 and COL1A1. SLIT3 knockdown significantly alleviated ECM accumulation in LX-2 cells. Conversely, SLIT3-overexpressing further increased the protein levels of α-SMA, COL1A2, and COL1A1 in LX-2 cells.
HSCs have the characteristics of high proliferation and mobility after activation. Previous studies have shown that Slit3 knockdown can significantly inhibit the proliferation and migration of fibroblasts after TGF-β treatment (Gong et al. 2020). The above results are consistent with our experimental results. Compared with LX-2 cells transfected with siNC, Slit3 knockdown cells showed lower cell viability and migration ability under TGF-β treatment, while Slit3 overexpression further enhanced cell viability and promoted cell migration. In conclusion, knockdown of Slit3 can attenuate the response of HSCs to TGF-β.
The transcription factor YAP has been identified as an important mediator of mechanical transduction signals in fibroblasts and extensively regulates the biological activities of fibroblasts(Gong et al. 2020; Haak et al. 2019). Previous studies have demonstrated that SLIT3 regulates YAP signaling pathway activation in fibroblasts (Gong et al. 2020). Therefore, the potential association between SLIT3 and YAP in HSCs was examined. The results were consistent with previous reports that YAP phosphorylation levels were significantly increased in SISlit3-transfected LX-2 cells, indicating inhibition of TGF-β-induced YAP activation. Activation of HSCs is associated with upregulation of several genes involved in matrix remodeling, actin cytoskeleton, cell proliferation, and immune processes. Ctgf, for example, is a direct target of YAP (Zhubanchaliyev et al. 2016). It has been reported that YAP regulates SMad2/3 and participates in tissue fibrosis (Zhang et al. 2022). Therefore, the protein expression level of CTGF and the phosphorylation level of Smad2/3 were detected by Western blotting. The results showed that SLIT3 knockdown inhibited CTGF expression and Smad2/3 phosphorylation. In conclusion, our results demonstrated that loss of SLIT3 attenuated the effects of TGF-β stimulation on HSCs by down-regulating YAP and its downstream target genes.
Availability of data and materials
The authors declare that all data supporting the findings of this study are available within the paper and any raw data can be obtained from the corresponding author upon request.
References
Angulo P et al (2015) Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 149:389-397.e310
Asrani SK, Devarbhavi H, Eaton J, Kamath PS (2019) Burden of liver diseases in the world. J Hepatol 70:151–171
Bruno S et al (2020) HLSC-derived extracellular vesicles attenuate liver fibrosis and inflammation in a murine model of non-Alcoholic Steatohepatitis. Mol Ther J Am Soc Gene Ther 28:479–489
Bruschi FV et al (2020) PNPLA3 I148M Up-regulates hedgehog and yap signaling in human hepatic stellate cells. Int J Mol Sci 21:8711
Challa TD et al (2019) Liver ASK1 protects from non-alcoholic fatty liver disease and fibrosis. EMBO Mol Med 11:e10124
Del Campo JA, Gallego P, Grande L (2018) Role of inflammatory response in liver diseases: Therapeutic strategies. World J Hepatol 10:1–7
Gong L et al (2020) SLIT3 deficiency attenuates pressure overload-induced cardiac fibrosis and remodeling. JCI Insight. https://doi.org/10.1172/jci.insight.136852
Haak AJ et al (2019) Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci Trans Med 11:6296
Hiemer SE, Szymaniak AD, Varelas X (2014) The transcriptional regulators TAZ and YAP direct transforming growth factor β-induced tumorigenic phenotypes in breast cancer cells. J Biol Chem 289:13461–13474
Hinz B (2015) The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol J Int Soc Matrix Biol 47:54–65
Kim BJ et al (2018) Osteoclast-secreted SLIT3 coordinates bone resorption and formation. J Clin Investig 128:1429–1441
Koyama Y, Brenner DA (2017) Liver inflammation and fibrosis. J Clin Investig 127:55–64
Li C, Zhang R, Zhan Y, Zheng J (2021) Resveratrol inhibits hepatic stellate cell activation via the hippo pathway. Mediators Inflamm 2021:3399357
Liu T et al (2019) Alleviation of hepatic fibrosis and autophagy via inhibition of transforming growth factor-β1/Smads pathway through shikonin. J Gastroenterol Hepatol 34:263–276
Long H et al (2004) Conserved roles for Slit and Robo proteins in midline commissural axon guidance. Neuron 42:213–223
Makino Y et al (2018) CTGF mediates tumor-stroma interactions between hepatoma cells and hepatic stellate cells to accelerate HCC progression. Can Res 78:4902–4914
Marcellin P, Kutala BK (2018) Liver diseases: A major, neglected global public health problem requiring urgent actions and large-scale screening. Liver Int Off J Int Assoc Study Liver 38(Suppl 1):2–6
Massagué J (2012) TGFβ signalling in context. Nat Rev Mol Cell Biol 13:616–630
Mehta KJ, Farnaud SJ, Sharp PA (2019) Iron and liver fibrosis: mechanistic and clinical aspects. World J Gastroenterol 25:521–538
Peng Y, Wu S, Li Y, Crane JL (2020) Type H blood vessels in bone modeling and remodeling. Theranostics 10:426–436
Plump AS et al (2002) Slit1 and Slit2 cooperate to prevent premature midline crossing of retinal axons in the mouse visual system. Neuron 33:219–232
PoililSurendran S, George Thomas R, Moon MJ, Jeong YY (2017) Nanoparticles for the treatment of liver fibrosis. Int J Nanomed 12:6997–7006
Powell EE, Wong VW, Rinella M (2021) Non-alcoholic fatty liver disease. Lancet (London, England) 397:2212–2224
Ramazani Y et al (2018) Connective tissue growth factor (CTGF) from basics to clinics. Matrix Biol J Int Soc Matrix Biol 68–69:44–66
Seo HY et al (2020) src inhibition attenuates liver fibrosis by preventing hepatic stellate cell activation and decreasing connetive tissue growth factor. Cells 9:558. https://doi.org/10.3390/cells9030558
Toosi AE (2015) Liver fibrosis: causes and methods of assessment, a review. Roman J Intern Med 53:304–314
Trivedi P, Wang S, Friedman SL (2021) The Power of plasticity-metabolic regulation of hepatic stellate cells. Cell Metab 33:242–257
Tsuchida T, Friedman SL (2017) Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 14:397–411
Weiskirchen R, Tacke F (2019) Relevance of autophagy in parenchymal and non-parenchymal liver cells for health and disease. Cells 8:16. https://doi.org/10.3390/cells8010016
Xiang D et al (2020) Physalin D attenuates hepatic stellate cell activation and liver fibrosis by blocking TGF-β/Smad and YAP signaling. Phytomed Int J Phytother Phytopharmacol 78:153294
Yanguas SC et al (2016) Experimental models of liver fibrosis. Arch Toxicol 90:1025–1048
Zhang K et al (2016) ω-3 PUFAs ameliorate liver fibrosis and inhibit hepatic stellate cells proliferation and activation by promoting YAP/TAZ degradation. Sci Rep 6:30029
Zhang T et al (2022) NUAK1 promotes organ fibrosis via YAP and TGF-β/SMAD signaling. Sci Trans Med 14:eaaz4028
Zhao Z, Lin CY, Cheng K (2019) siRNA- and miRNA-based therapeutics for liver fibrosis. Trans Res J Lab Clin Med 214:17–29
Zhubanchaliyev A et al (2016) Targeting mechanotransduction at the transcriptional level: YAP and BRD4 are novel therapeutic targets for the reversal of liver fibrosis. Front Pharmacol 7:462
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
XF and JC designed the study and carried them out, XF, JC, DJ, MZ and YM supervised the data collection, analyzed the data, interpreted the data, XF and JC prepare the manuscript for publication and reviewed the draft of the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Author Xiling Fu declares that he/she has no conflict of interest; author Jiabao Chang declares that he/she has no conflict of interest; author Damin Jiao declares that he/she has no conflict of interest;; author Mengying Zhu declares that he/she has no conflict of interest; author Yuqi Ma declares that he/she has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fu, X., Chang, J., Jiao, D. et al. SLIT3 knockdown inhibited TGF-β-induced hepatic stellate cells activation by down-regulating YAP signal. Mol. Cell. Toxicol. 20, 251–258 (2024). https://doi.org/10.1007/s13273-023-00336-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13273-023-00336-3