
Abstract
The effect of the chemical composition of the hydrotreated light cycle oil (HDT LCO) on the benzene, toluene, ethylbenzene, and xylene (BTEX) production by a hydrocracking (HCK) procedure, is presented. Six different types of HDT LCOs were obtained by submitting two types of LCOs to hydrotreating (HDT) with different catalysts and experimental conditions. The products were analyzed as mono-, di- and tri-aromatic compounds using the supercritical fluid chromatography (SFC) method (ASTM D5186). The HDT LCOs were subjected to HCK with a 50/50 in weight mixture of nickel-molybdenum on alumina (NiMo/Al2O3) and H-ZSM5 (NiMo/H-ZSM5, 50/50) at 375 °C, 7.5 MPa, 1.2 h−1, and 750 m3/m3 H2/Oil. The HCK products were analyzed by gas chromatography with a flame ionization detector (GC-FID) and divided into five groups: gas, light hydrocarbons (LHCs), BTEX, middle hydrocarbons (MHCs), and heavy hydrocarbons (HHCs).
The results showed that the BTEX formation ranged from 27.0 to 29.8 wt.% and it did not show a significant dependence on the mono-aromatic (59.9 and 75.6 wt.%), total aromatic (61.1–84.2 wt.%) contents or MHCs conversion (58.3–64.3 wt.%) from the departing HDT LCO feedstock. This result implies that, contrary to previous expectations, the BTEX formation does not directly depend on the amounts of total or mono-aromatic compounds when departing from real feedstocks. A GC-PIONA (paraffin, isoparaffin, olefin, naphthene, aromatic) characterization method (ASTM D6623) for mechanism understanding purpose was also carried out.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The present work is the continuation of the studies on benzene, toluene, and xylene (BTX) production from light cycle oil (LCO) [1,2,3,4]. LCO has lost market as a part of diesel feedstock because of its inherent low quality (high content of sulfur, nitrogen, and aromatics) [5,6,7], which makes the resulting fuel, usually as part of diesel, difficult to comply with strict environmental regulations around the world [8, 9].
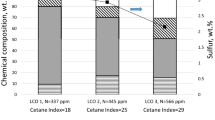
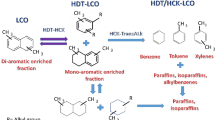
The chemical nature of LCO is being highly aromatic. LCO from Mexican refineries contains up to 90 wt.% of mono-, di-, and tri-aromatic compounds (Fig. 1) [7]. Most of the aromatic compounds are di-aromatic-type hydrocarbons, i.e., naphthalene derivatives (Fig. 1). Among the alternatives for LCO upgrading is the BTX production after successive hydrotreating (HDT) and hydrocracking (HCK) procedures (Fig. 2) [1, 2, 10]. Laredo et al. [2] presented a review showing several options to get high-quality gasoline or BTX enriched fractions from hydrotreated light cycle oil (HDT LCO) or similar feedstocks. In this context, the catalysts play a key role in the selective hydrogenation (partial saturation) of naphthalene-type compounds, giving place to mono-aromatic (tetrahydronaphthalene or tetralin) structures, followed by their cracking, producing one-ring-aromatic hydrocarbons with alkyl chains (alkylbenzenes) (Fig. 2). Bi-functional catalysts containing acidic supports and hydro/dehydrogenating metals are often used [1, 2, 11,12,13]. The group VI and VIII metal sulfide hydrocracking catalysts generally perform well the required hydro-dehydrogenation function [11]. Notwithstanding, strong acidity cracks molecules excessively into light gaseous hydrocarbons and accelerates coke deposition [11]. Wu et al. [12] found out that strong acidity resulted in intense interaction between acid and metal centers, which can decrease the catalyst hydrogenation activity. On the other hand, low catalytic acidity resulted in high hydrogenation activity (high hydrogenation reaction rates) with minimal selective-hydrogenation activity, which means that the consecutive hydrogenation of the first and second aromatic rings, i.e., from naphthalene to tetrahydronaphthalene (tetralin), and finally to decahydronaphthalene (decalin) type compounds, can occur without giving place to the hydrocracking of intermediate mono-aromatic (tetralin) type hydrocarbons for BTEX production. Therefore, moderate acidity showed the best balance between hydrogenation and cracking activity to achieve the highest BTX yields [13]. Another important point was the metal types on the catalysts. Wu et al. [12] found out that the hydrogenation rates from the different metals decreased in the following order: NiW > NiMo > CoMo > CoW, and these results were consistent with previous studies.

Hydrocarbon composition, sulfur, and nitrogen content, and cetane index of some LCO samples from Mexican refineries

Reaction scheme for obtaining BTEX and other hydrocarbons from naphthalene derivatives in LCO. HDT hydrotreating, HCK hydrocracking, HYD hydrogenation, DTR disproportionation/transalkylation
In a recent review, Laredo et al. [13] studied the effect of the catalyst and experimental conditions on the total BTX yields when departing from model molecules, like tetralin and naphthalene derivatives, and from real feedstocks. It was observed that BTX yields higher than 53–55% of the total were never achieved by the hydrocracking of tetralin and naphthalene derivatives, neither by changing the catalyst nor the experimental conditions. It was concluded that higher yields were never attained due to the inevitable gas formation and other produced hydrogenated hydrocarbons (naphthene derivatives) and C9+-alkylbenzenes. Additionally, after submitting real feedstocks to consecutive HDT and HCK procedures, a total BTX yield higher than 35% was never reached by using any type of HCK catalyst, even when the presence of aromatic compounds that could deliver BTX was very different. It seems that some types of aromatic hydrocarbons can form BTX, and some other types cannot.
According to the context described above, the main objective of this work is to study the effect of the chemical composition, in terms of total and mono-aromatic contents or composition of middle hydrocarbons (MHCs) in six different HDT LCO samples, on the benzene, toluene, ethylbenzene, and xylene (BTEX) yield. The HCK reaction was carried out by using a 50/50 in weight mixture of NiMo/Al2O3 and H-ZSM5 (HCK 50/50), for it had been tested before with model compounds and real feedstocks, showing suitable acid and metal properties for attaining the desired goal [3, 4].
Materials and methods
Materials
Ultra-high purity hydrogen (99.99 vol.%, Infra-México) was employed in the experiments. Pemex kindly provided two LCO samples [4, 14] (Table 1). The catalysts used in this work, either for HDT and HCK, were commercial catalysts that were tested in previous works [3, 4, 14]. Their characteristics are shown in Table 2. The catalysts were named as follows: HDT-1 was the CoMo/Al2O3 catalyst, HDT-2 was the NiMoP/Al2O3 catalyst, and HCK 50/50 was the 50/50 in weight mixture of NiMo/Al2O3 and H-ZSM5 [3, 4]. The catalysts were crushed, sieved (60 mesh), and dried at 120 °C for 2 h before loading.
Characterization methods of the liquid phase
The physical and chemical characteristics obtained by ASTM procedures [15] are shown in Table 1. This type of feedstock and product characterizations were already explained in Laredo et al. [6] and Laredo et al. [7]. On the other hand, the special gas chromatography chemical characterization methods employed in this work were carried out as follows:
Determination of aromatic and polynuclear aromatic contents in diesel fuels by ASTM D5186
The characterization of all feedstocks and products was carried out in an Agilent 1260 Infinity II SFC System following ASTM D5186 [16], usually known as the SFC (superfluid chromatography) method.
Fast chemical characterization by GC-FID
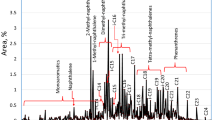
The chemical characterization of the HDT and HCK products was carried out using a Bruker gas chromatography-flame ionization detector (GC-FID) system, with the following set of columns: WCOT: 105 m × 320 mm i.d., 50 m × 320 mm i.d., and 105 m × 320 mm i.d. Helium was used as carrier gas (3 mL/min). The temperature program was as follows: 40 °C (10 min), 10 °C/min up to 300 °C (1 min). The detector and injector were set at 320 and 330 °C, respectively. Split/splitless range 1–10,000. The retention times used for the different fractions or lumps are shown in Table 3. Figure 3 displays a graphic example of the method. It is known that the response factor of the FID in the case of hydrocarbons without heteroatoms can be considered close to the unity [17]. Therefore, the obtained area of each signal was considered as the weight percent of the hydrocarbons present in this type of samples. For simplicity purposes, the resulted chromatogram was divided into lumps light hydrocarbons (LHCs) consisting mostly of paraffin, isoparaffin and decalin derivatives and BTEX represented by benzene, toluene, ethylbenzene, and xylenes. Middle hydrocarbons (MHCs) formed mostly by C9+-alkylbenzenes, tetralin, and naphthalene derivatives, and some longer chain paraffins, and isoparaffins, and finally, the HHC fraction formed mainly by some phenanthrene derivatives and heavy paraffins and isoparaffins.

Examples of the analysis of the chromatograms from HDT LCO (HL.2.1.330) and its hydrocracked product (HC.2.1.330) by the GC-FID method developed in this work
Chemical characterization by using ASTM D6623
Two adequate samples, obtained after the HDT (HL.2.1.330) and HCK (HC.2.1.330) processes (Tables 4, 5) were subjected to a detailed chemical characterization according to the ASTM D6623-01 procedure for gasoline distillates, usually known as GC-PIONA or GC-PIANO [18], for the possible distinction between paraffin (P), isoparaffin (I), olefin (O), naphthenic (N), and aromatic (A) hydrocarbons present in the samples. For the correct characterization using this method, the sample required to have a final boiling point around 300 °C. The procedure was carried out using an Agilent 6890 series chromatography system. The resulting chromatograms were analyzed by means of the ChemStation Hydrocarbon Expert™ PIANO software.
Experimental setup
The HDT and HCK experiments were carried out in bench-scale equipment with a fixed-bed trickle reactor (catalyst volume = 10 mL; internal diameter = 1.0 cm; downflow mode). Figure 4 is a simplified diagram of this pilot plant including some basic instrumentation and analytical peripherals. The reactor (R-101) was loaded with 10 mL of the required catalyst and placed in an electric furnace provided with three independent heating zones to attain an isothermal axial profile. The specific feedstock was introduced into the reactor by using a reciprocating pump (inlet flow: 0.10–0.17 cm3/s) and the hydrogen flow rate (3–8 stdL/h at 101.32 × 10–3 MPa and 20 °C) was measured with a 0–20 stdL/h range Brook 5850 series thermal mass flow controller. The reactor effluent was directed to a low-pressure separator (S-101). The liquid fraction leaving S-101 was sent to a second separator (S-102) and gas fractions from S-101 and S-102 were mixed and quantified in a gas meter and characterized by a UOP standard analysis for refinery gases [19]. The S-102 liquid product was collected in a container, weighed for mass balance purposes, and characterized as described as disclosed in Sect. “Characterization methods of the liquid phase”.

Simplified diagram of the bench-scale setup
Catalytic activity tests
In both cases, the HDT and HCK processes, the crushed, sieved, and dried HDT-1, HDT-2, or HCK mixture (HCK 50/50) (Table 2) catalysts were loaded into the reactor and then sulfided in situ with kerosene spiked with dimethyl-disulfide (2.5 wt.% S) at 210–315 °C, 4.0 MPa, 1.0 h−1, and H2/oil ratio of 315–378 m3/m3 for 18 h. After the sulfiding step, a soaking period was performed with kerosene at 315 °C, 4.0 MPa, 1.0 h−1, and 378 m3/m3 for 48 h. Next, a blank run was performed with n-hexadecane as feedstock at 350 °C, 3.72 MPa, 1.30 h−1, and 794 m3/m3; the presence of aromatic species in the condensed liquid fraction was not detected. After this test, the experimental setup was ready for the HDT or HCK tests.
Hydrotreating process
The LCO samples, LCO-1 or LCO-2, were used as feedstocks for the HDT process by means of either CoMo/Al2O3 (HDT-1) or NiMo/Al2O3 (HDT-2) as catalyst, under the experimental conditions described in Table 4. The experiment was carried out until 96 h were attained to prepare enough material for the next step. The HDT products (Table 4) were described by the following type of acronyms: HL.X.Y.ZZZ, where X represents either LCO-1 or LCO-2 as feedstock, Y refers to HDT catalysts, HDT-1 or HDT-2, and finally, ZZZ is 320 or 330 °C, the temperature employed for the process. After each experiment, gas and liquid fractions were separated and weighed for mass balance purposes. The gas phase was measured using a gas meter and characterized by a UOP standard analysis for refinery gases [19]. The HDT LCO liquid fraction was received in a container, weighed, and characterized according to the ASTM methods described in Table 1 and with the gas chromatography methods featured in this work (Sects. “Determination of aromatic and polynuclear aromatic contents in diesel fuels by ASTM D5186” and “Fast chemical characterization by GC-FID”). The results are shown in Table 4. Sample HL.2.1.330 obtained after the HDT process was subjected to a detailed chemical characterization according to ASTM D6623-01, as described in Sect. “Chemical characterization by using ASTM D6623”. The result is shown in Table 6. For the figures, both gas and liquid phases were considered and normalized to 100 wt.%.
Hydrocracking process
Each one of the six HDT LCOs prepared as described in Sect. “Hydrotreating process” (Table 4) was used as feedstock for the HCK process under the following experimental conditions: 375 °C; 7.6 MPa, 1.2 h−1 LHSV, and 750 m3/m3 H2/oil ratio, using the catalyst that consisted of a 50/50 mixture per weight of NiMo/Al2O3//H-ZSM5 (HCK 50/50). After each run, the setup operation achieved a steady state for at least 24 h; hence, the liquid product was collected and analyzed every 8 h. Once each experiment was performed, a mass balance was carried out, where gas and liquid fractions were separated and weighed. The HCK products (Table 5) were named by the following acronyms: HC.X.Y.ZZZ, where the values described the previous HDT products, and as explained before, X refers to either LCO-1 or LCO-2 (feedstock), Y represents the HDT catalyst, HDT-1 or HDT-2, and finally, ZZZ is 320 or 330 °C, the temperature employed for the first HDT process. The gas phase was received in a gas meter and characterized by means of a UOP standard analysis for refinery gases [19]: in this fraction, the presence of aromatic species was not detected. The HCK liquid products were received in a container and characterized according to the ASTM method described in Table 1 and with the gas chromatographic methods presented in this work (Sects. “Determination of aromatic and polynuclear aromatic contents in diesel fuels by ASTM D5186” and “Fast chemical characterization by GC-FID”). The results are shown in Table 5. Sample HC.2.1.330, obtained after the HCK process, was subjected to a detailed chemical characterization according to ASTM D6623-01, as previously described. The result is shown in Table 6. For the figures, both gas and liquid phases were considered and normalized to 100 wt.%.
Results and discussion
Description of the GC-FID method for the liquid phase characterization of the HDT and HCK products
The GC-FID characterization method featured in this work was developed to know, as soon as the experiment was performed, the effect of the different feedstocks and experimental conditions on the BTEX yield in the liquid phase obtained after the HCK process. Figure 3 shows the main signals for benzene, toluene, ethylbenzene, meta- and para-xylenes (overlap), and o-xylene. The rest of the signals were divided according to the description in Table 3. The fraction of light hydrocarbons (LHCs) was mainly composed of C4-C8- paraffins and isoparaffins (alkanes and iso-alkanes). The middle hydrocarbons (MHCs) were the fundamental constituents of the feedstock and were composed mainly of ≥ C9 alkylbenzenes, tetralin, and naphthalene derivatives, and some higher molecular weight paraffins and isoparaffins. Finally, the heavy hydrocarbon fraction (HHC) was represented by phenanthrene derivatives and some higher molecular weight paraffins and isoparaffins.
Effect of the hydrotreating process on the LCO composition.
Gas and liquid phase products were obtained from this process (Table 4). In all cases, the gas phase accounted for 2 wt.% or less of the total balance and was formed mainly by hydrogen (55–79 wt.%) and hydrogen sulfide (19–34 wt.%). Small amounts (< 0.5 wt.%) of methane, ethane, propane, and butane were present in some of the experiments. The two main products, hydrogen, and hydrogen sulfide were removed for the mass balance.
Regarding the liquid phase (98 wt.%), depending on the employed catalyst and temperatures of 320 and 330 °C, keeping the other parameters constant (7.5 MPa, 1.71 h−1 of LHSV, and 708 m3/m3 H2/Oil), LCO 1 departed from 2.7 wt.% (27,000 mg/kg) of sulfur content to 400 and 240 mg/kg with HDT-1 (CoMo/Al2O3), respectively (Table 4). With HDT-2 (NiMoP/Al2O3), at 330 and 370 °C, the obtained sulfur contents were 150 and 120 mg/kg. Under the experimental conditions described above, the nitrogen content went from 255 mg/kg to 18.5, 7.8, 0.3 and 0.3 mg/kg (Table 4). The NiMoP/Al2O3 catalyst turned out to be a superior hydrodesulfurization and hydrogenation catalyst than CoMo/Al2O3 [20, 21]. In the case of LCO 2, only the HDT-1 catalyst (CoMo/Al2O3) was used, and the sulfur content went from 4.3 wt.% (43,000 mg/kg) to 300 and 130 mg/kg when the used temperatures were 320 and 330 °C and the nitrogen contents ranged from 196 to 1.3 and 0.3 mg/kg (Table 4). Since generally, the catalysts employed in HCK processes are sulfides of group VI and VIII metals, remaining sulfur can be useful for keeping the catalyst active, however, the presence of nitrogen compounds is known to be harmful to the HCK catalyst [22, 23]. Therefore, about 200 mg/kg of sulfur must remain in the HDT LCO to maintain the catalyst activity for the next HCK process. Notwithstanding, it is more important to reduce the amount of nitrogen compounds because it is known that their presence poisons the acidic sites of the HCK catalyst required for the next step [5, 22].
The chemical composition obtained in the HDT products varied with the feedstock, catalyst, and experimental conditions (Fig. 5). HDT-2 (NiMoP/Al2O3) was exerted higher hydrogenation capabilities than HDT-1 (CoMo/Al2O3); therefore, the amount of mono-, di-, tri-, and total aromatics was lower. In this context, HDT-2 favored the formation of more hydrogenated products which are harmful for the BTEX formation after the HCK process. Therefore, as it was pointed out, the CoMo-based catalyst (HDT-1) exhibited lower hydrogenation capabilities than the NiMoP-based catalyst, HDT-2 [5, 20, 21] being this catalyst a better choice for the HDT. From Table 4, the HDT LCO liquid phase samples consisted mostly of MHC- type fractions in all cases (from 93 to 96 wt.%). They contained some BTEX (from 2.3 to 6.7 wt.%) and a fair amount of C9+-alkylbenzenes (C9+-AlkBzs) from the hydrocracking process that occurred during the hydrotreating [24]. The presence of the C9+-AlkBzs was only noticeable in the HDT product, HL.2.1.330, after the GC-PIONA analysis [18] that will be discussed in the following section.

Effect of the catalyst and the hydrotreating conditions on LCO-1 and LCO-2
Hydrocracking products from different hydrotreated LCOs
Effect of the chemical composition of the HDT LCO
Gas and liquid phase products were obtained from this process (Table 5). The gas phase accounted for 8–13 wt.% of the total balance after disregarding hydrogen, hydrogen sulfide carbon dioxide, and other impurities, leaving only the hydrocarbon-type molecules. The gas phase was usually formed by methane (1.4–8.3 wt.%), ethane (14.9–29.0 wt.%), propane (35.3–43.1 wt.%), butane (9.5–14.3 wt.%), n-pentane (0.06–0.47 wt.%), i-pentane (3.0–10.1 wt.%), and hexanes (5.4–35.0 wt.%).
The figures for the analysis of the products were prepared by considering the gas and liquid phases and afterward normalizing them to a 100 wt.%. As the mono-and total aromatic compounds increased, the presence of MHCs slightly increased, the formation of gas and LHCs decreased while the HHCs remained unaltered. Regarding our study, the formation of benzene, toluene, ethylbenzene, and xylene (BTEX) seemed not to be affected in a positive way. The BTEX formation values ranged from 25.2 to 29.2 wt.%. An unexpected result contrary to the assumptions made in previous reports [4, 13, 14] that the higher the amounts of mono-aromatic compounds, the higher the BTEX formation was obtained. It is noteworthy to mention from Figs. 6, 7 that the MHC content in the HCK product slightly increased as the mono- and total aromatic content increased. Figure 8 shows the MHC conversion versus the difference between BTEX products and feedstock. It is obvious that the MHC presence even in excess did not increase the BTEX yield, meaning that not all the MHCs present in this fraction were suitable for BTEX production.

Effect of the mono-aromatic content from the feedstock on the chemical composition of the products after hydrocracking the HDT LCOs

Effect of the total aromatic content from the feedstock on the chemical composition of the products after hydrocracking the HDT LCOs. Fractions: LHCs light hydcrocarbons, BTEX benzene, toluene, ethylbenzene, and xylenes, MHCs middle hydrocarbons, and HHCs heavy hydrocarbons

Effect of the middle hydrocarbon (MHC) conversion on the BTEX production after hydrocracking the HDT LCOs
To increase the understanding of the resulting chemical composition, two suitable samples were analyzed by GC-PIONA [18]. The samples presented adequate distillation properties for performing the task (Table 6). The whole product composition can be seen in Fig. 9a comparing the hydrotreated LCO (sample HL.2.1.330) and hydrocracked product (sample HC.2.1.330). The presence of paraffins, isoparaffins, BTEX, naphthenes, and olefin derivatives increased sharply in the hydrocracked product. It is important to observe the almost total disappearance of tetralin, naphthalene, and indane derivatives, and the alkylbenzenes (AlkylBz) derivatives decreased from 30.9 to 10.0 wt.%, however, it did not mean that the resulting products were only BTEX. A closer look at the AlkylBz composition by carbon number is given in Fig. 9b. Except for C9-AlkylBzs, C10- to C13-AlkylBzs showed reductions from their original amounts. However, this reduction may not only be related to BTEX (C8-AlkylBzs) production, but to other types of hydrocarbons like naphthenes, paraffins, and isoparaffins by further hydrogenation (Fig. 10).

Comparison by GC PIONA of the HL.2.1.330 and HC.2.1.330 samples: a all type groups, b alkylbenzene compounds

Suggested reaction scheme for the HCK of the HDT LCO. kHCK = reaction rate constant for the HCK process, kHYD = reaction rate constant for the hydrogenation process, kDTR = reaction rate constant for the disproportionation/transalkylation step
The effect of the HCK 50/50 catalyst mixture (NiMo/Al2O3//H-ZSM5, 50/50 wt.%) on the HCK reaction in tetralin as a model compound, was already discussed by Laredo et al. [3]. The maximum pore diameter of H-ZSM-5, according to the Database of zeolite structures [25], is 0.636 nm. The Brönsted acid sites present in this type of zeolite are particularly important for HCK purposes. [3, 4, 26]. The presence of the metallic function (NiMo) in adequate amounts is necessary to reduce the catalyst deactivation by coke formation [3, 4]. Nevertheless, as a study carried out before by Laredo et al. [13], when departing from real feedstocks like the HDT LCO, a total BTX yield higher than 35% was never attained by using a HCK catalyst of any type. In real feedstocks, this can be due to the presence of another type of competing hydrocarbons [7], the inevitable gas formation and the formation of other C9+-AlkylBzs.
Mechanisms involved in the hydrocracking of the HDT LCO.
BTEX can be produced either by the direct HCK process or by disproportionation and transalkylation (DTR) processes [3, 4, 27,28,29,30,31] from more complex alkyl-aromatic compounds obtained earlier by the same direct hydrocracking of naphthalene, tetralin, and indene derivatives as shown in Fig. 10.
Disproportionation and transalkylation (DTR) are the two major practical processes for the interconversion of alkylaromatics, especially for the production of dialkylbenzenes (Fig. 11) [28, 29].

Major routes for alkyl-transfer reactions
The DTR selectivity, considering the thermodynamic dependence of methylbenzenes, was extensively studied according to the feed composition when using the following catalysts: chlorinated aluminum, hydrogen fluoride, boron trifluoride, alumina, and amorphous and crystalline aluminosilicates [27]. As an example, the results of the disproportionation of several C7–C11 methylbenzenes using aluminum oxide as a catalyst are shown in Table 7. Based on this reference [27], the composition of di-, tri-, and tetramethylbenzenes was close to thermodynamic equilibrium. Figure 12 was prepared by considering this example, which shows the simulated composition of benzene (Bz), toluene (Tol), xylenes or dimethylbenzenes (DMBzs), trimethyl- to hexamethylbenzenes (TMBx, TeMBz, PMBz, and HMBz) as DTR products when departing from the composition of the alkylbenzenes from the HDT LCO of this work. It seemed, as suggested, that the final composition after several cycles led to thermodynamic equilibrium [27]; therefore, not all the BTEX formation after the HCK procedure can be attributed to be reached following this path.

Simulated disproportionation product composition when departing from the alkylbenzenes in the HDT LCO
In the case of zeolites as catalysts [28, 29], the transalkylation of alkylbenzenes may proceed by intermolecular alkyl transfer involving dealkylation-alkylation steps with surface alkoxy species as reaction intermediates or through the formation of bulkier diaryl intermediates (Fig. 13) [29,30,31]. The contribution of these two mechanisms to the overall process depends on the relative stability of the alkylaromatic and bulkier diaryl cationic intermediates and is largely influenced by the reactant size, zeolite pores, and the presence of channel intersections or cavities [31]. Therefore, the cavities and pore channels must be large enough to accommodate the required transition state, especially for alkyl-aromatics with sterically hindered alkyl groups (bimolecular intermediate) [29,30,31], otherwise the formation of the relevant di- or poly-alkyl aromatics should be accommodated, for example, by a dealkylation/alkylation mechanism [29]. An important condition that must be fulfilled is that the alkyl-containing aromatic molecule (the one that must lose the alkyl group) must have unlimited access to the active sites (steric constraints, catalyst choice) for effective transalkylation reactions [29]. When studying the disproportionation-transalkylation of methyl-ethylbenzene (MEB), Ali et al. [32] concluded that ZSM5, a 10 × 10 MR pore zeolite, showed low to medium MEB conversion (20–60%), remarkably high dealkylation selectivity (60–70%), and very low disproportionation and transalkylation selectivity. These authors concluded that beta zeolite presented the best performance for the transalkylation process of MEB. In this work, the use of the zeolite H-ZSM5 did not seem to favor this path due to the difficulty of the sterically hindered C9+-diaryl intermediate formation inside the relatively small channels associated with this option.

Simplified disproportionation and transalkylation mechanisms of alkylbenzenes in zeolites [31]
Regarding the process temperature, transalkylation is favored at temperatures higher than 300 °C, while reaction temperatures below 350 °C will selectively favor the coupling of alkyl-aromatics and, consequently, disproportionation. Temperatures below 300 °C will lead to rapid deactivation by molecular retention and might not favor the formation of products, and extremely high temperatures will lead to dealkylation and consequently rapid deactivation by deposition of carbonaceous material [29].
In this case, it is not possible to make a clear identification of the mechanism followed for achieving the product distribution from the HCK of HDT LCO for improving the BTEX yield without additional experiments (i.e., kinetic studies or isotope tracking). If the options can be (Fig. 10) direct hydrocracking (kHCK) and disproportionation/transalkylation (kDTR), a combination of both processes seems to be the most plausible answer.
Conclusions
The effect of the feedstock composition on the BTEX yield was studied. Six different samples of hydrotreated light cycle oils (HDT LCOs) with different compositions were obtained from two different light cycle oils (LCOs) using two different catalysts: CoMo/Al2O3 (HDT-1) and NiMoP/Al2O3 (HDT-2). The second one presented higher hydrogenation capabilities to produce a feedstock with lower content of sulfur, nitrogen, and aromatic compounds. The hydrotreated LCO samples obtained with different amounts of mono-, di-, tri- and total aromatic contents were used as feedstock for the following hydrocracking process with 50/50 in weight mixture of NiMo/Al2O3 and /H-ZSM5 (NiMo/H-ZSM5 (50/50)) at 375 °C, 7.5 MPa, 1.2 h−1 as LHSV and 750 m3/m3 H2/oil ratio. It was observed that the feedstock composition, neither as mono- and total aromatic contents nor as middle hydrocarbon fraction (MHC) presented a significant effect on the BTEX formation. A maximum amount of 29.8 wt.% of BTEX was obtained. A closer look showed that after hydrocracking, all the tetralin and naphthalene derivatives were consumed, although the overall presence of C9+-alkylbenzenes (C9+-AlkBzs) also decreased; through a more detailed analysis of this group, it was found that the formation of C9-AlkylBzs remained unchanged while the C10+-AlkylBzs decreased sharply.
Nevertheless, with the available information, the understanding of product formation and feedstock composition, and therefore, ways to improve the BTEX yield is not possible at this moment.
References
Laredo GC, Romo PP, Escobar J, García-Gutiérrez JL, Vega-Merino P (2017) Light cycle oil upgrading to benzene, toluene and xylenes by hydrocracking: studies using model mixtures. Ind Eng Chem Res 56:10939–10948. https://doi.org/10.1021/acs.iecr.7b02827
Laredo GC, Vega-Merino PM, Schacht-Hernández P (2018) Light cycle oil upgrading to high quality fuels and petrochemicals: a review. Ind Eng Chem Res 57:7315–7321. https://doi.org/10.1016/j.cattod.2015.09.046
Laredo GC, Pérez-Romo P, Vega-Merino PM, Árzate-Barbosa E, García-López A, Águeda-Rangel R, Martínez-Moreno VH (2019) Effect of the catalytic system and operating conditions on BTX formation using tetralin as a model molecule. Appl Petrochem Res 9:185–198. https://doi.org/10.1007/s13203-019-00237-4
Laredo GC, Pérez-Romo P, Agueda-Rangel R, García-López A (2020) Effect of the experimental conditions for BTX formation from hydrotreated light cycle oil. Appl Petrochem Res 10:21–34. https://doi.org/10.1007/s13203-020-00242-y
Stanislaus A, Marafi A, Rana MS (2010) Recent advances in the science and technology of ultra low sulfur diesel (ULSD) production. Catal Today 153:1–68. https://doi.org/10.1016/j.cattod.2010.05.011
Laredo GC, Figueroa Y, Cano JL, Mares MT, Castillo J (2002) Estudio de la composición del aceite cíclico ligero provenientes de crudos mexicanos. J Mex Chem Soc 46:115–119
Laredo GC, Pérez-Romo P, Águeda-Rangel R, Escobar J, Vega-Merino P (2020) Detailed characterization of light cycle oil for BTX production purposes. Int J Petrol Petrochem Eng 6:1–12. https://doi.org/10.20431/2454-7980.0603001
Norma Oficial Mexicana NOM-016-CRE-2016, Especificaciones de calidad de los petrolíferos. Diario Oficial de la Federación. 29/09/2016
https://www.epa.gov/diesel-fuel-standards/diesel-fuel-standards-rulemakings
Bisht D, Petri J (2014) Considerations for upgrading light cycle oil with hydroprocessing technologies. Indian Chem, Eng 56:321–335. https://doi.org/10.1080/00194506.2014.927179
Park J-I, Ali SA, Alhooshani K, Azizi N, Miyawaki J, Kim T, Lee Y, Kim H-S, Yoon S-H, Mochida I (2013) Mild hydrocracking of 1-methyl naphthalene (1-MN) over alumina modified zeolite. J Ind Eng Chem 19:627–632. https://doi.org/10.1016/j.jiec.2012.09.014
Wu T, Chen SL, Yuan GM, Xu J, Huang LX, Cao YQ, Fan TT (2018) High-selective-hydrogenation activity of W/Beta catalyst in hydrocracking of 1-methylnaphthalene to benzene, toluene, and xylene. Fuel 234:1015–1025. https://doi.org/10.1016/j.fuel.2018.07.133
Laredo GC, García-Gutiérrez JL, Pérez-Romo P, Olmos-Cerda EH (2021) Effect of the catalyst in the BTX production by hydrocracking of light cycle oil. Appl Petr Res. https://doi.org/10.1007/s13203-021-00266-y
Olmos-Cerda EH, Laredo GC, Pérez-Romo P, Águeda-Rangel R, García-López A (2021) Selective hydrogenation of light cycle oil for BTX and gasoline production purposes. Int J Chem React Eng. https://doi.org/10.1515/ijcre-2020-0144
www.astm.org. (Accessed in April. 2021)
ASTM D5186-20. Standard test method for determination of the aromatic content and polynuclear aromatic content of diesel fuels by supercritical fluid chromatography. https://www.astm.org/Standards/D5186.htm
Ito S (2013) Analysis of the aromatic hydrocarbons on gasoline and naphtha with the Agilent 6820 series gas chromatograph and a single polar capillary column. Agilent Technologies. Yokogawa Analytical Systems, Inc. Mitaka Bldg. 2-11-13. Nakacho, Musashino-shi. Tokyo, 180-0006. https://www.agilent.com/cs/library/applications/5988-9261EN.pdf
ASTM D6623-01 Test method for determination of individual components in spark ignition engine fuels by high resolution gas chromatography. https://www.astm.org/Standards/D6623.htm
UOP539-12, Refinery gas analysis by GC. https://www.astm.org/Standards/UOP539.htm
Bose D (2015) design parameters for a hydro desulfurization (HDS) unit for petroleum naphtha at 3500 barrels per day. World Sci News 9:99–111 (WSN-9-2015-99-111)
Ledoux MC, Djelloudi B (1990) Comparative hydrodenitrogenation activity of molybdenum, Co–Mo and Ni–Mo alumina-supported catalysts. Appl Catal 67:81–91. https://doi.org/10.1016/S0166-9834(00)84433-4
Shin J, Oh Y, Choi Y, Lee J, Lee JK (2017) Design of selective hydrocracking catalyst for BTX production from diesel-boiling-range polycyclic aromatic hydrocarbons. Appl Catal 547:12–21. https://doi.org/10.1016/j.apcata.2017.08.019
Jin N, Wang G, Yao L, Hu M, Gao J (2016) Synergistic process for FCC light cycle oil efficient conversion to produce high-octane number gasoline. Ind Eng Chem Res 55:5108–5115. https://doi.org/10.1021/acs.iecr.6b00360
Robinson PR, Dolbear GE (2006) Hydrotreating and hydrocracking: fundamentals. In: Hsu CS, Robinson PR (eds) Practical advances in petroleum processing. Springer, New York. https://doi.org/10.1007/978-0-387-25789-1
Database of zeolite structures. https://www.iza-structure.org/databases/ (Accessed on March 2021)
Townsend AT, Abbot J (1993) Catalytic reactions of tetralin on H-ZSM5 zeolite. App Catal A 95:221–236. https://doi.org/10.1016/0926-860X(93)85076-2
Aromatic Transalkylation. https://nefthim.com/manual/aromatics-transalkylation
Tsai T-C, Lin S-B, Wang I (1999) Disproportionation and transalkylation of alkylbenzenes over zeolite catalyst. Appl Catal A Gral 181:355–398. https://doi.org/10.1016/S0926-860X(98)00396-2
Mokoena K. Alkyl transfer (transalkylation) reactions of alkylaromatics on solid acid catalysis. PhD Thesis. University of the Witwatersrand, Johannesburg. http://wiredspace.wits.ac.za/bitstream/handle/10539/1751/ALKYL_TRANSDFER__TRANSALKYL.PDF?sequence=1&isAllowed=y. (Accessed on September 10, 2020)
Huang J, Jiang Y, Marthala VRR, Hunger M (2008) Insight into the mechanisms of the ethylbenzene disproportionation: transition state shape selectivity on zeolites. J Am Chem Soc 130(38):12642–12644
Margarit VJ, Osman M, Al-Khattaf S, Martínez C, Boronat M, Corma A (2019) Control of the reaction mechanism of alkylaromatics transalkylation by means of molecular confinement effects associated to zeolite channel architecture. ACS Catal 9:5935–5946
Ali SY, Ogunrombi KE, Al-Khattaf SS (2013) Kinetics of dealkylation-transalkylation of C9 alkyl-aromatics over zeolites of different structures. Chem Eng Res Dev 91:2601–2616. https://doi.org/10.1016/j.cherd.2013.04.014
Acknowledgements
The authors are grateful for the financial support provided by the Instituto Mexicano del Petróleo (IMP) via the research project D.61065. Eli H. Olmos Cerda is grateful for the PhD scholarship provided by CONACYT, México.
Author information
Authors and Affiliations
Contributions
GCL conceived and wrote the paper; RAR, AGL and EHOC carried out the experiments, and JLGG revised the information.
Corresponding author
Ethics declarations
Conflict of interest
The author(s) certify that they have no Conflict of Interest regarding the subject matter or materials discussed in this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Laredo, G.C., Águeda-Rangel, R., García-López, A. et al. Effect of the chemical composition of six hydrotreated light cycle oils for benzene, toluene, ethylbenzene, and xylene production by a hydrocracking process. Appl Petrochem Res 11, 249–263 (2021). https://doi.org/10.1007/s13203-021-00276-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-021-00276-w