
Abstract
The study of a light cycle oil (LCO) upgrading alternative involving hydrotreating and hydrocracking/transalkylation procedures for obtaining a benzene, toluene and xylene (BTX) enriched fraction is presented. The research work was focused on the effect of the experimental conditions on the hydrocracking of an hydrotreated light cycle oil (HDT LCO) in order to produce the highest amounts of BTX, when the catalysts consisted of a mixture (50/50 in weight) of nickel–molybdenum on alumina (NiMo/Al2O3) and ZSM-5 materials (NiMo/ZSM-5 (50)). It was found that 7.4 MPa, up to 375 °C, LHSV of 1.2 h−1 and a H2/Oil value of 442 m3/m3 were the optimal experimental conditions for producing an enriched BTX fraction (31%). In order to facilitate the analysis, the study was carried out considering four types of hydrocarbons as lumps for the feed and HCK products: light hydrocarbons (LHC) composed by C4–C7 non-aromatic compounds, BTX, middle hydrocarbons (MHC) consisting of C7–C10 paraffins and isoparaffins, alkylbenzenes, tetralin and naphthalene derivatives and a small amount of high molecular weight hydrocarbons (HHC). Based on this description, HDT LCO used as feedstock for the hydrocracking (HCK) procedure, presents a 99% of a MHC fraction. The HCK conversion, BTX selectivity and yields were obtained from the chromatographic analysis of the products. A simple kinetic model considering only the MHC conversion was carried out. The obtained activation energy confirmed the endothermic nature of the HCK process. The activity decay of the catalytic mixture was also studied by varying the time on stream.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
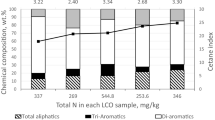
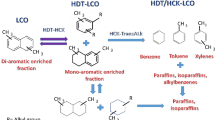
The continuation of the work regarding the production of benzene, toluene and xylene (BTX) from light cycle oil (LCO) is presented in these papers [1,2,3]. LCO, a middle distillate from FCC (fluid catalytic cracking), has lost market as a part of the diesel feedstock due to its inherent low quality (high sulfur, nitrogen and aromatic contents) [4, 5], which makes the resulting fuel difficult to comply with the stringent worldwide environmental regulations [6, 7]. In this sense, LCO from Mexican refineries contains up to 90 wt% of mono-, di- and tri-aromatic compounds (Fig. 1) [5], where most of these compounds are di-aromatic hydrocarbons, i.e. naphthalene derivatives. Among the alternatives for LCO upgrading, the production of BTX after successive hydrotreating (HDT) and hydrocracking (HCK) procedures (Fig. 2) [1, 2, 8] is found. These types of studies are usually carried out using model mixtures [1]. However, when a catalyst or a process is almost ready for scaling up or commercial applications, experiments with real feedstocks are mandatory [2, 9].

Hydrocarbon composition, sulfur and nitrogen content and cetane index of some LCO samples from Mexican refineries

Reaction scheme for obtaining BTX, LPG and naphtha from naphthalene derivatives in the LCO
Additionally, it is well known [1,2,3, 10] that catalysts play a key role in the hydrogenation (partial saturation) of naphthalene followed by the selective cracking of naphthenic structures, producing one-ring-aromatic hydrocarbons with alkyl chains. For this purpose, bi-functional catalysts with acid (support) and metal (hydrogenation-dehydrogenation) functions are frequently used. However, an excess of hydrogenation activity and strong acidity may crack molecules excessively into light gaseous hydrocarbons and accelerate coke deposition. In this sense, hydrocracking catalysts with metal (groups VI and VIII) sulfides perform, in general, the hydro-dehydrogenation and hydrocracking of heavy oil feedstocks. For this reason, metallic-sulfide-supported catalysts are considered more effective materials than metals [11].
Sheng et al. [12] patented the hydrocracking process of a hydrogenated fraction in the presence of copper and chromium oxides or silver on an acid-modified mordenite with a high Si/Al ratio and stabilizing metals like palladium or nickel. The process was carried out at 430 °C, 6.9 MPa, liquid hourly space velocity (LHSV) of 2 h−1 and with a H2/Oil ratio of 3 m3/m3, obtaining 29% of selectivity to monocyclic aromatic hydrocarbons (MAH).
On the other hand, Kim et al. [13] patented a process, where a BTX rich fraction was produced from LCO after being subjected to the HCK process in a FCC reactor using a common FCC catalyst at 600 °C, 0.24 MPa, weight hourly space velocity (WHSV) of 27.2 h−1 and a catalyst-to-oil ratio of 10 m3/m3. From this product, BTX, olefins and poly-aromatic compounds were separated. The poly-aromatic rich fraction was submitted to hydrogenation (HYG: NiMo/Al2O3, 300 °C, 9.8 MPa, LHSV of 1.5 h−1 and H2/Oil ratio of 500 m3/m3) and recycling in the LCO used as feedstock in the first step.
Meanwhile Iwasa et al. [14] claimed a method for producing MAH, preferably BTX, from HDT LCO by means of cracking and reforming steps. BTX selectivity up to 45% was obtained using a Pt/Beta zeolite catalyst at pressures of 0.1–2.0 MPa, temperatures of 400–650 °C and contact times of 5–80 s.
Likewise, Yanagawa et al. [15] patented a method for producing MAH from HDT LCO with GaP/ZSM-5 and/or ZnP/ZSM-5 catalysts. BTX conversions of 49–51 wt% were achieved when 50/50 vol/vol mixtures of LCO and HDT oil were processed with a GaP/ZSM-5 catalyst at 0.3 MPa, 540 °C and LHSV of 0.4 h−1.
As for Upare et al. [16], they used CoMo (0.5)/Beta (β = SiO2/Al2O3 of 25) for the selective hydrocracking of a low-boiling-point fraction of a pyrolysis fuel oil (PFO) [17] for obtaining an enriched MAH fraction such as BTX, using a fixed-bed reactor system. The best experimental conditions for producing the highest MAH yield of 54.8% at 99.1% conversion of the PFO fraction were 370 °C, 8 MPa, H2/feed (m3/m3) of 1250 and LHSV of 0.2 h−1. Considering that the PFO was distilled to 55 wt%, the conversion from the departing PFO was about 54.5% and, therefore, the overall MAH yield was 30.1% after separating the heavy aromatic compounds.
Oh et al. [18] presented a detailed work on the design of a specific catalyst for BTX production departing from optimized HDT LCO. By means of a HCK Mo-S/BZ (BZ = H-Beta + H-ZSM-5) catalyst, the authors obtained a BTX enriched fraction with a MAH yield close to the theoretical one (61.3 compared to 67.6%) at the following experimental conditions: 357 to 450 °C, 6 MPa, WHSV of 2 h−1 and a H2/Oil ratio of 1972 m3/m3.
In a previous study, Laredo et al. [3] worked on the hydrocracking of tetralin for BTX production using mixtures of NiMo/Al2O3 and ZSM-5 catalysts. In this work, a suitable mixture of the materials (i.e. 20/80 to 50/50) for a HCK catalyst capable of transforming tetralin into BTX with convenient conversions (79–96%) at 450–500 °C, 3.9–5.9 MPa, 1.3 h−1 and 168–267 H2/feed (m3/m3) in a bench-scale trickle-bed reactor was described. These mixtures presented the required hydrogenation capabilities for reducing the catalyst deactivation by carbon deposition [19, 20] and acidic functions for hydrocracking purposes [1, 2]. A 50/50 mixture proved to have good hydrocracking/transalkylation activities due to the acidic sites of the ZSM-5 material coupled to a low deactivation behavior associated with the hydrogenation function exerted by the metal catalysts. Then, the next step was to test this catalytic mixture using a real feedstock.
Additionally, the study of the reaction rates is required for the development of profitable manufacturing processes. The goal is to describe the process behavior in terms of macroscopically observed quantities such as temperature, pressure, composition and so on. This empirical approach has been the basis for chemical reactor technology [21]. A review regarding HCK kinetic modeling for reaction design, optimization and simulation when using real feedstocks was published by Ancheyta et al. [22]. According to this work, the kinetic modeling of real feedstocks is difficult because of the analytical complexity and computational limitations. The more compounds a model includes, the more kinetic parameters are needed to be estimated; consequently, more experimental information is required. In these cases, the analysis of a real feedstock in a process is frequently very difficult to be performed due to its real inherent complexity. The problem can be simplified if the chemical species are grouped into lumps and each lump is considered as an independent entity [23]. This approximation assumes that all the compounds in each fraction present the same average reactivity. Another approach can be to treat them as a distribution function.
Therefore, the main objectives of this work are as follows: (1) to find the optimal experimental conditions for a hydrocracking process employed for producing higher amounts of BTX from HDT LCO (Fig. 2), using a 50/50 NiMo/Al2O3//ZSM-5 mixture, from this point on known as NiMo/ZSM-5 (50), (2) to develop a method for feedstock and product characterization as lumps to facilitate the interpretation of results, (3) to estimate kinetic parameters based on the mentioned characterization method and pilot plant data and (4) to study the product distribution depending on the catalyst activity during time on stream (TOS) of 192 h.
Materials and methods
Materials
Ultra-high purity hydrogen (99.999 vol%, Infra-Mexico) was employed in the experiments. Two commercial catalysts were used: NiMo/Al2O3 (IMP-DSD-3 + . IMP) and ZSM-5 (PZ 2/25, Zeochem).
Characterization methods for both materials and data (Table 1) were published in a previous paper by Laredo et al. [3]. Nitrogen adsorption–desorption isotherms and specific surface areas for both materials were obtained at 77 K on a Micromeritics ASAP-2000 analyzer in accordance with the BET method. The acid sites of both materials were determined by FTIR-pyridine desorption using an FTIR spectrometer Thermo Nicolet model Magna 560. For the analysis, a pure-catalyst-made disk was treated under vacuum at 1.33 × 10–3 Pa and 500 °C for 5 h, which was then cooled down to room temperature and later treated with pyridine vapor and afterwards heated at 150 °C under high vacuum for 30 min. IR spectra were collected at different temperatures, i. e., ambient temperature, 100, 200, 300 and 400 °C. The amount of Brönsted and Lewis acid sites was calculated via integration of the area of the absorption bands, showing maximum intensity values at 1450 and 1550 cm−1, respectively. The integrated absorbance of each band was obtained by using the appropriate software (OMNIC) and applying the corresponding extinction coefficient, normalized by the weight of the samples. The calibration of this method was performed periodically by using a faujasite-type zeolite (NIST 8850) as external standard.
The NiMo/Al2O3 metallic composition was obtained by elemental analysis, using a Perkin-Elmer Model 3100 Atomic Absorption Spectrophotometer (Table 1).
Both catalysts were crushed, sieved (60 mesh) and dried at 120 °C for 2 h prior to loading.
PEMEX kindly provided the light cycle oil (LCO, Table 2) required for the first part of the process for obtaining the hydrotreated light cycle oil (HDT LCO) to be used as feedstock for the subsequent HCK procedure [3].
In order to study the residual HCK activity of the NiMo/ZSM-5 (50) catalytic mixture after 400 °C, 7.4 MPa and LHSV of 0.8 h−1during 192 h of time on stream (TOS) a thermogravimetric analysis-differential scanning calorimetry (TGA–DSC) study was carried out. The procedure was performed using a Setsys 12 analyzer for the measurement of the materials that covered the catalyst surface after the HCK experiments. About 20 mg of the NiMo/ZSM-5 (50) sample were heated in the presence of a gas mixture containing 15% O2 in N2 flowing at a rate of 60 mL/min. The mass loss was monitored as a function of temperature. A linear heating rate of 5 °C/min was used throughout the analyses up to 900 °C. In the 200–600 °C interval, an exothermic event was detected, which corresponded to the combustion of carbonaceous material in the catalyst (Wc).
Experimental setup
The bench-scale unit used for both the HDT and HCK processes was equipped with a trickle bed reactor (Catalyst volume = 6.62 mL; internal diameter = 1.0 cm; down-flow mode). A simplified diagram of this pilot plant including some basic instrumentation and analytical peripherals is shown in Fig. 3. The reactor (R-101) was loaded with 5.0 g of fresh catalyst (NiMo/Al2O3 and the NiMo/Al2O3//ZSM-5 mixture in a 50/50 weight mixture in the case of the HDT and HCK processes, respectively) and heated with an electrical furnace featuring three independent heating zones to attain an isothermal axial profile. The required feedstock (LCO for the first HDT step and HDT LCO for the subsequent HCK step) was introduced into the reactor by using a reciprocating pump (Inlet flow rate: 0.10–0.17 cm3/s) and the hydrogen flow rate (Flow rate: 3–8 STD L/h at 101.32 × 10–3 MPa and 20 °C) was measured with a 0–20 STD L/h ranges Brook 5850 series thermal mass flow controller. The reactor effluent was directed to a low-pressure high-temperature separator (S-101). The liquid fraction leaving the S-101 was sent to a second separator (S-102,) and the gas fractions from S-101 and S-102 were mixed and quantified in a gas meter. The liquid product from S-102 was collected in a recipient, weighed for mass balance purposes and characterized by gas chromatography. All the percentages described in this work are in weight.

Simplified diagram of the bench-scale setup
Catalytic activity tests
In both cases, the HDT and HCK processes, the crushed, sieved and dried NiMo or NiMo/ZSM-5 (50) were loaded into the reactor and then pre-sulfided in situ for 18 h with kerosene spiked with dimethyl-disulphide (S: 2.5 wt%) at 210–315 °C, 4.0 MPa, 1.0 h−1 and inlet hydrogen-to-oil ratio (H2/oil ratio): 315–378 m3/m3. After the pre-sulfiding step, a soaking period was performed with kerosene at 315 °C, 4.0 MPa, 1.0 h−1 and 378 m3/m3 for 48 h. Next, a blank run feeding pure n-hexadecane was carried out at 350 °C, 3.72 MPa, 1.30 h−1 and 794 m3/m3; the presence of aromatic species in the condensed liquid fraction was not detected. After this trial, the experimental setup was ready for the HDT or HCK tests.
HDT process
LCO used as feedstock was submitted to HDT using NiMo/Al2O3 at 330 °C, 5.5 MPa and LHSV of 2.5 h−1 for 240 h in order to get enough material (HDT LCO) for the next step. Chemical characteristics of both feedstock and product are shown in Table 2.
HCK process
HDT LCO (Table 2) used as feedstock was submitted to the experimental conditions that can be seen in Table 3 in order to study the subsequent HCK process. Essentially, the temperature was varied from 350 to 425 °C; pressures of 4.9, 7.4 and 9.8 MPa and LHSVs of 0.8, 11 and 1.7 h−1 were used, keeping the H2/Oil ratio at 442 m3/m3. After each run, the setup operation accomplished steady-state for at least 24 h; hence, the liquid product was collected and analyzed every 8 h.
HCK catalyst activity study
HDT LCO used as feedstock was continuously submitted to the following experimental HCK conditions: 400 °C, 7.4 MPa and LHSV of 0.8 h−1, until 192 h were completed. During the experiments, samples were taken every 8 h and analyzed as follows.
Chemical characterization of feedstock and products
The chemical characterization of the LCO, HDT LCO and HCK products was carried out with a flame ionization detector-gas chromatograph (FID-GC) and the following set of columns: WCOT: 105 m × 320 mm i.d., 50 m × 320 mm i.d. and 105 m × 320 mm i.d. Helium was used as carrier gas (3 mL/min). The temperature program was as follows: 40 °C for 10 min, 10 °C/min ramp up to 300 °C and then 1 min at 300 °C. The detector and injector temperatures were set at 320 and 330 °C. Slip/sliptless range 1–10,000. Retention times used for describing the different fractions as lumps are shown in Table 4.
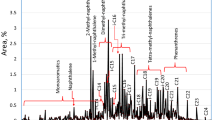
The lumps were composed as follows: light hydrocarbons (LHC) were conformed mostly by C4–C6 alkanes, isoalkanes and cycloalkanes; BTX by benzene, toluene and xylenes; middle hydrocarbons (MHC) by C7–C10 alkanes and isoalkanes, ethylbenzene, C9+ alkylbenzenes, 1,2,3,4-tetrahydronaphthalene (tetralin) and naphthalene derivatives. Examples of the chromatograms obtained for both the HDT LCO used as feedstock and the HCK product are shown in Fig. 4. All the percentages described in this work are in weight.

Examples of chromatograms of feedstock (a) and product (b)
Equations
The liquid products were weighed and characterized by the FID-GC method described in Sect. 2.1. The following equation was used:
- A)
MHC conversion (MHCC)
$$ {\text{MHC}}_{{\text{C}}} ,\% = 100\left[ {\frac{{{\text{MHC}}_{{{\text{feed}}}} - {\text{MHC}}_{{{\text{prod}}}} }}{{{\text{MHC}}_{{{\text{feed}}}} }}} \right], $$(1)where MHCfeed and MHCprod are the percentages of middle hydrocarbons in the feedstocks and products.
- B)
Hydrocracking yield (HCKy)
$$ {\text{HCK}}_{Y} ,\% = {\text{MHC}}_{C} \frac{LF}{{100}}, $$(2)where LF is the liquid fraction percentage in weight.
- C)
BTX yield (BTXy)
$$ {\text{BTX}}_{{\text{Y}}} ,\% = \frac{{{\text{LF}}*{\text{BTX}}_{{{\text{prod}}}} }}{100}, $$(3)where BTXprod is the BTX selectivity in the products in weight.
- D)
Catalyst activity
From the experiments at 400 °C, 7.4 MPa and LHSV of 0.8 h−1, until 192 h were completed, the catalyst activity (A) was calculated using a non-linear equation fitting of the empirical Eq. (4) [21]. The mathematical fitting was carried out by using the PolymathTM 6.1 software:
MHCC (Eq. 1) and A and n are fitting parameters that are functions of the feedstock.
Results and discussion
FID-GC characterization of the HDT-LCO used as feedstock
The HDT-LCO used as feedstock (first line, Table 3, Fig. 4a) was composed mostly of middle hydrocarbons (MHC) 99.1%. The rest was heavy hydrocarbons (HHC, 0.6%) with slight presence of benzene, toluene or xylene (BTX). Light hydrocarbons (LHC) were not found.
Effect of the experimental conditions on the HCK yield and BTX selectivity using the NiMo/ZSM-5 (50) catalyst
The results are shown in Table 3. Under all the experimental conditions (temperature, pressure, LHSV and H2/oil ratio), the temperature increase caused an increment in the hydrocracking conversion and gas yield. As it was expected, as a result of the HCK process, a decrement in the MHC fraction led to an increment in the LHC and BTX fractions (Figs. 5 and 6). It is very important to note that the linear increment in temperature was not proportional to increments in LHC or in BTX. At temperatures above 400 °C, the increment was very small. These results showed that increments in temperature above 400 °C were not advisable because the yield gain was negligible, considering the operational costs and the probability of forming coke [19, 20].

Effect of the temperature and LHSV on the chemical composition of the liquid product using NiMo/ZSM-5 (50) at 7.5 MPa and H2/oil = 442 m3/m3

Effect of the temperature and pressure on the chemical composition of the liquid product using NiMo/ZSM-5 (50) at H2/OIL = 442 m3/m3: a LHSV = 1.7 h−1 and b LHSV 2.7 h−1
The LHSV effect on the HCK yield at different temperatures is shown in experiments 1–12. The HCK yield increased with the LHSV and temperature from 33.4 to 80.1% from 350 to 425 °C at LHSV of 0.8 h−1, to 29.4–64.6 range at 1.1 h−1 and 22.9–33.4% range at 1.7 h−1. The gas fraction followed a similar behavior with very slight increments. As it can be expected, at lower LHSVs, the conversion augmented due to higher contact time between the catalyst and the feedstock. It is important to stress that for increasing the HCK yield and consequently the BTX yield, it was necessary to increase the severity although the LHC fraction also augmented.
Regarding the pressure (data from experiments 5–8 and 13–16 at LHSV of 1.1 h−1 and from experiments 9–12 and 17–20 at LHSV of 1.7 h−1), the HCK yield increased with the increasing pressure and temperature, which results in the BTX and LHC increment with the consequent decrease in the MHC fraction. At low LHSV, the increments in LHC and BTX were quite important when the temperature and pressure were increased; however, when the LHSV was high, the increments in LHC and BTX formation were less important (Fig. 6a, b).
Similar results were described by Upare et al. [16] during the tetralin HCK using a CoMo (0.5)/Beta catalyst. When the temperature was increased from 340 to 380 °C, the HCK yield was also increased, reaching a plateau at 380 °C. Regarding the pressure increments (0–8 MPa), a steady HCK yield was reached at 8 MPa. Finally, the increase in LHSV from 0.4 to 1.6 h−1 produced a reduction in the tetralin HCK yield, as it was observed in this work. Gutiérrez et al. [24] found that by using a Pt–Pd/HY catalyst and increasing the pressure, the hydrodesulfurization and hydrocracking activity augmented and the naphtha selectivity was raised, having a negligible over-cracking. The concentration of aromatic hydrocarbons in the obtained naphtha was 30 wt%. While maintaining the pressure at 6 MPa and WHSV of 2 h−1, Oh et al. [18] observed an increment in HDT LCO hydrocracking by increasing the temperature from 375 to 425 °C, which was evidenced by the amount of the gas phase formed.
Effect of the experimental conditions on the BTX yield
Figure 7 shows a comparison between experimental conditions and the BTX yield. It is quite remarkable that as the temperature was increased up to 400 °C (Fig. 7a), BTXY was also increased; however, higher temperatures did not show such a high improvement and only coke formation and other undesirable effects could be triggered [19, 20]. The pressure effect on the BTX yield showed that a maximum was reached at 7.4 MPa (Fig. 7b). Finally, from Fig. 7c, it can be seen that the LHSV effect on the BTX yield was quite strong, giving the best values at low LHSV.

Effect of the experimental conditions and catalyst on the BTX yield: a Temperature, b Pressure and c) LHSV
The same behavior was observed by Upare et al. [16] using a CoMo (0.5)/Beta catalyst. The increment in temperature produced an increase in the BTX selectivity (340–380 °C) until reaching a plateau at 380 °C. Maximum BTX selectivity was attained at 6 MPa, when a pressure interval ranging from 0 to 8 MPa was studied. However, the decrease in LHSV did not show the same effect on increasing the BTX selectivity as it happened in our work. Oh et al. [18] observed an increment in the BTX selectivity during the HCK process by increasing the temperature from 375 to 425 °C, at 6 MPa and WHSV of 2 h−1. However, due to the decrease in the liquid phase yield caused by the excessive HCK, a moderate BTX yield was produced.
Therefore, in our case, optimum conditions were 400 °C, 7.4 MPa and LHSV of 0.8 h−1, using the NiMo/ZSM-5 (50) material for a 31.0% BTX enriched fraction (overall BTX yield of 30.1%, due to the 97% liquid fraction). This value was lower than the 68.5% BTX enriched fraction obtained by Oh et al. [18]; however, the overall BTX yield obtained by them was 33.1% due to over-cracking (liquid fraction of 48.4%).
Finally, it is necessary to emphasize that HCK solely could not produce benzene, toluene and xylenes directly, but by a transalkylation process [25, 26] from more complex alkyl-aromatic compounds obtained by the direct hydrocracking of tetralin derivatives. Additionally, the presence of aromatic compounds, including BTX, can be the result of bimolecular processes involving hydrogen transfer reactions catalyzed by ZSM − 5, as Townsend and Abbot [25] have suggested. Transalkylation reactions were favored at lower LHSVs and higher temperatures [27]. The pressure effect did not seem to be of great importance.
Simple model for the kinetics of the overall LCO hydrocracking
The overall rates for the HDT LCO hydrocracking were based on the following assumptions (Fig. 8, Eq. 5): the feed was composed mostly by MHC (99.1 wt%) and the MHC lump conversion for the formation of BTX, LHC and Gas presented a simple first-order kinetic behavior when the experiments were carried out at 7.4 MPa [28].

Scheme of the HCK process of the MHC lump from the LCO
then
and after integration;
where MHCi and MHCf are the initial and final middle hydrocarbon concentration, k is the rate constant, and LHSV is the liquid hourly space velocity.
The first-order assumption was proved correct as is shown in Fig. 9. The plot depicts a linear relationship; therefore, the hydrocracking might follow a first-order rate behavior:

First-order plot for the hydrocracking of middle hydrocarbons (MHC) present in the HDT LCO
No change in the system hydrogen concentration can be assumed, since the experiments were carried out at a constant pressure of 7.4 MPa. Figure 10 shows the plot of the ln k versus the reciprocal of the absolute temperature according to the Arrhenius equation:

Arrhenius equation plot for the hydrocracking of middle hydrocarbons (MHC) present in the HDT LCO
where Ea is the activation energy and R is the gas constant equal to 8.314472 J/K mol; the obtained activation energy was 139.5 kJ/mol. Although the used catalyst and studied feedstock were different, it was interesting to compare the value obtained by Qader and Hill [28] for the hydrocracking of gas oil at 10.3 MPa using a nickel-tungsten catalyst, which was 175.4 kJ/mol.
Following a similar procedure, but using the Eyring equation (Fig. 11)
where ∆H is the activation enthalpy, kb, is the Boltzmann constant (1.3806488 × 10–23 J/K), h is Plank´s constant (6.626070150 × 10–34 J/s) and ∆S is the activation entropy; the value of ∆H was 134.6 kJ/mol. The positive sign shows that the LCO hydrocracking is an endothermic reaction. The value of ∆S was − 46.4 J/mol K. The negative sign indicates that the disorder of the isolated system has decreased. According to the previous discussion, it is expected because the HCK reaction is endothermic and the temperature increase results in an augmented conversion producing LHC and BTX compounds. In the same way, when the LHSV was decreased, the residence time increased; consequently, the conversion was higher because the reagents were in contact with the catalyst for more time. However, since the gas fraction is undesirable because it affects the desirable liquid fraction, a compromise is required for increasing the BTX selectivity without over-cracking the feedstock. The H2–oil relationship was also a parameter that played a key role because it was used to hydrogenate polynuclear aromatics and reduce the aromatic condensation that can lead to coke formation [19, 20]. Moreover, the operating pressure exerted no negligible effect on the conversion and an optimum value of 7.4 MPa was found.

Eyring equation plot for the hydrocracking of middle hydrocarbons (MHC) present in the HDT LCO
Effect of the time on stream on the chemical composition.
The effect on the chemical composition of the hydrocracked product obtained during a TOS of 192 h is shown in Fig. 12. According to the figure, the catalytic activity was stable for BTX production for 112 h at 400 °C, 7.4 MPa and LHSV of 0.8 h−1. At the beginning of the experiment (48 h), the HCK product presented an average chemical composition similar to that obtained after 24 h under the same experimental conditions (Fig. 5), being the LHC fraction the main product, followed by BTX, MHC and Gas lumps. It is very important to point out that when the activity began to diminish after 48 h of TOS, the LHC products began to decrease, but the MHC presence decreased until reaching a minimum after 72 h and then began to increase. The BTX production remained steady until 112 h have passed as a result of the changes of the other products. This type of hydrocracked product distribution is not rare, as Ahmed et al. [29] have shown. In this case, however, the changes in MHC may be the result of the disappearance of tetralin and naphthalene derivatives at the expense of the formation of alkylbenzenes, and when the deactivation began, the observed process reversed (Fig. 13). A detailed study regarding the composition changes by group type is in course.

Effect on the chemical composition of the HDT LCO hydrocracked products after 192 h of TOS at 400 °C, 7.4 MPa and LHSV of 0.8 h−1

Chromatograms of the HDT-LCO and hydrocracked products after 72, 126 and 192 h at 400 °C, 7.4 MPa and LHSV of 0.8 h−1
The observed chemical composition also implied that the BTX formation required some specific sites of the catalyst mixture for the transalkylation reaction [26, 27], while the LHC formation depended on other type of sites, which are more prone to deactivation. Therefore, just only when these sites began to be depleted, the specific sites for the BTX formation began to be attacked.
Bi-functional catalysts like NiMo/ZSM-5 (50) depend on the metal–acid balance. The hydrogenation power of a metallic function is necessary to minimize excessive cracking and polymerization reactions that can poison the acid sites and finally lead to catalyst deactivation [30, 31]. Zeolites have shown remarkable cracking and transalkylation properties associated with their acidic nature and topology [26, 27, 32]. ZSM-5, in its hydrogenated form, has Brönsted and Lewis acidity with acid OH groups at channel intersections [33] and alumina presents high surface area, bigger pores and the presence of acid OH on the surface [34]. Gosselink and Stork [35] concluded that the increasing coke deposition in the pores accounts for the increase in pore diffusion limitation effects. According to them, other parameters related to the feedstock such as heaviness, aromatics and organic nitrogen contents affect the deactivation reaction. Detailed chemical characterization studies of the feedstock, products, catalyst and longer TOS experiments will be required for confirming if a deactivation process took place on the NiMo/ZSM-5 (50) catalyst.
By using Eq. 4, regarding the activity rate for the MHC lump and the values found for this experiment, two scenarios can be considered:
- a)
When n was permitted to change during the estimation, and the attained value for n was 2.08, the value obtained for A was 2.96 × 10–5 with a correlation coefficient (R2) of 0.9502.
- b)
When the n value reported in a previous paper (2.28) [3] was used for tetralin hydrocracking with the same catalysts, a direct comparison was possible and the obtained value for A was 1.106 × 10−5 with a correlation coefficient (R2) of 0.9438. This value is quite similar to 1.02 × 10–5 obtained for tetralin HCK (Corr. Coef. R2 of 0.9752), demonstrating that in both feedstocks, the deactivation mechanism was the same and coke formation was involved.
TGA-DTA studies on the used catalyst
The coke deactivation mechanism of a catalyst is both chemical and physical; being the most important, the strong adsorption of polymerized carbon molecules on the acid sites. As polymerized carbon accumulates, the catalyst pores can be partially or totally blocked. Strong acid sites induce the formation of coke precursors, which undergo condensation reactions that produce large polynuclear aromatic molecules that can physically cover the catalytic surface [19, 20].
Figure 14 shows the TGA mass loss curve for the NiMo/ZSM-5 (50) catalyst recovered after the experiment under the following conditions: 400 °C, 7.4 MPa and LHSV of 0.8 h−1, until 192 h were completed. The DSC results are superimposed, which show the temperature difference. The decomposition began at 50 °C and ended at 600 °C. After 600 °C, no significant changes were observed. From 100 to 200 °C, an endothermic event was observed with a noticeable weight loss of 28 wt%, which corresponded to the desorption of hydrocarbons physisorbed on the catalyst during the reaction. In the 200–600 °C interval, an exothermic event was detected with a less pronounced weight loss of 10 wt%, which corresponded to the combustion of carbonaceous material on the catalyst (Wc). It is important to note that a slight weight loss (2 wt%) was detected after 600 °C with the corresponding small exothermic peak that corresponds to more polymeric carbon (coke), which was probably due to the complexity of the real feedstock (HDT LCO).

DTA/DSC curve of the used NiMo/Al2O3 catalyst in nitrogen atmosphere after the experiment at 400 °C, 7.4 MPa and LHSV of 0.8 h−1 until 192 h were completed
The total weight loss was 40 wt%, which is a higher value than the one observed in the previous work using tetralin as a model feedstock [3], which can be explained by the presence of more complex compounds contained in the feedstock. The temperature interval that allowed the combustion of the organic material showed that no refractory coke was formed, but coke precursors that covered the acid sites of the catalyst support [36]. The observed mass loss versus temperature pattern (TGA) was very similar to the behavior described in our previous work [3] with the same catalytic mixture, but using tetralin as model feedstock. These results are also similar to those observed by Upare et al. [17] who analyzed Mo/Beta and CoMo/Beta catalysts and were attributed to water (< 200 °C) and coke or related hydrocarbons (> 350 °C).
Conclusions
A series of experiments was carried out for attaining higher HCK yields coupled to benzene, toluene and xylene (BTX) selectivity. The tested catalyst was 50/50 NiMo/ZSM-5 (50) with hydrogenation/hydrocracking/transalkylation capabilities. The experimental conditions that allowed to reach a higher BTX yield were low LHSV (1.2 h−1 or less), up to 400 °C, and 7.4 MPa. The HCK conversion showed a strong dependence on the temperature and LHSV. From the HCK yield data, a kinetic model was obtained. A first-order HCK behavior for the conversion of the middle hydrocarbon lump (MHC) to light hydrocarbons (LHC) and BTX was assumed and from the application of Arrhenius equation, the obtained activation energy (Ea) was 139.5 kJ/mol. By applying the Eyring equation, a positive value of 134.6 kJ/mol, from the activation enthalpy (∆H), proved that the HCK process is endothermic. Additionally, an entropy value (∆S) of − 46.4 J/mol K was obtained.
The NiMo/ZSM-5 (50) catalyst activity during 192 h of time on stream affected the chemical composition of the HCK products. These changes can be due to the catalyst properties, location of acid sites and feedstock chemical composition.
References
Laredo GC, Perez RP, Escobar J, Garcia-Gutierrez JL, Vega-Merino P (2017) Light cycle oil upgrading to benzene, toluene and xylenes by hydrocracking: studies using model mixtures. Ind Eng Chem Res 56:10939–10948. https://doi.org/10.1021/acs.iecr.7b02827
Laredo GC, Vega-Merino PM, Schacht-Hernández P (2018) Light cycle oil upgrading to high quality fuels and petrochemicals: a review. Ind Eng Chem Res 57:7315–7321. https://doi.org/10.1016/j.cattod.2015.09.046
Laredo GC, Pérez-Romo P, Vega-Merino PM, Arzate-Barbosa E, García-López A, Agueda-Rangel R, Martínez-Moreno VH (2019) Effect of the catalytic system and operating conditions on BTX formation using tetralin as a model molecule. Appl Petrochem Res. https://doi.org/10.1007/s13203-019-00237-4
Stanislaus A, Marafi A, Rana MS (2010) Recent advances in the science and technology of ultra-low sulfur diesel (ULSD) production. Catal Today 153:1–68. https://doi.org/10.1016/j.cattod.2010.05.011
Laredo GC, Figueroa Y, Cano JL, Mares MT, Castillo J (2002) Estudio de la composición del aceite cíclico ligero provenientes de crudos mexicanos. J Mex Chem Soc 46:115–119
Environmental Protection Agency (2019) Diesel fuel standards and rulemakings. https://www.epa.gov/diesel-fuel-standards/diesel-fuel-standards-and-rulemakings. Accessed on March 2020.
European Automobile Manufacturers Association. Worldwide fuel charter—ACEA. https://fliphtml5.com/twsl/iert. Accessed on March 2020.
Bisht D, Petri J (2014) Considerations for upgrading light cycle oil with hydroprocessing technologies. Indian Chem, Eng 56:321–335. https://doi.org/10.1080/00194506.2014.927179
Ancheyta J (2017) Chemical reaction kinetics: concepts, methods and case studies. Wiley, New York
Park J-I, Ali SA, Alhooshani K, Azizi N, Miyawaki J, Kim T, Lee Y, Kim H-S, Yoon S-H, Mochida I (2013) (2013) Mild hydrocracking of 1-methyl naphthalene (1-MN) over alumina modified zeolite. J Ind Eng Chem 19:627–632. https://doi.org/10.1016/j.jiec.2012.09.014
Baudon A, Lemberton JL, Guisnet M, Marchal N, Mignard S (1996) Hydrocracking of n-heptane on a sulfide NiMo-Y zeolite catalyst: effect of the sulfidation method. Catal Lett 36:245–247. https://doi.org/10.1007/BF00807627
Sheng MN, Mameniskis WA, Ryan PW (1977) Production of monoaromatics from light pyrolysis fuel oil. US 4,022,681 (05/10/1977)
Kim CJ, Kim TJ, Kim DW, Kim SW, Oh SH, Park SR, Oh SH, Lee YK, Kim GR, Jung HS, Kim EK, Lee BI, Choo DH (2011) Method for producing high value aromatics and olefins from light cycle oil produced by a fluidized catalytic process. EP 2,351,820 A2 (03/08/2011).
Iwasa Ykobayashi M (2016) Methods for producing monocyclic aromatic hydrocarbons. US 2016/0024400 A1 (01/28/2016)
Yanagawa S, Aoki Y, Hayasaka K (2016) Method for producing aromti hydrocarbons. US 9,243,192 (01/26/2016)
Upare DP, Park S, Kim MS, Jeon Y-P, Kim J, Lee D, Lee J, Chang H, Choi S, Choi W, Park Y-K, Lee CW (2017) Selective hydrocracking of pyrolysis fuel oil into benzene, toluene and xylene over CoMo/beta zeolite catalyst. J Ind Eng Chem 46:353–363. https://doi.org/10.1016/j.jiec.2016.11.004
Upare DP, Park S, Kim MS, Kim J, Lee D, Lee J, Chang H, Choi W, Choi S, Jeon Y-P, Park Y-K, Lee CW (2016) Cobalt promoted Mo/beta zeolite for selective hydrocracking of tetralin and pyrolisis fuel into monocyclic aromatic hydrocarbons. J Ind Eng Chem 35:99–107. https://doi.org/10.1016/j.jiec.2015.12.020
Oh Y, Shin J, Noh H, Kim C, Kim Y-S, Lee Y-K, Lee JK (2019) Selective hydrotreating and hydrocracking of FCC light cycle oil into high-value light aromatic hydrocarbons. Appl Catal A 577:86–98. https://doi.org/10.1016/j.apcata.2019.03.004
Argyle MD, Bartholomew CH (2005) Heterogeneous catalyst regeneration and reactivation: a review. Catalyst 5:145–269. https://doi.org/10.3390/catal5010145
Guisnet M, Costa L, Ramoa-Ribeiro R (2009) Prevention of zeolite deactivation by coking. J Mol Cat A: Chem 305:69–83. https://doi.org/10.1016/j.molcata.2008.11.012
Fogler HS (2010) Essential of chemical reaction engineering. Prentice Hall International Series in the Physical and Chemical Engineering Sciences. ISBN-10: 0137146124, ISBN-13: 978–0137146123.
Ancheyta J, Sanchez S, Rodriguez MA (2005) Kinetic modeling of hydrocracking of heavy oil fractions: a review. Cat Today 106:76–92
Weekman AW (1979) Lumps, model and kinetics in practice. AIChE monograph series. ISBN-10: 0816900418, ISBN-13: 978-08169004
Gutiérrez A, Arandes JM, Castaño P, Olazar M, Bilbao J (2012) Effect of pressure on the hydrocracking of light cycle oil with a Pt-Pd/HY Catalyst. Energy Fuels 26:5897–5904. https://doi.org/10.1021/ef3009597
Townsend AT, Abbot J (1993) Catalytic reactions of tetralin on HZSM-5 zeolite. App Catal A 95:221–236. https://doi.org/10.1016/0926-860X(93)85076-2
Tsai T-C, Lin S-B, Wang I (1999) Disproportionation and transalkylation of alkylbenzenes over zeolite catalyst. Appl Catal A Gral 181:355–398. https://doi.org/10.1016/S0926-860X(98)00396-2
Ali SY, Ogunrombi KE, Al-Khattaf SS (2013) Kinetics of dealkylation-transalkylation of C9 alkyl-aromatics over zeolites of different structures. Chem Eng Res Dev 91:2601–2616. https://doi.org/10.1016/j.cherd.2013.04.014
Qader SA, Hill GR (1968) Hydrocracking of gas oil. Ind Eng Chem Proc Des Dev 8:98–105
Ahmed HS, Shaban SA, Menoufy MF, El Kady FY (2013) Effect of catalyst deactivation on vacuum residue hydrocracking. Egypt J Pet 22:367–372. https://doi.org/10.1016/j.ejpe.2013.10.006
Sato K, Iwata Y, Miki Y, Shimada H (1999) Hydrocracking of tetralin over NiW/USY zeolite catalyst for the improvement of heavy oils upgrading catalyst. J Catal 186:45–56. https://doi.org/10.1006/jcat.1999.2546
Shin J, Oh Y, Choi Y, Lee J, Lee JK (2017) Design of selective hydrocracking catalyst for BTX production from diesel-boiling-range polycyclic aromatic hydrocarbons. Appl Catal 547:12–21. https://doi.org/10.1016/j.apcata.2017.08.019
Chon H, Woo SI, Park S-E (1996) Recent advances and new horizons in zeolite science and technology. Elsevier, Amsterdam
Lee J, Choi Y, Shin J, Lee JK (2016) Selective hydrocracking of tetralin for light aromatic hydrocarbons. Catal Today 265:144–153. https://doi.org/10.1016/j.cattod.2015.09.046
Ballinger TH, Yates JT (1991) IR spectroscopy detection of Lewis acid sites on Al2O3 using adsorbed CO Correlation with Al–OH group removal. Langmuir 7:3041–3045
Gosselink JW, Stork WHJ (1997) Coping with catalyst deactivation in hydrocracking: catalyst and process development. Ind Eng Chem Res 36:3354–3359. https://doi.org/10.1021/ie9605995
Matsushita K, Hauser A, Marafi A, Koide R, Stanislaus A (2004) Initial coke deposition on hydrotreating catalysts. Part 1. Changes in coke properties as a function of time on stream. Fuel 83:1031–1038. https://doi.org/10.1016/j.fuel.2003.10.015
Acknowledgements
The authors are grateful for the financial support provided by the Mexican Petroleum Institute, Mexico.
Author information
Authors and Affiliations
Contributions
GCL conceived and designed the experiments; AG and RAR performed the experiments; GCL and PPR wrote the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Laredo, G.C., Pérez-Romo, P., Agueda-Rangel, R. et al. Effect of the experimental conditions on BTX formation from hydrotreated light cycle oil. Appl Petrochem Res 10, 21–34 (2020). https://doi.org/10.1007/s13203-020-00242-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-020-00242-y