
Abstract
Androgens are thought to cause prostate cancer, but the underlying mechanisms are unclear. Data from animal studies suggest that for androgens to cause prostate cancer, they must be aromatized to estrogen and act in concert with estrogen metabolites. We tested the hypothesis that androgen-receptor and estrogen receptor-mediated effects of androgen and estrogen are necessary, as well as genotoxicity of estrogen metabolites. NBL rats were treated with androgenic and estrogenic compounds for 16–75 weeks through slow-release silastic implants or pellets. Testosterone alone induced cancer in the prostate of 37% of rats. 5α-Dihydrotestosterone, which cannot be converted to estradiol or testosterone, did not cause a significant prostate cancer incidence (4%). Addition of estradiol to 5α-dihydrotestosterone treatment did not markedly enhance prostate cancer incidence (14%), unlike adding estradiol to testosterone treatment which induced a 100% tumor incidence. Testosterone plus estradiol treatment induced a DNA adduct detectable by 32P-postlabeling, oxidative DNA damage (8-hydroxyguanosine), and lipid peroxidation at the site within the prostate where this treatment causes cancers, preceding later cancer formation. The non-estrogenic 4-hydroxy metabolite of estradiol, when combined with testosterone, induced prostatic dysplasia within 16 weeks and, after long-term treatment, a very low incidence of prostate cancer (21%). When an estrogen that cannot be hydroxylated (2-fluoroestradiol) was added to this combined treatment with testosterone and 4-hydroxyestradiol, dysplasia frequency after 16 weeks was doubled. These results strongly support the hypothesis, but additional definitive studies are needed which may identify new targets to interfere with these mechanisms that are clinically feasible in humans.
Similar content being viewed by others


Introduction
Prostate cancer is the most frequently diagnosed cancer and the second most frequent cause of death due to cancer in US males. Although the causes of prostate cancer are not clear, hormones, particularly androgens and estrogens, are thought to be critically involved in human prostatic carcinogenesis [1]. The basis for this assumption is that the prostate gland is an androgen-dependent tissue and that prostate cancer is an androgen-dependent malignancy [1]. The underlying mechanism has been postulated to be androgenic stimulation of cell proliferation resulting in an increased risk of oncogenic genetic alterations [2]. However, there are no human data of the effects of androgen treatment on prostatic cell proliferation, but treatment of men with 5α-dihydrotestosterone (DHT) did not alter androgen-regulated gene expression in one study [3]. Chronic testosterone (T) treatment of rats via subcutaneous silastic tubing implants or cholesterol pellets results in development of prostate adenocarcinomas in at least five different strains at incidences ranging from 7 to 19% [4, 5].
While prostate cancer risk increases steeply with age [6], circulating androgen levels decrease with aging. Estrogen levels remain unchanged or increase slightly resulting in an increase in the ratio of 17β-estradiol (E2) to T with age, which has been suggested to point to a role of estrogen in prostate carcinogenesis [7]. Intraprostatic levels of DHT decrease with aging in epithelial cells, but not in stromal cells, while levels of E2 and estrone increase with age in the stromal compartment but not in the epithelium [8]. These observations suggest that prostate cancer risk may be associated with elevated concentrations of circulating and/or intraprostatic estrogens, but there is no direct evidence for such an association [9,10,11].
T can be converted to E2 by the enzyme aromatase, which is expressed in fat tissue and in the human and rodent prostate [12]. However, there is no evidence of an association between prostate cancer risk and single nucleotide polymorphisms in the aromatase (CYP19A1) gene that are associated with altered serum levels of total and free E2 [13]. Nonetheless, animal experiments suggest that estrogen may be involved in the aforementioned induction of prostate cancer by T in rats. We have shown that T treatment combined with E2 induces a prostate cancer incidence of 100% in NBL rats [14, 15]. These results have led us to hypothesize that estrogen and aromatization of T to E2 play a critical role in prostate carcinogenesis, at least in the rat. Of note, estrogen treatment alone results in shutdown of LH production and endogenous androgen production, resulting in prostatic atrophy. Therefore, experiments to determine whether E2 can cause prostate cancer require joint administration of androgen to restore physiological levels of testosterone.
E2 and estrone (E1) can be enzymatically metabolized to 2- and 4-hydroxyestradiol and 2- and 4-hydroxyestrone by CYP1A1 and CYP1B1. These so-called catecholestrogens can be converted to highly reactive estrogen semiquinones and estrogen quinones by the process of redox cycling [16, 17]. These reactive intermediates can adduct to DNA and redox cycling additionally causes generation of reactive oxygen species (ROS). ROS can induce lipid peroxidation, resulting in the formation of lipid hydroperoxides, and both can damage DNA and potentially lead to mutations [16, 17]. The 4-hydroxyestradiol (4OH-E2)-quinone-DNA adducts rapidly result in apurinic sites in the DNA which, when repaired by error-prone DNA repair mechanisms, can lead to mutations [18]. Detection of quinone-DNA adducts has been difficult, because they depurinate with a very short half-life, leading to apurinic sites that are difficult to detect. However, with highly sensitive analytical methods (LC-MS/MS), such adducts have been observed after estrogen treatment of DNA, cells, and tissues [19,20,21]. In the rodent [22] and human prostate [E. cavalieri & E. Rogan, personal communication], and in studies of urinary levels of estrogen metabolites and adducts in men with or without prostate cancer [23, 24], evidence has been found of enzymatic conversion of E2 and E1 by CYP1A1 and CYP1B1 to the catecholestrogens and subsequently the quinones.
We hypothesized that for T to be carcinogenic to the rat prostate, it must be aromatized to E2 which acts as a chemical carcinogen through the above described metabolism to catecholestrogen and subsequent redox cycling leading to DNA damage [25]. We have demonstrated that these reactions can take place in the rat prostate [22] and we identified a major DNA adduct by 32P-postlabeling selectively in the periurethral area of their prostates, the site of later cancer development [26], following treatment for 16 weeks with T plus E2 of NBL rats. A low level of this adduct was also found at this location in control rats, perhaps indicating sensitivity of this tissue to DNA damage. In the present paper, studies are described that provide further evidence that T is carcinogenic for the prostate and that aromatization of T to E2 and estrogen genotoxicity are involved in hormonal prostate carcinogenesis in the NBL rat model.
Methods
Animals and Animal Care
Male NBL/Cr rats 4–5-week-old were obtained from Charles River (Kingston, NY). The animals were housed in solid bottom cages, 2–3 to a cage under conventional conditions (22 ± 2 °C, 40–70% relative humidity, 12 h light/12 h dark); they had free access to tap water and an open formula, natural ingredient diet, NIH 07 (Zeigler Bros., Gardners, PA). Body weights were recorded weekly. The experiments were reviewed and approved by the NYU School of Medicine Institutional Animal Care and Use Committee.
Hormone Treatments
At 8–9 weeks of age, the animals received the following hormone treatment: two silastic implants (Dow Corning (Midland, MI), type 602 305, I.D. 1.6 mm; O.D. 3.2 mm, sealed with General Electric (Waterford, NY), RTV-108 silicone rubber adhesive sealant), each containing a tightly packed filling of T or DHT (Steraloids, Wilton, NH) over a length of 2 cm. E2 was administered via one such implant containing a tightly packed 1-cm-long filling of the steroid (Steraloids). The implants were placed subcutaneously on the back of the rats. T and E2 implants were replaced at 6-month intervals; at the end of each interval, the implants still contained a substantial amount of the steroids. This treatment was previously shown to stably increase circulating E2 levels 148% from 3.3 ± 0.7 to 49 ± 4.0 pg/ml similar to peak estradiol levels in female rats, while keeping T concentrations at normal levels [27]. Control rats received empty implants or control pellets. Ninety-day (~ 13 week) slow release pellets containing 4OH-E2 or 2F-E2 (obtained from Steraloids) were made for this project by Innovative Research of America (Sarasota, FL) and replaced every 13 weeks.
Autopsy and Tissue Sampling Procedures
Animals were euthanized by a pentobarbital overdose and exsanguination after 13 or 16 weeks or, in long-term experiments when they became moribund or at the end of the experiments at 75 weeks after initiation of treatment. Immediately following death, the entire accessory sex gland complexes together with the urinary bladder were removed in to and either fixed in 4% neutral buffered aqueous formaldehyde solution or rapidly snap frozen in liquid nitrogen following micro-dissection to separate the dorsolateral, ventral, and anterior prostate, periurethral area, and seminal vesicles the secretion of which was emptied before freezing. These tissues were stored at − 80 °C until they were analyzed following pulverization using a Microdismembrator-II (Braun, Melsungen, Germany) in liquid nitrogen cooled stainless steel ball containing capsules.
Histology and Immunohistochemistry Methods
The accessory sex glands were dissected after fixation as described previously [28] to separate the ventral lobes, anterior prostate (= coagulating gland) plus seminal vesicle complex, and dorsolateral prostate complex, including the periurethral area plus prostatic urethra. These tissues were processed to and embedded in a paraffin wax, the dorsolateral prostate complex cut in two halves. Five-micrometer-thick sections were prepared and stained with hematoxylin and eosin (H&E). Accessory sex glands were step sectioned at approximately 250-μm intervals, producing six sections per tissue for the dorsolateral prostate complex and coagulating gland plus seminal vesicles and one section for the ventral prostate.
For staining of 8-OHdG, a mouse monoclonal antibody was obtained from the Japan Institute for the Control of Aging (Fukuroi City, Shizuoka, Japan). Sections were autoclaved with 0.62 M ZnSO4 at 100 °C for 15 min and then treated with 2 M HCl at 37 °C for 30 min. Sections were pre-incubated with 10% normal horse serum for 30 min at 37 °C and then treated with an avidin/biotin block (Vector Laboratories, Burlingame, CA) before overnight incubation at 4 °C with the primary antibody against 8-OHdG (diluted 1/40). After incubation with a secondary peroxidase conjugated anti-mouse antibody (DakoCytomation, Glostrup, Denmark), immunoreactivity was visualized with DAB (DakoCytomation). Human seminal vesicle was used as a positive control tissue [29].
Measurement of 4-OHE2-Induced DNA Adducts
4-OHE2-induced DNA adducts were measured essentially as described by Li et al. [20] with some modifications. Briefly, tissues were ground to a powder in liquid nitrogen and suspended in 50 mM ammonium acetate, pH 4.4, containing 1 mg/ml ascorbic acid, with a final concentration of 30% methanol. They were passed through a Certify II solid phase extraction column and eluted with 70% methanol in the buffer. The eluant was evaporated to dryness and resuspended in 0.2 ml methanol. LC/MS analyses were carried out with a Waters Aquity Ultra Pressure Liquid Chromatography (UPLC) system connected with a high-performance Quattro Micro triple quadrupole mass spectrometer designed for LC-MS/MS operation. Analytical separations on the UPLC system were conducted using Aquity UPLC BEH C18 1.7u column (1 × 100 mm) at a flow rate of 0.15 ml/min. The analytical gradient started with 80% A (0.1% formic acid in H2O) and 20% B (0.1% formic acid in CH3CN). Eighty percent A changed to 79% over 4 min,followed by a 6 min linear gradient to 45% A, resulting in total separation time of 10 min. The elutions from the UPLC column were introduced to the mass spectrometer.
The electrospray ionization (ESI) method was used in both positive ion (PI) and negative ion (NI) mode with a capillary voltage of 3.0 kV, an extractor cone voltage of 3 V, and a detector voltage of 650 V. Desolvation gas flow was maintained at 600 l/h. Cone gas flow was set at 60 l/h. Desolvation temperature and source temperature were set to 200 and 100 °C, respectively. Tandem mass spectrometry (MS/MS) was performed in a multiple-reaction monitoring (MRM) mode to produce structural information about a compound by fragmenting specific parent ions inside the mass spectrometer and identifying the resulting fragment ions. Pure standards were used to optimize the LC/MS conditions prior to analysis. Due to small amounts of tissue, prostate tissues within each group of this study were pooled together. Analyses were performed in duplicate and data are presented as the mean of two replicates. The limits of detection for DNA adducts varied between 1.82 × 105 and 4.94 × 105 pmol/g tissue.
Measurement of 8-Hydroxyguanosine (8-OHdG)
8-OHdG was measured by HPLC according to Hofer and Moller [30]. DNA was extracted and digested using a modified alkaline phosphatase-nuclease P1 method, adding DNase I, phosphodiesterases I and II, and an iron chelator, as described in detail elsewhere [31]. Quantitation of nucleosides and 8-OHdG was done using a 4.6 × 250 mm Microsorb C18 column (Varian, Palo Alto, CA) and reverse-phase ESA HPLC with electrochemical detection of 8-OHdG by a Coulochem II electrochemical detector (ESA Biosciences, Chelmsford, MA) and of nucleosides by spectrophometry according to Hofer and Moller [30]. Data presented are expressed as 8-OHdG/105 dG,
Measurement of Lipid Hydroperoxides
Lipid hydroperoxides were measured after isopropanol extraction of pulverized tissue (1:3, w/v) with a colorimetric assay [32] using a kit (LPO-CC) from Kamiya Biomedical (Seattle, WA) with cumene hydroperoxide as standard, as described by Wang and Liehr [33].
Measurement of DNA Adducts by 32P-Postlabeling
DNA adducts were detected and quantified by 32P-postlabeling as described previously in detail [26]. Briefly, DNA was isolated by phenol/chloroform extraction and enzymatic digestion according to Gupta [34]. DNA was assayed for adducts using the nuclease Pi-enhanced 32P-postlabeling procedure of Reddy and Randerath [35]. To quantify adduct levels, adduct spots were excised from chromatograms and subjected to Cerenkov counting and values were expressed as relative adduct labeling (RAL) according to Reddy and Randerath [36].
Statistical Analysis
Mortality data were analyzed using the Kruskall-Wallis test and Dunn’s multiple comparison test. For body weight data, analysis of variance followed by Dunnett’s multiple comparison test or a 2 sample t test was used. Tumor and lesion incidences were analyzed using the Fisher exact probability test. Data on tissue adduct and lipid peroxide levels were analyzed using a 2 sample t test or ANOVA followed by Student-Newman-Keuls or Tukey multiple comparison test. P values of ≤ 0.05 (one- or two-sided, as applicable) were considered significant.
Results
Testosterone Induced Prostate Cancer and Estradiol Enhanced the Incidence, but DHT with or Without Estradiol Did Not (Tables 1 and 2)
First, we tested the idea that T needs to be aromatized to E2 to be carcinogenic by treating NBL rats with the 5α-reduced T metabolite DHT, which cannot be aromatized or converted back to T. We used a DHT dose that increased ventral prostate weight following 16 weeks of treatment to a similar degree as treatment with the dose of T we used previously and in this study (Table 1). The incidence of prostate carcinomas induced by chronic T treatment was 37% (11 of 30 rats), but DHT induced prostate cancer in only one of 27 treated rats (4%) (Table 2). Based on our previous observation that T plus E2 induced prostate cancers in 100% of treated NBL rats [14], we expected that addition of E2 to the chronic DHT treatment would produce a high prostate cancer incidence but only 14% of animals developed these tumors (Table 2). However, DHT plus E2 did induce a 100% incidence of dysplastic lesions in the dorsal and particularly lateral prostate after 16 weeks (Table 1) and after long-term treatment (data not shown), as we have previously observed for T plus E2 treatment [14, 27].
E2 is known to induce pituitary adenomas in rats and we have previously found a 100% incidence of such tumors after T plus E2 treatment, markedly reducing survival [14]. E2 when combined with DHT, however, produced pituitary tumors in only 30% of rats (Table 2), even though pituitary weights were increased after 16 weeks of treatment (Table 1). Furthermore, compared to controls, the effects of DHT and the DHT plus E2 combination on weights of seminal vesicle/anterior prostate complex were fundamentally different from those of T and T plus E2 treatment for 16 weeks, slightly reducing rather than increasing seminal vesicle/anterior prostate weights and reducing testicular weights far more (Table 1).
The dysplastic lesions found after 16 weeks of E2 plus T or E2 plus DHT treatment were morphologically identical to those described in detail previously [27], as were the mostly microscopic-size adenocarcinomas originating in the periurethral ducts found following these treatments [14]. The adenocarcinomas found in the dorsolateral prostate (eleven) and anterior prostate (one) of rats treated with only T or DHT were grossly visible and histologically well to moderately differentiated, similar to those we described following treatment with a chemical carcinogen and T [28]. The precise origin of most of these large carcinomas was not clear as they involved both glands in the periphery of the prostate and prostatic ducts leading towards the prostatic urethra; in several cases, these tumors were so large that they caused urethral obstruction. One of the carcinomas concentrically involved the periurethral ducts spreading out to the periphery of the prostate, suggesting that larger tumors after hormone treatment may also have originated from these ducts. However, one of the carcinomas in T-treated rats was smaller and clearly confined to the periphery of the dorsal and lateral prostate and no small cancers were found in the periurethral ducts of rats treated with only T or DHT. Metastases were found in the spleen, stomach, pancreas, liver, and/or lungs of three of the ten rats (30%) with prostate carcinomas after treatment with only T.
Short-Term Treatment with Estradiol Combined with Long-Term Testosterone Administration Caused a High Incidence of Prostate Cancer (Table 2)
To determine whether a short course of E2 treatment would induce prostate cancer but not induce pituitary tumors and impact survival in rats chronically treated with T, we gave NBL rats treatment with E2 plus T for 16 or 8 weeks at which time the E2 implant was removed, but the T implants were left in place for the duration of the study, until the animals became moribund or died (Table 2). Animals lived 22–31 weeks longer than when the E2 implant was not removed, did not develop pituitary tumors, and they had carcinomas that originated in the periurethral ducts of the dorsolateral prostate (Table 2). In both treatment groups, some animals also developed carcinomas of the seminal vesicles and anterior prostate (Table 2) which we have never observed after continuous E2 treatment together with T. In the 16-week E2 group, the incidence of carcinomas of prostatic origin (75%) was driven by the presence of periurethral or dorsolateral prostate tumors. One animal from the 8-week E2 group developed a small adenocarcinoma in the periphery of the dorsolateral prostate and one rat from the 16-week E2 group had a periurethral adenocarcinoma that was grossly visible and extended into the dorsolateral prostate periphery. This tumor had metastasized to other abdominal organs. All of the seminal vesicle and six of the eleven anterior prostate tumors were grossly visible and many of the large tumors had metastasized, mostly to other abdominal organs. There was one undifferentiated sarcoma of the seminal vesicle in the 16-week E2 group (not listed in Table 2).
An additional group of rats received T plus E2 treatment for 16 weeks at which time both the T and the E2 implants were removed and the animals followed until moribund. The animals also lived longer than those in which T plus E2 treatment was continued, but only one animal developed a periurethral prostate carcinoma (Table 2).
Long-Term Treatment with the Catechol Estrogen 4-Hydroxyestradiol in Combination with Testosterone Induced Prostate Cancer and Dysplasia at Low Incidence, but Required Additional Estrogenic Stimuli to Produce a High Dysplasia Incidence (Table 3)
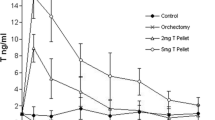
To circumvent aromatization of T and the antiandrogenic effects of E2, we exposed NBL rats to 4OH-E2, the catechol metabolite of E2, alone or together with T using silastic tubing implants containing T and custom-made slow release pellets for 4OH-E2 administration. Because we used slow-release pellets with 13-week release duration, we first compared 13 weeks of E2 plus T with 16 weeks of this hormone treatment and found an identical 100% dysplasia incidence and comparable degrees of inflammation in the lateral prostate (data not shown). The dose of 4OH-E2 used in these studies, 5 μg/day given by slow release pellets, was based on the result of a preliminary 21-day experiment in which we found that this dose resulted in the formation of depurinating DNA adducts detectable by mass spectrometry (Fig. 1). We identified the following depurinating adducts in lateral, dorsal, and anterior prostate tissue: 4OH-E2–1-N7-guanine, 4OH-E1-1-N7-guanine, 4OH-E2-1-N3-adenine, and 4OH-E1-1-N3-adenine. In the pooled prostate tissues of 4OH-E2-treated rats, we detected a total amount of these adducts of 2.10 pmol/g tissue, whereas in control prostates, we found a total of 0.45 pmol/g tissue, indicating that the 4OH-E2 dose we used did induce depurinating estrogen-DNA adducts.

4-OHE2-induced DNA adducts. Levels of DNA adducts in dorsal (DP), lateral (LP), and anterior prostate (AP) of NBL rats following treatment with 5 μg/day 4-OHE2 or placebo for 21 days. LC-MS/MS analysis of pooled prostate tissue. Data are expressed as mean of two pooled tissue samples from five animals each
T plus 4OH-E2 induced inflammation but no dysplasia after 13 weeks, while this treatment for 52 weeks induced carcinomas in 4 (28%) NBL rats, three in the prostate (21%) and one in the seminal vesicle. The latter treatment also induced a 43% incidence of dorsolateral prostate dysplasia, in addition to inflammation. One or two lesions classified as carcinoma in situ [28] were found in all 52-week treatment groups (Table 3). By contrast, we previously showed [14] that treatment with T plus E2 induced a 100% incidence of adenocarcinomas in the periurethral prostate and inflammation and dysplasia in the dorsolateral prostate of NBL rats (see Table 2). Treatment with only 4OH-E2 also induced dorsolateral prostate inflammation and dysplasia and a couple of carcinomas in situ and one prostate adenocarcinoma.
We then examined whether adding an estrogenic stimulus to the T plus 4OH-E2 treatment would potentiate their effects on the prostate. We used 2-fluoro-estradiol (2F-E2) which has been used successfully in the past to separate genotoxicity from estrogenicity in the male hamster kidney model to demonstrate that estrogen genotoxicity is required for estrogens to induce tumors in that classical model [37]. First, we identified a 2F-E2 dose (5 μg/day) that produced estrogenic activity in NBL rats when delivered by slow release pellets; this dose increased pituitary weights from 3.30 ± 0.24 to 4.45 ± 0.21 mg/100 g body weight (p = 0.005; 2 sided t test). Following 13 weeks of treatment, 2F-E2 alone increased the percentage of rats with dorsolateral prostate inflammation; and combined 2F-E2 plus T caused inflammation in 100% of rats and dorsolateral prostate dysplasia in 30%. The combination of 2F-E2 plus T plus 4OH-E2 produced inflammation in 90% of rats and dysplasia in 60%, short of the 100% incidence of both lesions observed previously after T plus E2 treatment [14]. However, although pituitary weights were increased by 2F-E2 alone (see above), addition to T or T plus 4OH-E2 reduced the weight to control levels (3.20 ± 0.93 and 3.67 ± 0.51 mg/100 g body weight, respectively) suggesting that this reduced estrogenicity of 2F-E2.
Induction by Testosterone, Estradiol, and Testosterone Plus Estradiol of DNA Adducts Detectable by 32P-Postlabeling, 8-Hydroxyguanosine, and Lipid Hydroperoxides (Table 4)
We previously identified by 32P-postlabeling the formation of a major DNA adduct selectively in the periurethral area of the NBL rat after treatment with E2 plus T for 16 weeks [14]. This finding demonstrated the induction of DNA damage by this hormone treatment at the precise site of later cancer development and preceding tumor development in time. Because we found that T alone caused a 37% incidence of prostate carcinomas, we measured the formation of this adduct in prostate tissues following treatment with just T and compared this with exposure to only E2 and E2 plus T. By 16-week treatment with E2 plus T, we confirmed the induction of a major DNA adduct spot (P1) that was chromatographically identical to the adduct that we had previously identified selectively in the periurethral prostate [26] (Supplemental Fig. 1A). Interestingly, T alone also induced this adduct at about half the amount and low levels of the adduct spot were found after E2 treatment and in control rats (Table 4). The adduct spot was not detectable in the dorsolateral prostate (Supplemental Fig. 1A) or the ventral prostate (data not shown).
As a measure of oxidative DNA damage, we determined the formation of 8-OHdG in the NBL rat prostate by HPLC and immunostaining. Following 16-week treatment with E2 plus T, we detected elevated levels of 8-OHdG by HPLC in the periurethral region of the prostate, which was the only prostatic area that had detectable levels in control animals (Table 4). The treatment also induced low levels of this oxidized DNA base in the dorsolateral prostate where the combined hormone treatment induced inflammation and dysplasia, but no cancer. 8-OHdG was undetectable by HPLC in the ventral prostate. Immunostaining essentially confirmed these findings (Supplemental Fig. 1B). Interestingly, the seminal vesicles stained strongly for 8-OHdG in control animals, as they do in humans [29], but E2 plus T treatment reduced staining intensity (data not shown); we have never observed lesions in the seminal vesicles of E2 plus T-treated rats.
The formation of lipid hydroperoxides, indicators of lipid peroxidation that can be induced by ROS, followed the same general pattern as the generation of 8-OHdG (Table 4). The highest levels were found in the periurethral and dorsolateral prostate of E2 plus T-treated rats, and lowest levels in the ventral lobe of control rats, while intermediate levels were found in the target regions of control animals, suggesting that these tissues are particularly sensitive to or poorly protected from oxidative DNA damage. The difference from control values was almost significant for the periurethral region and became significant when the data for the periurethral and dorsolateral prostate were combined.
Discussion
We obtained novel evidence that estrogen is involved in prostate cancer induction by T and strongly suggestive evidence of a critical contribution of a combination of androgen- and estrogen-receptor mediation as well as DNA damage, probably caused by estrogen metabolites; the latter notion is supported by our previous findings that generation of catecholestrogen and redox cycling can occur in the prostate of rats treated with estrogen [22]. The results indicate (a) that T plus E2 treatment causes DNA damage and lipid peroxidation in the rat prostate at the site of origin of carcinomas that develop later with continued hormone treatment; and (b) that E2 is essential for the high prostate cancer response to treatment with T plus E2, suggesting that aromatization of T to E2 may be critical for the tumor response to T alone. Unfortunately, we were not able conduct a study with T treatment plus an aromatase inhibitor. The results of combination treatments with 4OH-E2 and T, with or without addition of the estrogen 2F-E2 which cannot be converted to reactive species, provide evidence supporting the notion that catecholestrogen formation and redox cycling of 4-hydroxylated E2 are important factors and that estrogen receptor activation (by 2F-E2) is also necessary to generate prostatic dysplasia and cancer. However, the 2F-E2 dose we used was probably too low (inducing only a modest pituitary enlargement) to have a sufficiently strong estrogenic effect and result in a high tumor response; additional experiments with higher doses of this estrogenic compound are needed to definitively test the hypothesis that a combination of androgen and estrogen receptor mediation and estrogen metabolite-induced DNA damage are the mechanism by which T, with or without additional E2, causes prostate cancer in the NBL rat.
Aromatase knockout mice [38] and aromatase overexpressing mice [39, 40] suffer from androgen metabolism abnormalities that limit their potentially interesting use to address this hypothesis [41]. Aromatase knockout mice lack estrogen production, have elevated circulating T levels, and their prostates are enlarged, but they do not develop cancer [38]. In aromatase-overexpressing mice estrogen production is elevated, while T levels are considerably reduced and no neoplastic or preneoplastic prostate lesions develop [39, 40]. These observations are consistent with the idea that both hormones are necessary for prostate carcinogenesis.
Both estrogen receptors (ER)-α and -β are expressed in the rat and human prostate and they may mediate some or all of the prostatic effects of estrogens [42, 43]. When Thompson et al. [44] treated NBL rats with T plus E2 in combination with the pure antiestrogen ICI182,780, development of prostatic dysplasia (a putative preneoplastic lesion comparable to human prostatic intraepithelial neoplasia or PIN) was inhibited. It has long been known that E2 induces pituitary tumors that produce prolactin in rats. Lane et al. [45] demonstrated that bromocryptin treatment, which reduces prolactin secretion, reduces the induction of prostatic dysplasia by T plus E2 in NBL rats, suggesting that prolactin plays a role in the development of this lesion. Importantly, the dysplasia in NBL rats treated with E2 plus T occurs in a different region of the prostate (dorsolateral prostate) than the carcinomas we find following this treatment, which originate from the periurethral prostatic ducts [14], and this dysplasia rarely progresses to cancer [unpublished observations]. Thus, the findings of Thompson et al. [44] and Lane et al. [45] may not have a bearing on cancer development in the prostate of T plus E2-treated rats. This notion is supported by our observation that chronic treatment with T alone or T combined with E2 for the first 8–16 weeks induces prostate cancer in 37, 63, and 75% of rats, respectively, without inducing pituitary tumors. Overall, these data suggest that estrogen receptors and prolactin do not play a role in T plus E2 induction of prostate cancer in NBL rats, but conclusive experiments are still lacking.
We also obtained evidence to indicate that T alone induces cancer in the prostate of the NBL rat at a 35–40% incidence, which is considerably higher than reported previously by us and others for this and other rat strains [4, 5]. The mechanistic basis for the high susceptibility of NBL rats to prostate cancer induction by T is not known. These tumors were locally invasive adenocarcinomas that were capable of metastasizing. The T dose we used elevated circulating T non-significantly by 17%, well within the physiological range of T levels in NBL rats [5]. These findings indicate that T is a carcinogen for the rat prostate and suggest that this androgen could be the cause of human prostate cancer at relevant circulating concentrations [5]. The evidence for this from human epidemiological studies is weak; there are only two reports of a positive association between free serum T levels and risk of prostate cancer [46, 47], which would be consistent with our findings in rats. Interestingly, a recent report indicated that men with very low circulating free T levels have a low risk of prostate cancer [48].
Indirect evidence that androgens are involved in human prostate carcinogenesis is derived from clinical studies with the 5α-reductase inhibitors finasteride and dutasteride which both reduced risk of developing prostate cancer by 23–24% over a 4–7-year intervention period [49, 50]. However, the duration of the intervention was short in view of the known slow growth of prostate cancer, and the study subjects were middle-aged men who have a high frequency of small cancers in their prostates [51]. Thus, these studies are unlikely to provide much insight into whether androgens are involved in the process of prostate carcinogenesis as such or only influence growth and progression of pre-existing cancer.
The observation that DHT, which cannot be aromatized to E2 or converted back to T, is a very weak prostate carcinogen at best in the NBL rat suggests that E2 generated from T by aromatase (CYP19) is critically involved in the carcinogenic activity of T for the rat prostate. However, adding E2 to DHT treatment did not substantially increase its prostate cancer-inducing efficacy, unlike our previous findings with the combination of E2 with T [14, 15]. DHT also reduced the pituitary tumor-inducing effect of E2 considerably, from a 100 to a 30% incidence, and had effects on accessory sex gland weights that were quite different from those of T, regardless of whether the androgens were combined with E2. Conceivably, DHT treatment alters androgen and estrogen metabolism or action in ways that reduces their prostate cancer-inducing activity. Alternatively, 3β-androstanediol, the downstream metabolite of DHT, is known to bind and activate ER-β and inhibits human prostate cancer cell proliferation [52]. Of note, we observed by immunohistochemistry that the regions of the NBL rat prostate that are most susceptible to the carcinogenic effects of T + E2 have low ER-α expression and very high ER-β expression [unpublished observations].
In conclusion, our findings provide strong support for the hypothesis that for T to be carcinogenic to the rat prostate, it must be aromatized to E2 which acts as a chemical carcinogen through the above described metabolism to catecholestrogen and subsequent redox cycling leading to DNA damage. We also obtained evidence to suggest that estrogen receptor mediation is important for this carcinogenic effect, in addition to androgenic mechanisms. More research is warranted to provide further proof of the latter notion. These findings may identify druggable targets that are clinically feasible for prostate cancer chemoprevention.
References
Bosland MC (2000) The role of steroid hormones in prostate carcinogenesis. J Natl Cancer Inst Monogr 27:39–66
Henderson BE, Feigelson HS (2000) Hormonal carcinogenesis. Carcinogenesis 21:427–433
Page ST, Lin DW, Mostaghel EA, Marck BT, Wright JL, Wu J, Amory JK, Nelson PS, Matsumoto AM (2011) Dihydrotestosterone administration does not increase intraprostatic androgen concentrations or alter prostate androgen action in healthy men: a randomized-controlled trial. J Clin Endocrinol Metab 96:430–437
Noble RL (1982) Prostate carcinoma of the Nb rat in relation to hormones. Int Rev Exp Pathol 23:113–159
Bosland MC (2014) Testosterone treatment is a potent tumor promoter for the rat prostate. Endocrinology 155:4629–4633
Carter HB, Piantadosi S, Isaacs JT (1990) Clinical evidence for and implications of the multistep development of prostate cancer. J Urol 143:742–746
Vermeulen A, Kaufman JM, Goemaere S, van Pottelberg I (2002) Estradiol in elderly men. Aging Male 5:98–102
Krieg M, Nass R, Tunn S (1993) Effect of aging on endogenous level of 5 alpha-dihydrotestosterone, testosterone, estradiol, and estrone in epithelium and stroma of normal and hyperplastic human prostate. J Clin Endocrinol Metab 77:375–381
Eaton NE, Reeves GK, Appleby PN, Key TJ (1999) Endogenous sex hormones and prostate cancer: a quantitative review of prospective studies. Br J Cancer 80:930–934
Roddam AW, Allen NE, Appleby P, Key TJ (2008) Endogenous sex hormones and prostate cancer: a collaborative analysis of 18 prospective studies. J Natl Cancer Inst 100:170–183
Yao S, Till C, Kristal AR, Goodman PJ, Hsing AW, Tangen CM, Platz EA, Stanczyk FZ, Reichardt JK, Tang L, Neuhouser ML, Santella RM, Figg WD, Price DK, Parnes HL, Lippman SM, Thompson IM, Ambrosone CB, Hoque A (2011) Serum estrogen levels and prostate cancer risk in the prostate cancer prevention trial: a nested case-control study. Cancer Causes Control 22:1121–1131
Ellem SJ, Schmitt JF, Pedersen JS, Frydenberg M, Risbridger GP (2004) Local aromatase expression in human prostate is altered in malignancy. J Clin Endocrinol Metab 89:2434–2441
Travis RC, Schumacher F, Hirschhorn JN, Kraft P, Allen NE, Albanes D, Berglund G, Berndt SI, Boeing H, Bueno-de-Mesquita HB, Calle EE, Chanock S, Dunning AM, Hayes R, Feigelson HS, Gaziano JM, Giovannucci E, Haiman CA, Henderson BE, Kaaks R, Kolonel LN, Ma J, Rodriguez L, Riboli E, Stampfer M, Stram DO, Thun MJ, Tjønneland A, Trichopoulos D, Vineis P, Virtamo J, Le Marchand L, Hunter DJ (2009) CYP19A1 genetic variation in relation to prostate cancer risk and circulating sex hormone concentrations in men from the breast and prostate Cancer cohort consortium. Cancer Epidemiol Biomark Prev 18:2734–2744
Bosland MC, Ford H, Horton L (1995) Induction at high incidence of ductal prostate adenocarcinomas in NBL/Cr and Sprague-Dawley Hsd:SD rats treated with a combination of testosterone and estradiol-17 beta or diethylstilbestrol. Carcinogenesis 16:1311–1317
Ozten N, Horton L, Lasano S, Bosland MC (2010) Selenomethionine and alpha-tocopherol do not inhibit prostate carcinogenesis in the testosterone plus estradiol-treated NBL rat model. Cancer Prev Res (Phila) 3:371–380
Cavalieri E, Frenkel K, Liehr JG, Rogan E, Roy D (2000) Estrogens as endogenous genotoxic agents--DNA adducts and mutations. J Natl Cancer Inst Monogr 2000:75–93
Bolton JL, Thatcher GR (2008) Potential mechanisms of estrogen quinone carcinogenesis. Chem Res Toxicol 21:93–101
Mailander PC, Meza JL, Higginbotham S, Chakravarti D (2006) Induction of a. T to G.C mutations by erroneous repair of depurinated DNA following estrogen treatment of the mammary gland of ACI rats. J Steroid Biochem Mol Biol 101:204–215
Li K, Todorovic MR, Devanesan P, Higginbotham S, Kofeler H, Ramanathan R, Gross ML, Rogan EG, Cavalieri EL (2004) Metabolism and DNA binding studies of 4-hydroxyestradiol and estradiol-3,4-quinone in vitro and in female ACI rat mammary gland in vivo. Carcinogenesis 25:289–297
Saeed M, Rogan E, Fernandez SV, Sheriff F, Russo J, Cavalieri E (2007) Formation of depurinating N3Adenine and N7Guanine adducts by MCF-10F cells cultured in the presence of 4-hydroxyestradiol. Int J Cancer 120:1821–1824
Zhang F, Swanson SM, van Breemen RB, Liu X, Yang Y, Gu C, Bolton JL (2001) Equine estrogen metabolite 4-hydroxyequilenin induces DNA damage in the rat mammary tissues: formation of single-strand breaks, apurinic sites, stable adducts, and oxidized bases. Chem Res Toxicol 14:1654–1659
Cavalieri EL, Devanesan P, Bosland MC, Badawi AF, Rogan EG (2002) Catechol estrogen metabolites and conjugates in different regions of the prostate of Noble rats treated with 4-hydroxyestradiol: implications for estrogen-induced initiation of prostate cancer. Carcinogenesis 23:329–333
Markushin Y, Gaikwad N, Zhang H, Kapke P, Rogan EG, Cavalieri EL, Trock BJ, Pavlovich C, Jankowiak R (2006) Potential biomarker for early risk assessment of prostate cancer. Prostate 66:1565–1571
Yang L, Gaikwad NW, Meza J, Cavalieri EL, Muti P, Trock B, Rogan EG (2009) Novel biomarkers for risk of prostate cancer: results from a case-control study. Prostate 69:41–48
Bosland MC (2013) A perspective on the role of estrogen in hormone-induced prostate carcinogenesis. Cancer Lett 334:28–33
Han X, Liehr JG, Bosland MC (1995) Induction of a DNA adduct detectable by 32P-postlabeling in the dorsolateral prostate of NBL/Cr rats treated with estradiol-17 beta and testosterone. Carcinogenesis 16:951–954
Ofner P, Bosland MC, Vena RL (1992) Differential effects of diethylstilbestrol and estradiol-17 beta in combination with testosterone on rat prostate lobes. Toxicol Appl Pharmacol 112:300–309
McCormick DL, Rao KV, Dooley L, Steele VE, Lubet RA, Kelloff GJ, Bosland MC (1998) Influence of N-methyl-N-nitrosourea, testosterone, and N-(4-hydroxyphenyl)-all-trans-retinamide on prostate cancer induction in Wistar-Unilever rats. Cancer Res 58:3282–3288
Oberley TD, Zhong W, Szweda LI, Oberley LW (2000) Localization of antioxidant enzymes and oxidative damage products in normal and malignant prostate epithelium. Prostate 44:144–155
Hofer T, Möller L (1998) Reduction of oxidation during the preparation of DNA and analysis of 8-hydroxy-2′-deoxyguanosine. Chem Res Toxicol 11:882–887
Huang X, Powell J, Mooney LA, Li C, Frenkel K (2001) Importance of complete DNA digestion in minimizing variability of 8-oxo-dG analyses. Free Radic Biol Med 31:1341–1351
Ohishi N, Ohkawa H, Miike A, Tatano T, Yagi K (1985) A new assay method for lipid peroxides using a methylene blue derivative. Biochem Int 10:205–211
Wang MY, Ohkawa H, Miike A, Tatano T, Yagi K (1994) Identification of fatty acid hydroperoxide cofactors in the cytochrome P450-mediated oxidation of estrogens to quinone metabolites. Role and balance of lipid peroxides during estrogen-induced carcinogenesis. J Biol Chem 269:284–291
Gupta RC (1984) Nonrandom binding of the carcinogen N-hydroxy-2-acetylaminofluorene to repetitive sequences of rat liver DNA in vivo. Proc Natl Acad Sci U S A 81:6943–6947
Reddy MV, Randerath K (1986) Nuclease P1-mediated enhancement of sensitivity of 32P-postlabeling test for structurally diverse DNA adducts. Carcinogenesis 7:1543–1551
Reddy MV, Randerath K (1987) 32P-analysis of DNA adducts in somatic and reproductive tissues of rats treated with the anticancer antibiotic, mitomycin C. Mutat Res 179:75–88
Liehr JG (1983) 2-Fluoroestradiol. Separation of estrogenicity from carcinogenicity. Mol Pharmacol 23:278–281
McPherson SJ, Wang H, Jones ME, Pedersen J, Iismaa JP, Wreford N, Simpson ER, Risbridger GP (2001) Elevated androgens and prolactin in aromatase-deficient mice cause enlargement, but not malignancy, of the prostate gland. Endocrinology 142:2458–2467
Fowler KA, Gill K, Kirma N, Dillehay DL, Tekmal RR (2000) Overexpression of aromatase leads to development of testicular leydig cell tumors : an in vivo model for hormone-mediated TesticularCancer. Am J Pathol 156:347–353
Li X, Nokkala E, Yan W, Streng T, Saarinen N, Warri A, Huhtaniemi A, Santti R, Makela S, Poutanen M (2001) Altered structure and function of reproductive organs in transgenic male mice overexpressing human aromatase. Endocrinology 142:2435–2442
Jarred RA, McPherson SJ, Bianco JJ, Couse JF, Korach KS, Risbridger GP (2002) Prostate phenotypes in estrogen-modulated transgenic mice. Trends Endocrinol Metab 13:163–168
Leav I, Lau KM, Adams JY, McNeal JE, Taplin ME, Wang J, Singh H, Ho SM (2001) Comparative studies of the estrogen receptors beta and alpha and the androgen receptor in normal human prostate glands, dysplasia, and in primary and metastatic carcinoma. Am J Pathol 159:79–92
Lau KM, Leav I, Ho SM (1998) Rat estrogen receptor-alpha and -beta, and progesterone receptor mRNA expression in various prostatic lobes and microdissected normal and dysplastic epithelial tissues of the Noble rats. Endocrinology 139:424–427
Thompson CJ, Tam NN, Joyce JM, Leav I, Ho SM (2002) Gene expression profiling of testosterone and estradiol-17 beta-induced prostatic dysplasia in Noble rats and response to the antiestrogen ICI 182,780. Endocrinology 143:2093–2105
Lane KE, Leav I, Ziar J, Bridges RS, Rand WM, Ho SM (1997) Suppression of testosterone and estradiol-17beta-induced dysplasia in the dorsolateral prostate of Noble rats by bromocriptine. Carcinogenesis 18:1505–1510
Travis RC, Key TJ, Allen NE, Appleby PN, Roddam AW, Rinaldi S, Egevad L, Gann PH, Rohrmann S, Linseisen J, Pischon T, Boeing H, Johnsen NF, Tjønneland A, Overvad K, Kiemeney L, Bueno-de-Mesquita HB, Bingham S, Khaw KT, Tumino R, Sieri S, Vineis P, Palli D, Quirós JR, Ardanaz E, Chirlaque MD, Larrañaga N, Gonzalez C, Sanchez MJ, Trichopoulou A, Bikou C, Trichopoulos D, Stattin P, Jenab M, Ferrari P, Slimani N, Riboli E, Kaaks R (2007) Serum androgens and prostate cancer among 643 cases and 643 controls in the European prospective investigation into Cancer and nutrition. Int J Cancer 121:1331–1338
Gann PH, Hennekens CH, Ma J, Longcope C, Stampfer MJ (1996) Prospective study of sex hormone levels and risk of prostate cancer. J Natl Cancer Inst 88:1118–1126
Watts EL, Appleby PN, Perez-Cornago A, Bueno-de-Mesquita HB, Chan JM, Chen C, Cohn BA, Cook MB, Flicker L, Freedman ND, Giles GG, Giovannucci E, Gislefoss RE, Hankey GJ, Kaaks R, Knekt P, Kolonel LN, Kubo T, Le Marchand L, Luben RN, Luostarinen T, Männistö S, Metter EJ, Mikami K, Milne RL, Ozasa K, Platz EA, Quirós JR, Rissanen H, Sawada N, Stampfer M, Stanczyk FZ, Stattin P, Tamakoshi A, Tangen CM, Thompson IM, Tsilidis KK, Tsugane S, Ursin G, Vatten L, Weiss NS, Yeap BB, Allen NE, Key TJ, Travis RC (2018) Low free testosterone and prostate Cancer risk: a collaborative analysis of 20 prospective studies. Eur Urol 74:585–594
Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, Lieber MM, Cespedes RD, Atkins JN, Lippman SM, Carlin SM, Ryan A, Szczepanek CM, Crowley JJ, Coltman CA Jr (2003) The influence of finasteride on the development of prostate cancer. N Engl J Med 349:215–224
Andriole GL, Bostwick DG, Brawley OW, Gomella LG, Marberger M, Montorsi F, Pettaway CA, Tammela TL, Teloken C, Tindall DJ, Somerville MC, Wilson TH, Fowler IL, Rittmaster RS (2010) Effect of dutasteride on the risk of prostate cancer. N Engl J Med 362:1192–1202
Sakr WA, Grignon DJ, Crissman JD, Heilbrun LK, Cassin BJ, Pontes JJ, Haas GP (1994) High grade prostatic intraepithelial neoplasia (HGPIN) and prostatic adenocarcinoma between the ages of 20-69: an autopsy study of 249 cases. In Vivo 8:439–443
Bremmer F, Jarry H, Unterkircher V, Kaulfuss S, Burfeind P, Radzun HJ, Ströbel P, Thelen P (2018) Testosterone metabolites inhibit proliferation of castration- and therapy-resistant prostate cancer. Oncotarget 9:16951–16961
Acknowledgments
The authors acknowledge dedicated contributions of the staff of the animal and histology facilities of the Department of Environmental Medicine at the Nelson Institute in Tuxedo, NY.
Funding
This work was supported in part by the National Institutes of Health (CA159385, CA104334, CA75293, and CA48084 to MCB, CA104334-S1, to KV) and the Prevent Cancer Foundation (to MCB).
Author information
Authors and Affiliations
Author notes
Joachim Liehr is deceased. This paper is dedicated to his memory.
- Joachim Liehr
Corresponding author
Ethics declarations
The animal experiments described in this paper were reviewed and approved by the NYU School of Medicine Institutional Animal Care and Use Committee and all applicable national and institutional guidelines for the care and use of animals were followed.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
This paper is dedicated to the memory of Joachim G. Liehr who died in 2003. He developed the notion that estrogen genotoxicity is an essential component of estrogen carcinogenesis and created a substantive body of evidence in support of this mechanism.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Figure 1
(DOCX 1157 kb)
Rights and permissions
About this article

Cite this article
Ozten, N., Vega, K., Liehr, J. et al. Role of Estrogen in Androgen-Induced Prostate Carcinogenesis in NBL Rats. HORM CANC 10, 77–88 (2019). https://doi.org/10.1007/s12672-019-00360-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-019-00360-7