
Abstract
The G protein-coupled estrogen receptor-1, GPER-1, coordinates fibronectin (FN) matrix assembly and release of heparan-bound epidermal growth factor (HB-EGF). This mechanism of action results in the recruitment of FN-engaged integrin α5β1 to fibrillar adhesions and the formation of integrin α5β1-Shc adaptor protein complexes. Here, we show that GPER-1 stimulation of murine 4 T1 or human SKBR3 breast cancer cells with 17β-estradiol (E2β) promotes the formation of focal adhesions and actin stress fibers and results in increased cellular adhesion and haptotaxis on FN, but not collagen. These actions are also induced by the xenoestrogen, bisphenol A, and the estrogen receptor (ER) antagonist, ICI 182, 780, but not the inactive stereoisomer, 17α-estradiol (E2α). In addition, we show that GPER-1 stimulation of breast cancer cells allows for FN-dependent, anchorage-independent growth and FN fibril formation in “hanging drop” assays, indicating that these GPER-1-mediated actions occur independently of adhesion to solid substrata. Stable expression of Shc mutant Y317F lacking its primary tyrosyl phosphorylation site disrupts E2β-induced focal adhesion and actin stress fiber formation and abolishes E2β-enhanced haptotaxis on FN and anchorage-dependent growth. Collectively, these data demonstrate that E2β action via GPER-1 enhances cellular adhesivity and FN matrix assembly and allows for anchorage-independent growth, cellular events that may allow for cellular survival, and tumor progression.
Similar content being viewed by others


Introduction
Fibronectin (FN) plays a major role in cellular adhesion, growth, and survival, and it is important for processes such as wound healing [1], vascular growth [2], and embryonic development [3]. On the contrary, altered expression of FN, or perturbations in the specific recognition of FN by integrin α5β1, has been associated with the development of cancer and fibrosis [4, 5]. FN is synthesized in a soluble form as a dimeric glycoprotein that is assembled into an insoluble fibrillar matrix in a complex, dynamic cell-mediated process that is initiated by its specific recognition by integrin α5β1 via individual Arg-Gly-Asp (RGD)-binding sites on each monomer thereby facilitating integrin clustering. Upon FN engagement, integrin α5β1 undergoes conformational alterations associated with increased receptor affinity [6]. FN-occupied integrin α5β1 is then recruited to sites of close cell matrix contact known as “focal adhesions” that are enriched in tyrosyl-phosphorylated proteins and actin stress fibers where robust anchorage to FN occurs. The local concentration of integrin-bound FN increases, allowing bound FN molecules to more readily interact with one another and form short FN fibrils between cells, thus beginning the process of fibrillogenesis. Conversion of soluble FN to insoluble fibrils proceeds when cryptic FN-binding sites are exposed along the length of bound FN by contractile forces that stretch FN by pulling on their FN-bound integrin receptors [7] and partially unfolding FN, unmasking cryptic FN-binding sites [8, 9], and allowing nearby FN molecules to associate. This FN-FN interaction enables the soluble, cell-associated fibrils to branch and stabilize into an insoluble FN matrix. Fragmentation of FN uncovers non-RGD-binding sites leading to enhanced integrin α4β1 adhesion and FN matrix contractility [10], illustrating the influence of matrix proteases on provisional FN matrix assembly.
A number of studies have shown that FN is critical to normal homeostasis of the mammary gland and is associated with the development of breast cancer. Namely, the addition of exogenous FN negatively impacts acinar differentiation in the mammary gland and creates a microenvironment conducive to the growth of mammary epithelia [5]. Integrin α5β1 and FN are prominently expressed in the mammary gland, and their basal expression is increased during active proliferation of mammary gland tissue in mice suggesting that this FN-integrin interaction may be required for hormone-dependent proliferation in the mammary gland [11]. In addition, transgenic mice expressing dominant-negative integrin β1 show disrupted mammary gland development that is associated in a loss of AKT activation and Shc-dependent extracellular regulated kinase-1 and -2 (Erk-1/-2) activation [12]. Moreover, successful implantation of human mammary tumor xenografts in immunocompromised mice is facilitated by coadministration of exogenous FN, indicating a survival advantage for tumor cells that interact with FN [13]. This observation is supported by studies that have shown that mammary adenocarcinoma cells are capable of converting soluble FN into fibrils [14] resulting in increased responsiveness to growth factors and enhanced anchorage-independent growth [15]. The survival of tumor cells under these imposed in vitro growth conditions is reflective of their capacity to assemble a provisional extracellular matrix [14] and to circumvent death signals promoted by mechanosensors that report reduced tensile forces [16]. As measured in a two-dimensional environment, ligation of integrin α5β1 to FN-coated substrata is sufficient to promote intracellular signals associated with cellular growth and survival, including activation of Src, focal adhesion kinase (FAK), B/AKT, and Erk-1/-2 [17–20]. Under circumstances where either integrin density or its binding sites on adhesive proteins are limiting, serum-derived factors have been shown to facilitate the recruitment of tyrosyl-phosphorylated proteins (FAK, Src, vinculin, and paxillin) into focal adhesion plaques and induce the formation of RhoA-dependent actin stress fibers [21]. However, matrix engagement by integrin is not sufficient to promote subsequent cell growth responses, and FN polymerization is a critical requirement for measurable adhesion-dependent growth on planar surfaces [22]. Regulatory roles for phosphatidylinositol 3-OH kinase, FAK, Src-like kinases, and phospho-paxillin in FN matrix assembly have been suggested [23, 24]. Studies evaluating FN assembly in a three-dimensional environment have shown that exogenous FN can facilitate fibrillogenesis [25] but that this cellular activity is not sufficient for anchorage-independent growth by mammary adenocarcinoma cells, as they also must become responsive to external growth factors [14, 15, 26].
Shc is a nonenzymatic adaptor protein that participates in kinase signaling cascades and generally functions as a signaling intermediary to determine growth factor responsiveness and extracellular matrix (ECM) engagement [27]. Shc protein is expressed as three isoforms (p66Shc, p52Shc, and p46Shc) that are synthesized as a result of differential ribosomal initiation start sites on the same genetic locus. Their shared carboxyl terminus encodes PTB and SH2 phosphotyrosine-binding domains that are separated by an intervening collagen homologous (CH) 1 domain. Shc proteins are recruited to tyrosine kinase receptors and nonreceptor tyrosine kinases via interactions between their PTB and SH2 binding domains and phosphotyrosines [27]. On the other hand, Shc proteins physically associate with integrin via an indirect linkage involving the SH3 domain of Src-like kinases and the conserved proline-rich collagen homologous (CH1) domain on Shc that lies between PTB and SH2 [28]. While all three Shc isoforms are recruited to focal adhesions and share a primary tyrosine phosphorylation site and common functional motifs that promote their interaction with other signaling effectors, different cell biological roles have been assigned for the long and short forms of Shc. p52Shc and p46Shc have been linked to signals that promote cellular survival and proliferation, primarily through their ability to couple to the Raf-Mek-Erk protein kinase signaling axis via Grb-2/Sos/Ras [27]. In contrast, p66Shc uncouples the Ras-to-Erk protein kinase cascade and is associated with RhoA-dependent anoikis [29] and thus, is best known as a proapoptotic protein. This observation is consistent with the fact that p66Shc is poorly expressed in hematopoietic lineage cells that are considered to be anchorage-independent and insensitive to substrate stiffness [30–33]. Similarly, lung carcinoma cell lines that lack p66Shc display aggressive metastatic behavior, anchorage-independent growth, and bypass anoikis [34]. Finally, patients whose breast or colon cancers express higher ratios of total tyrosyl-phosphorylated Shc to p66Shc are linked to poor prognosis [35, 36], further suggesting that failure to regulate Shc is associated with more advanced cancer.
Tumors that arise from the mammary gland exhibit biological behaviors that are described dichotomously as either estrogen- or growth factor-dependent. This categorization is largely derived from analysis of known receptors for estrogen (ERs) and epidermal growth factor (EGFR)-related receptors in breast tumor biopsies and the fact that there is a strong inverse relationship between expression of ER and EGFR [37]. G protein-coupled estrogen receptor-1 (GPER-1)/GPR30 represents a newly appreciated estrogen receptor whose expression in primary breast tumors is directly linked to tumor size and metastasis [38–41], a relationship diametrically opposed to the one shared between ER and these same tumor progression variables. This observation suggests that GPER-1 plays a distinct role from ER in breast cancer biology and is consistent with the fact that GPER-1 and ER are structurally distinct receptors that promote estrogen-mediated signals measured with different metrics and kinetics. While ER bears structural homology shared by the members of the nuclear steroid hormone receptor superfamily and functions as a hormone-inducible transcription factor, GPER-1 belongs to the most broadly studied class of cell surface receptors, the G protein-coupled receptor (GPCR) superfamily. GPER-1 promotes rapid signals attributed to this receptor class, including stimulation of adenylyl cyclase [42] and EGFR activation via the release of membrane-tethered heparan-bound epidermal growth factor (HB-EGF) [43]. We have previously shown that EGFR transactivation by GPER-1 requires activation of the FN receptor, integrin α5β1, in breast cancer cells, as measured by its recruitment to fibrillar adhesions, the conversion of soluble FN to a detergent-insoluble form, and the association of integrin α5β1 with the signaling adaptor, Shc [44].
Here, the influence of this GPER-1-integrin α5β1-Shc-dependent signaling mechanism on breast cancer cell adhesion was further evaluated by measuring its influence on cancer cell cytoarchitecture, adhesion, and haptotactic responses on immobilized FN and by evaluating their role in fibrillogenesis and growth in anchorage-independent conditions.
Materials and Methods
Cell Culture
SKBR3 (ERα-, ERβ-, GPER-1+) breast cancer cells were obtained from the American Type Culture Collection (ATTC) (Manassas, VA). SKBR3 variants expressing dominant-negative Shc (Shc317Y/F) and dominant-negative GPER (GPERΔ154) were generated as described previously [44]. ER-negative murine 4 T1 breast cancer cells were obtained from the ATCC. 4 T1 cells expressing GPERΔ154 were generated as described previously [44]. All cells were grown in phenol red-free (PRF) DMEM/Ham’s F12 media (1:1) with 5 % fetal bovine serum and 25 μg/ml gentamicin.
Growth Factors, Estrogens, Antiestrogens, and Matrix Proteins
Water-soluble 17β-estradiol (E2β), 17α-estradiol (E2α), and angiotensin II (ATII) were purchased from Sigma (St. Louis, MO). Bisphenol A (BPA) was a kind gift from the Hixon Lab at Brown University (Providence, RI). ICI 182, 780 was purchased from Tocris Bioscience (Ellisville, MO). Bovine, human, and rat FN were purchased from EMD Millipore (Milford, MA).
Antibodies
mAB IC3 specific for rat FN was a kind gift from the Schwarzbauer Lab at Princeton University (NJ) and has been previously described [45]. Phosphotyrosine-specific mAB, 4G10, was purchased from Upstate Biotechnology, Inc. Rabbit polyclonal antibodies (AB1949) specific for the cytoplasmic tail of integrin α5 subunit protein was purchased from Chemicon. Inhibitory rat anti-mouse integrin α5β1 monoclonal antibody, clone BMB4, was purchased from Millipore. Alexa fluor dye-conjugated secondary antibodies were purchased from Molecular Probes, Inc. (Eugene, OR)/Invitrogen.
Cellular Stimulation and Protein Extraction
Conditions for quiescence, cell stimulation, and protein extraction were discussed previously [43].
Immunofluorescence
Focal adhesions were visualized in 4 T1 and SKBR3 cells that were seeded onto glass coverslips in PRF-DMEM/F12 medium containing FN-reduced serum and allowed to adhere overnight at 37 C. The following day, serum was removed by washing in PRF-DMEM/F12. Cells were then cultured in the same media in the absence of serum for an additional 30 h. Serum-starved cells were fed 2-μg/ml rat FN in the absence or presence of ligand (10 nM E2α, 10 nM E2β, 1 μM ICI 182, 780, 10 nM BPA) for 2 h. Cells were then washed, fixed for 5 min in 4 % paraformaldehyde, permeabilized in 0.05 % Triton for 60 s, and blocked in 5 % BSA/PBS for 30 min. Cells were incubated with phosphotyrosine-specific 4G10 antibody diluted 1:500 in PRF-DMEM/F12 containing 5 % BSA for 60 min. Coverslips were washed in PRF-DMEM/F12, and cell-associated antibodies were detected using Alexa 594-conjugated anti-mouse IgG diluted 1:1,000 and delivered in PRF-DMEM/F12 containing 5 % BSA for 30 min. After staining, coverslips were washed and mounted on glass slides in Vectashield/4′6-diamidino-2-phenylindole (Vector Laboratories, Inc., Burlingame, CA). FN fibril formation was examined in SKBR3, SKBR3 GPERΔ154, and SKBR3 Shc317Y/F, and 4 T1 cells that were seeded onto glass coverslips in PRF-DMEM/F12 medium containing FN-reduced serum. Starved cells were fed rat plasma FN (25 μg/ml) in PRF-DMEM/F12 medium in the presence of ligand (10 nM E2α, 10 nM E2β, 1 μM ICI 182, 780, 10 nM BPA) for 18 h and then fixed and prepared for immunostaining as above. Fixed cells were stained with IC3 ascites diluted 1:1,000 and delivered in PBS containing 1 % BSA for 60 min. IC3 mAB was detected by staining with Alexa 594-conjugated anti-mouse IgG (1:1,000) and processed for microscopy. Integrin α5β1 and stress fibers were visualized in SKBR3, SKBR3 GPERΔ154, and SKBR3 Shc317Y/F cells that were seeded onto glass coverslips in PRF-DMEM/F12 medium containing FN-reduced serum and allowed to adhere overnight at 37 C. After adhesion, serum was removed by washing 3× with PRF-DMEM/F12, and the cells were then cultured in the same media in the absence of serum for an additional 30 h. Serum-starved cells were fed 2-μg/ml rat FN in the absence or presence of E2β (10 nM) for 2 h. Cells were then washed, fixed for 5 min in 4 % paraformaldehyde, permeabilized in 0.05 % Triton for 60 s, and blocked in 5 % BSA/PBS for 30 min. Cells were incubated with rabbit polyclonal antibodies (AB1949) specific for the cytoplasmic tail of integrin α5 subunit protein diluted 1:500 and TRITC-phalloidin diluted 1:500 in PRF-DMEM/F12 containing 5 % BSA for 60 min. Coverslips were washed in PRF-DMEM/F12, and cell-associated antibodies were detected using Alexa 594-conjugated anti-rabbit IgG diluted 1:1,000 and delivered in PRF-DMEM/F12 containing 5 % BSA for 30 min. After staining, coverslips were washed and mounted on glass slides in Vectashield/4′6-diamidino-2-phenylindole (Vector Laboratories, Inc., Burlingame, CA). All immunofluorescent images were visualized with a Nikon Eclipse 80i microscope (Nikon, Inc., Melville, NY) equipped with a Nikon Plan Fluor 100x0.5–1.3 Oil Iris with differential interference contrast and epifluorescent capabilities. Digital images were captured using a QImaging Retiga 2000R digital camera and Nikon imaging software (Elements Basic Research 3.0).
Adhesion Assay
Forty-eight-well plates were coated with 200 μl of PRF-DMEM/F12 containing 2-μg/ml human FN or 10-μg/ml collagen overnight. Wells were blocked with 5 % BSA PRF-DMEM/F12 for 1 h. SKBR3, SKBR3 GPERΔ154, and SKBR3 Shc317Y/F cells were seeded in triplicate, left untreated or treated with 10 nM E2β, and allowed to adhere for 2 h. Nonadherent cells were gently washed away with PRF-DMEM/F12. Adherent cells were fixed and stained with 0.4 % crystal violet and 4 % ethanol in water for 5 min, and then washed 2× in large volumes of water. Crystal violet dye was extracted with 10 % acetic acid, and absorbance was measured spectrophotometrically at 550 nm. All data points were determined from triplicate assays and expressed as the mean ± standard deviation. Nonspecific adhesion was subtracted as determined from cells that were seeded in BSA in the absence of substratum.
Boyden Chamber Migration Assay
Haptotaxis assays were conducted using modified Boyden chambers consisting of a porous polycarbonate membrane (6.5-μm thickness, 8-μm pores; Transwells, CoStar corporation, Cambridge, MA) [46]. The lower surfaces of the Transwell membrane were coated by adding 500 μl of serum free, PRF-DMEM/F12 containing 2-μg/ml human FN, or 10-μg/ml collagen to the lower reservoir overnight. The underneath surface of the membrane was then blocked in 5 % BSA in for 1 h. SKBR3, SKBR3 GPERΔ154, and SKBR3 Shc317Y/F (105) cells were placed in the upper reservoirs of the Transwell in serum free, PRF-DMEM/F12 and left untreated or treated with 10 nM E2β and allowed to migrate overnight at 37 C. Nonmigrated cells were removed from the upper surface of the membrane using a Q-tip, and cells remaining attached to the lower surface were fixed in ethanol and stained with 0.4 % crystal violet in sodium borate buffer, pH 9.2 for 5 min, and then washed 2× in large volumes of water. Dye was eluted from the migrant cells using acetic acid and measured spectrophotometrically at 550 nM. Each data point was measured in triplicate and measured as the mean plus or minus the standard deviation. Nonspecific migration was subtracted as determined from cells that were seeded in BSA in the absence of substratum.
Anchorage-Independent Growth
SKBR3, SKBR3 GPERΔ154, SKBR3 Shc317Y/F, 4 T1 vector, and 4 T1 GPERΔ154 cells (1 × 104) were seeded into PRF DMEM-F12 media in 0.35 % agarose in the absence or presence of E2β (10 nM) and 10 % fetal bovine serum which was FN-depleted using gelatin-conjugated sepharose as described by Pierschbacher et al. [47] and supplemented with exogenous FN (2 μg/ml). Cells were grown for 10 days at 37 C in a humidified chamber at 5 % CO2. In some assays, inhibitory rat anti-integrin mouse α5β1 monoclonal antibody, clone BMB4 from Millipore, or control nonimmune rat antibodies were incorporated in the agar overlay. Cultures were weighed every 2 days, and evaporated water was replaced as needed. Images of colonies were captured at × 100 magnification (Brightfield). Colonies of greater than 20 cells were enumerated by direct counting.
Anchorage-Independent Fibrillogenesis
Anchorage-independent fibrillogenesis was determined from “hanging drop” assays as previously described [48–50]. Briefly, 4 T1 vector or 4 T1 GPERΔ154 cells were placed in suspension in serum-free media supplemented with or without agonist (10 nM E2α, 10 nM E2β, 10 nM ATII, 1 μM ICI 182, 780, RGD, RGE) and rhodamine-labeled bovine FN (30 μg/ml). For each treatment, 10 × 15-μl aliquots were distributed onto the underside of a 100-mm Petri dish lid in a humidified chamber. Hanging drop cultures were incubated for 18 h at 37 C. Cells were fixed, stained with DAPI, and transferred to a glass slide. Images were captured using a Nikon 80i inverted fluorescent microscope fitted with a Retiga color camera at × 100 magnification. Multiple Z-axis sections were reconstructed into three-dimensional images using Nikon imaging software.
Results
Estradiol-Induced Mobilization of α5β1 into Focal Adhesions, the Formation of Actin Stress Fibers, and FN Fibril Formation Require GPER-1 and the Shc Signaling Adapter Protein
Src-like kinases, integrin α5β1, and Shc have been identified as integral components of a signaling pathway leading to E2β-mediated transactivation of the EGFR and FN matrix assembly [43, 44]. In the latter study, we showed that stimulation of human SKBR3 cells with E2β results in the recruitment of FN-occupied integrin α5β1 into fibrillar adhesions at the cell periphery [44] but did not address the role of GPER-1 or Shc in the formation of focal adhesion plaques, presumed precursory adhesion structures which give rise to fibrillar adhesions. To gain knowledge as to whether GPER-1 action was necessary for E2β-induced clustering of integrin α5β1 into focal adhesions and the formation of actin stress fibers, these cellular activities were compared in vector control SKBR3 cells and in a derivative line of SKBR3 cells expressing a dominant-negative form of GPER-1 (GPERΔ154) (Fig. 1). In these experiments, SKBR3 vector control and SKBR3 GPERΔ154 cells were seeded onto coverslips, serum-starved, and stimulated with E2β in the presence of exogenous FN for 2 h. Cells were fixed and stained with integrin α5β1-specific antibodies (green) and rhodamine-conjugated phalloidin to identify polymerized actin stress fibers (red). While vector control SKBR3 cells demonstrated prominent actin stress fibers that colocalized with integrin α5β1 in focal adhesions, neither actin stress fibers nor integrin α5β1-enriched focal adhesions were observed in unstimulated cells (Fig. 1a). Similarly, SKBR3 cells expressing a dominant-negative form of GPER-1 (GPERΔ154) that were stimulated with E2β were also unable to form actin stress fibers or concentrate integrin α5β1 into focal adhesions (Fig. 1a) suggesting that GPER-1 was required for the cellular activation of events that recruit integrin α5β1 to focal adhesions and promote actin stress fiber formation. As previously demonstrated, GPER-1 stimulation with E2β resulted in the formation of FN fibrils (red), while dominant-negative GPER-1 compromised FN fibril formation (Fig. 1a).

E2β-induced recruitment of integrin α5β1 to focal adhesions and the formation of actin stress fibers and FN fibrils are GPER- and Shc-dependent. a (top panel) SKBR3 cells expressing vector or GPERΔ154 and b (top panel) SKBR3 cells expressing vector or Shc317Y/F were seeded onto coverslips, serum starved, and stimulated with E2β (10 nM) and exogenous rat FN (2 μg/ml). Cells were incubated for 2 h, fixed with 4 % paraformaldehyde, permeabilized with detergent, and stained with integrin α5β1-specific antibodies (green) and Alexa-594-phalloidin (red). Nuclei were stained with DAPI (blue). a (bottom panel) SKBR3 cells expressing vector or GPERΔ154 and b (bottom panel) SKBR3 cells expressing vector or Shc317Y/F were seeded onto coverslips, serum starved, and stimulated with E2β (10 nM) and exogenous rat FN (25 μg/ml). Cells were incubated for 18 h, fixed with 4 % paraformaldehyde, and stained with rat FN-specific mAB, IC3 (red). Nuclei were stained with DAPI (blue)
To establish the requirement of Shc in E2β-induced integrin α5β1 recruitment to focal adhesions and the formation of actin stress fibers and FN fibril formation, these cellular events were evaluated in SKBR3 cells expressing control vector or a mutant Shc protein lacking its primary tyrosyl phosphorylation site, Shc317Y/F (Fig. 1b). Focal adhesions and actin stress fibers were not detectable in quiescent, unstimulated control or Shc317Y/F cells. Following exposure to E2β, integrin α5β1 was recruited into prominent focal adhesions that coaligned with the termini of actin stress fibers, while SKBR3 Shc317Y/F cells showed an impaired ability with regards to these integrin activation events. Expression of Shc317Y/F in SKBR3 cells negatively affected E2β-induced clustering of integrin α5β1 into focal adhesions and showed less prominent focal adhesion plaques that appeared to be disordered with regards to their alignment with the termini of focal adhesions (Fig. 1b). Likewise, E2β-induced FN fibril formation observed in control SKBR3 cells was prohibited in SKBR3 Shc317Y/F cells.
Collectively, these results indicate that Shc signaling following GPER-1 activation is required to promote the recruitment of integrin α5β1 to focal adhesions and to induce actin stress fiber formation and consequent FN fibril formation. We have previously shown that Shc317Y/F accumulates relative to Shc wild-type protein on integrin α5β1 and cells expressing this mutant Shc protein fail to form fibrillar adhesions [44]. Thus, our current observations may further suggest that the primary tyrosyl phosphorylation site on Shc is not required for entry of integrin α5β1 into focal adhesions but that this site is required for its subsequent recruitment to fibrillar adhesions.
The Influence of the Xenoestrogen, Bisphenol A, or the ER Antagonist, ICI 182, 780, on the Recruitment of Tyrosyl-Phosphorylated Proteins to Focal Adhesions and the Production of Fibronectin Fibrils
Xenoestrogens and ER antagonists act as GPER-1 agonists [40, 43, 51, 52]. To determine whether these estrogenic steroids also influence GPER-1-dependent formation of focal adhesions and FN fibrils, these integrin activation events were assessed in SKBR3 cells expressing vector or GPERΔ154 protein (Fig. 2a). For these experiments, cells were made quiescent by seeding them onto glass coverslips in FN-reduced serum followed by serum deprivation. Quiescent cells were left untreated or exposed to E2β; its inactive stereoisomer (E2α), the xenoestrogen, BPA, or the ER antagonist, ICI 182, 780 and focal adhesions were assessed by measuring the clustering of tyrosyl-phosphorylated proteins by immunostaining with the phosphotyrosine (pptyr)-specific monoclonal antibody, 4G10. Stimulation with E2β, BPA, or ICI 182, 780 resulted in the recruitment of tyrosyl-phosphorylated proteins into focal adhesion plaques (Fig. 2a). Focal adhesions were similarly measured in murine 4 T1 breast cancer cells that were stimulated with the ER antagonist, ICI 182, 780, or the xenoestrogen, BPA (data not shown). Focal adhesions were neither measured in quiescent cells nor in cells that were stimulated with E2α (Fig. 2a) nor EGF (data not shown). These results suggest that estrogen-stimulated enrichment of tyrosyl-phosphorylated proteins to focal adhesion plaques occurs independently of the ER and does not require EGF stimulation.

E2β stimulation of human ER-negative breast cancer cells induces the formation of focal adhesions and FN fibrils. a Human SKBR3 cells grown on glass coverslips were serum-starved and stimulated with either E2β (10 nM), E2α (10 nM), BPA (10 nM), or ICI 182, 780 (1 μM), and incubated for 2 h in the presence of exogenous rat FN (2 μg/ml). Following incubation, cells were fixed in 4 % paraformaldehyde, permeabilized with detergent, and focal adhesions were detected using phosphotyrosine-specific antibodies (red). Nuclei were stained with DAPI (blue). b SKBR3 cells grown on glass coverslips were serum-starved and stimulated as described above. Cells were incubated for 18 h in the presence of exogenous rat FN (25 μg/ml), fixed in 4 % paraformaldehyde, and FN fibrils were detected using rat FN-specific mAB, IC3 (red). Nuclei were stained with DAPI (blue)
Previously, we have shown that stimulation of human SKBR3 cells with E2β results in the recruitment of FN-occupied integrin α5β1 to the cell periphery into fibrillar adhesions, specialized adhesion structures at which FN fibrils form [44]. Here, we addressed the capacity of SKBR3 cells to form FN fibrils in response to stimulation with ICI 182, 780 or BPA. As shown in Fig. 2b, either ICI 182, 780 or BPA as well as E2β resulted in the formation of FN fibrils that were detected at the periphery of SKBR3 cells. FN fibril formation was not observed in SKBR3 cells that were left untreated or stimulated with E2α (Fig. 2b) or EGF (data not shown). Similar observations were measured in murine 4 T1 breast cancer cells. In both cell types, expression of GPERΔ154 protein prohibited FN fibril formation (Fig. 2b and data not shown), demonstrating that these estrogenic hormones are capable of promoting FN fibril formation (fibrillogenesis) in ER-negative breast cancer cells that are attached to planar surfaces coated with this ECM protein.
Stimulation of GPER-1 Selectively Enhances the Adhesivity of Human Breast Cancer Cells for FN-Coated Substrata by a Mechanism that Requires the Primary Tyrosyl Phosphorylation Site on Shc
In many instances, agonists that employ G protein-coupled receptors promote enhanced cellular adhesive interactions by modulating the affinity of integrins for their cognate ECM proteins and also by inducing the recruitment of integrins to focal adhesion plaques, a process referred to as “inside-out” integrin signaling [53, 54]. To examine the influence of GPER-1 stimulation by E2β on the adhesion of SKBR3 vector control breast cancer cells for immobilized adhesive ligand, SKBR3 or SKBR3 GPERΔ154 cells were detached and exposed to E2β or left untreated and seeded into polystyrene wells coated with various concentrations of FN or collagen I (COLL) (Fig. 3). Following a 2-h incubation time at 37 C, cells that were not firmly attached were gently washed away, adherent cells were fixed, cellular attachment was assessed by staining the remaining adherent cells with crystal violet, and attachment was measured as a function of eluted dye recovered from the resulting adherent cells. Unstimulated SKBR3 cells adhered to both FN- and COLL-coated substrata in a dose-dependent fashion with maximum cell adhesion measured at coating concentrations of 20 and 10 μg/ml of FN and COLL, respectively (data not shown). A 2-fold increase (p = 0.034) in the capacity of E2β-stimulated versus unstimulated SKBR3 cells to adhere to wells coated with suboptimal concentrations of FN (2 μg/ml) was observed (Fig. 3a), which was associated with increased cellular spreading. In contrast, more modest differences in enhanced E2β-mediated adhesion to COLL-coated substrata were measured (Fig. 3) with no discernible difference in cellular spreading (data not shown). E2β-increased adhesivity was eliminated in SKBR3 cells expressing GPERΔ154 suggesting that E2β promotes adhesion of SKBR3 breast cancer cells in a GPER-1-dependent manner. Similarly, Shc was tested for its involvement in GPER-1-enhanced adhesivity by comparing the relative capacity of SKBR3 and SKBR3 Shc317Y/F cells to adhere to immobilized FN or COLL (Fig. 3b). There was no significant increase (p = 0.46) in adhesion between the E2β-stimulated and untreated SKBR3 Shc317Y/F cells, suggesting that Shc is also involved in the signaling events that lead to increased adhesion to FN.

GPER-1 stimulation promotes SKBR3 cell adhesion onto FN-coated, but not collagen-coated, substrata in a Shc-dependent manner. a SKBR3 cells expressing vector or GPERΔ154 and b SKBR3 cells expressing vector or Shc317Y/F were seeded onto 48-well plates coated with FN (2 μg/ml) or collagen (10 μg/ml) and allowed to attach for 2 h at 37 C in the absence or presence of E2β (10 nM). After adhesion, unattached cells were removed by gentle washing, and the remaining adherent cells were fixed and stained with crystal violet. Excess crystal violet was washed away, and cell-associated crystal violet was extracted with 10 % acetic acid. Absorbance was measured at 550 nm. Each data point represents the mean ± standard deviation of triplicate samples. Nonspecific adhesion as measured on BSA-coated wells has been subtracted
Cellular adhesion to planar surfaces is greatly strengthened by cell spreading [55] and is often associated with increased cellular motility as measured in haptotactic responses on immobilized adhesive ligands. To examine whether GPER-1 and Shc promote increased migration on FN-coated substrata, SKBR3 vector control, SKBR3 GPERΔ154, or SKBR3 Shc317Y/F cells were seeded in the presence of 10 nM E2β or left untreated) into the upper reservoirs of a modified Boyden chambers containing a porous polycarbonate membrane (10-μm thickness, 8-μm pore) whose undersurface was coated with adhesive ligand (2 μg/ml FN or 10 μg/ml COLL) (Fig. 4). Cell migration was measured by determining the number of cells that were capable of migrating from the upper reservoir across the membrane to its undersurface. On FN-coated membranes, SKBR3 vector control cells stimulated with E2β showed a 6.5-fold increase in their capacity to migrate compared with unstimulated cells in this assay (Fig. 4). SKBR3 cells that were plated onto membranes that were coated with higher concentrations of FN (5–20 μg/ml) did not show increased haptotaxis when stimulated with E2β (data not shown), suggesting that GPER-1-enhanced migration was the product of increased recruitment of FN receptors to cellular adhesion sites (data not shown). E2β-enhanced haptotaxis on FN was abrogated in SKBR3 GPERΔ154 cells demonstrating that this migratory response was dependent upon GPER-1 action (Fig. 4). Likewise, expression of Shc317Y/F also impeded E2β-enhanced haptotaxis on FN (Fig. 4). SKBR3 cells migrated equally well on COLL-coated substrata independent of E2β stimulation, and Shc317Y/F had no impact on COLL migration suggesting that GPER-1 signaling did not enhance migration on this ECM protein (data not shown).

GPER-1 stimulation enhances Shc-dependent haptotaxis of human SKBR3 breast cancer cells on FN-coated, but not collagen-coated, substrata. SKBR3 vector, GPERΔ154, or Shc317Y/F cells were left untreated or treated with E2β (10 nM) and seeded into the transwells of modified Boyden chambers containing a porous polycarbonate membrane (10-μm thickness, 8-μm pore) that were left untreated or coated with either FN (2 μg/ml) or collagen (10 μg/ml) and incubated overnight at 37 C. Nonmigrant cells were removed from the top of each chamber, and migrant cells on the lower surface of the membranes were fixed and stained with crystal violet. The number of migrated cells were counted and images were taken at × 40 magnification
GPER-1 Promotes Anchorage-Independent Growth by Promoting Fibrillogenesis in a Three-Dimensional Environment
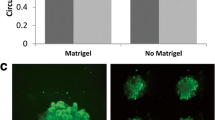
Since the conversion of soluble FN into fibrils by mammary adenocarcinoma cells has been linked to enhanced anchorage-independent growth [14] and GPER-1 action results in fibrillogenesis on planar surfaces (Figs. 1 and 2) [44], the possibility that GPER-1-mediated fibrillogenesis is required for anchorage-independent growth was evaluated (Figs. 5 and 6). To address this hypothesis, human SKBR3 or murine 4 T1 breast cancer cells were seeded into semisolid media supplemented with FN-depleted fetal bovine serum in the presence of increasing amounts of exogenous FN. Under conditions of FN depletion, neither SKBR3 nor 4 T1 cells were able to form colonies whereas both cell types readily formed colonies in the presence of 10 % fetal bovine serum which had not been FN-depleted (data not shown and Fig. 6a). However, either cell line readily formed colonies in FN-reduced conditions with 2-μg/ml exogenous FN (Figs. 5 and 6) provided that E2β was also present. Neither cell line grew in the presence of exogenous E2α and FN. Expression of GPERΔ154 in either cell background effectively prohibited E2β-dependent, anchorage-independent growth (Figs. 5 and 6b), suggesting that these growth properties are GPER-1-dependent. The specificity of GPERΔ154 for inhibiting GPER-1 action in this assay was demonstrated by the observation that the substitution of E2β for exogenous angiotensin II (ATII), which acts through its dedicated cognate G protein-coupled receptor, could restore FN-dependent, anchorage-independent growth (data not shown and Fig. 6b). Expression of Shc317Y/F in the SKBR3 cell background had an inhibitory effect on colony formation suggesting that Shc is also required for E2β-dependent growth in semisolid media (Fig. 5). Inclusion of inhibitory anti-integrin α5β1 antibodies in semisolid media containing murine 4 T1 cells showed that integrin α5β1 engagement is necessary for anchorage-independent growth under these conditions of limiting FN and exogenous E2β (Fig. 6c). Collectively, these data indicate that GPER-1 stimulation and exogenous FN is required for estradiol-dependent, anchorage-independent growth.

GPER-1 enhances FN-dependent, anchorage-independent growth of SKBR3 cells. SKBR3 vector, GPERΔ154, or Shc317Y/F cells were seeded into phenol red-free DMEM-F12 media containing 2 % FN-reduced serum in 0.35 % agarose in the absence or presence of E2β (10 nM) and supplemented with exogenous FN (2 μg/ml). Cells were grown for 10 days at 37 C in a humidified chamber. Cultures were weighed every 2 days, and water was replaced as needed. Images of colonies were captured at × 10 magnification (brightfield). Examples shown above are representative views of multiple experiments

E2β stimulation alters colony morphology of ER-negative mouse breast cancer cells grown in soft agar. a 4 T1 cells were seeded into phenol red-free DMEM-F12 media containing 2 % FN-reduced serum in 0.35 % agarose in the absence or presence of E2β (10 nM) and supplemented with exogenous FN (2 μg/ml). Cells were grown for 10 days at 37 C in a humidified chamber. Cultures were weighed every 2 days, and water was replaced as needed. Images of colonies were captured at × 10 magnification (brightfield). Examples shown above are representative views of multiple experiments. b 4 T1 vector or 4 T1 GPERΔ154 cells were seeded into phenol red-free DMEM-F12 media containing 2 % serum in 0.35 % agarose and left untreated or stimulated with E2β (10 nM) or ATII (10 nM) in the presence or absence of exogenous FN (2 μg/ml). Growth conditions were the same as above. c 4 T1 cells were seeded into phenol red-free DMEM-F12 media containing 2 % serum in 0.35 % agarose and left untreated or stimulated with E2β (10 nM), ICI 182, 780 (1 μM), IgG and E2β (10 nM), or anti-integrin α5β1 and E2β (10 nM) in the presence or absence of exogenous FN (2 μg/ml). Growth conditions are the same as above
To directly address the capacity of GPER-1 to promote fibrillogenesis in a three-dimensional environment, murine 4 T1 vector control or 4 T1 GPERΔ154 breast cancer cells were cultured in hanging drop assays in which rhodamine-labeled FN was incorporated [48–50]. As demonstrated in Fig. 7, 4 T1 cells that were cultured in suspension (in the absence of a substratum) overnight in the presence of E2β were capable of forming FN fibrils in this anchorage-independent assay (Fig. 7a). Similarly, FN fibrils were also measured by 4 T1 cells that were stimulated with ATII. FN fibril formation was not measured in untreated or E2α-treated cells. GPERΔ154 specifically inhibited fibrillogenesis in hanging drop cultures of 4 T1 cells that were stimulated by E2β but had no effect on ATII-induced fibrils (Fig. 7a). The ER antagonist, ICI 182, 780, also induced the formation of FN fibrils in the hanging drop assay (Fig. 7b), and this cellular activity was inhibited by GPERΔ154 (data not shown). Moreover, hanging drop cultures of E2β-stimulated 4 T1 breast cancer cells that were supplemented with soluble RGD peptide were unable to form FN fibrils; however, control RGE peptide did not have a negative effect on anchorage-independent fibrillogenesis (Fig. 7b). Taken together, the data in Figs. 5, 6 and 7 suggests that GPER-1 promotes anchorage-independent growth through its ability to synthesize FN fibrils.

GPER-1 promotes FN fibril formation of mouse 4 T1 breast cancer cells cultured in hanging drops. a 4 T1 vector or GPERΔ154 cells were placed in suspension in serum-free media supplemented with or without E2α (10 nM), E2β (10 nM), or angiotensin II (ATII) (10 nM), and rhodamine-labeled bovine FN (30 μg/ml). For each treatment, 10 × 15-μl aliquots were distributed onto the underside of a 100-mm Petri dish lid in a humidified chamber. Hanging drop cultures were incubated for 18 h at 37 C. Cells were fixed, stained with DAPI, and transferred to a glass slide. Images were captured using a Nikon 80i inverted fluorescent microscope fitted with a Retiga color camera at × 100 magnification. Multiple Z-axis sections were reconstructed into three-dimensional images using Nikon imaging software. Examples shown above are representative views of multiple experiments. b 4 T1 vector cells were placed in serum-free media supplemented with or without ICI 182, 780 (1 μM), or E2β (10 nM) with RGD or RGE, and rhodamine-labeled bovine FN (30 μg/ml). Hanging drops were incubated and analyzed as described in a
Discussion
Evidence is provided here that estrogenic hormones act via GPER-1 to enhance integrin α5β1-dependent adhesion of breast cancer cells to FN via a signaling mechanism that requires tyrosyl phosphorylation of the Shc adaptor protein. Specifically, we show that GPER-1 stimulation promotes the following: (i) the formation of focal adhesions leading to the reorganization of actin stress fibers; (ii) enhanced cellular adhesivity and haptotaxis on immobilized FN; (iii) anchorage-independent FN fibril formation; and (iv) FN-dependent, anchorage-independent growth. We have previously reported that integrin α5β1 and Shc are integral components of a signaling pathway leading to estrogen-mediated EGFR transactivation and FN matrix assembly on planar surfaces [44]. Collectively, these data support a model suggesting that GPER-1 coordinates two key cellular events required for the survival of breast cancer cells that escape the confines of glandular epithelia and invade the surrounding tissue parenchyma, namely, responsiveness to soluble growth factors and the capacity to form a provisional ECM (Fig. 8). Our findings are consistent with studies in mice that have shown a requirement for exogenous FN for efficient tumor cell implantation [56] and integrin β1 and Shc for homeostasis of the mammary gland [12].

Proposed mechanism of E2β activation of integrin α5β1 via GPER-1 signaling. E2β binding to GPER-1 at the plasma membrane promotes the dissociation of heterotrimeric G proteins into an α subunit and a βγ subunit on the cytoplasmic side of the plasma membrane. The βγ subunit promotes activation of Shc and its recruitment to integrin α5β1 and the formation of focal adhesions. Dissociation of Shc from integrin α5β1 is required for its subsequent centripetal movement to fibrillar adhesions and the release of membrane-tethered proHB-EGF
Estrogenic hormones regulate mammary gland homeostasis and, in certain instances, influence the cellular behavior of tumors that are derived from this tissue. However, the mechanism by which estrogen regulates FN-adhesive function has remained unclear. A direct influence of estrogen or the xenoestrogen, resveratrol, on cell behavior has been shown by studies measuring cytoarchitectural alterations in estrogen receptor-negative cells cultured in serum in response to hormone stimulation [57, 58]. Similar cytostructural changes, including enhanced actin stress fiber formation and the establishment of prominent focal adhesions, have been measured in long-term cultures of ER-positive MCF-7 breast cancer cells [59, 60] or endometrial cancer cells [57] stimulated with tamoxifen or the pure ER antagonist, ICI 182, 780, suggesting that alternative estrogen receptors may influence the interaction of these cancer cells derived from the female reproductive tract with their ECM. Here, we provide direct evidence that ICI 182, 780 or BPA act via the membrane estrogen receptor, GPER-1, to promote focal adhesion plaque formation, actin stress fiber assembly, and fibrillogenesis (Figs. 1 and 2). A result that is consistent with other reports that BPA acts via GPER-1 to induce rapid signaling effects [61] and gene transactivation [52] by breast cancer cells.
Modulation of integrin affinity for their adhesive ligands is commonly accomplished as the result of intracellular signals initiated by the interaction of external soluble mediators with GPCRs, a process referred to as inside-out integrin signaling [53]. For example, affinity upregulation of integrin αllbβ 3 for its ligand fibrinogen, a key event in thrombus formation, occurs in response to stimulation of platelets with ADP, epinephrine, or thrombin, whose receptors are GPCRs [62]. Likewise, β1 and β2 integrins on leukocytes exhibit increased affinity for their adhesive proteins in response to a broad array of immunomodulatory substances that act through GPCRs, including, but not limited to, f-Leu-Met-Phe, chemokine, and complement cascade products [63]. Similarly, our findings here indicate that estrogen action via GPER-1 activates integrin α5β1 resulting in its recruitment to focal adhesions (Figs. 1 and 2), increased adhesion and haptotaxis on planar surfaces coated with FN (Figs. 3 and 4), and FN matrix assembly in two- and three-dimensional environments (Figs. 1, 2, and 5, 6 and 7) by breast cancer cells. Since the experiments presented here do not directly address conformational alterations in the external domains of integrin α5β1 associated with FN affinity, it is not possible to formally conclude whether GPER-1-enhanced cellular adhesivity occurs via inside-out signaling. However, our findings are consistent with prior observations that have shown that GPCR activation promotes allosteric changes within the ligand-binding domains of β1, β2, and β3 integrins resulting in enhanced adhesive function [63–65]. Our findings suggest that enhanced integrin α5β1 adhesive action of breast cancer cells for FN does not appear to be solely relegated to signaling by GPER-1, as angiotensin II stimulation of breast cancer cells also enhanced fibrillogenesis (Fig. 6).
The adaptor protein Shc is intrinsically involved in intracellular signaling events that determine growth factor responsiveness and bidirectional integrin signaling [27]. Our work presented here provides additional support for this idea by showing that GPER-1 mediates enhanced integrin α5β1-Shc-dependent adhesivity and further supports the concept that integrin α5β1 and Shc form a signaling node that regulates FN matrix assembly and the release of membrane-tethered HB-EGF by SKBR3 breast cancer cells [44]. Further support for integrin α5β1 and Shc as an integration point in growth factor responsiveness and ECM signaling is demonstrated by the report that vascular endothelial cells coordinate FN-dependent adhesivity and vEGF receptor activation [66]. Similar to the work by Sweet and colleagues in vascular cells [66], we show that Shc is required for enhanced adhesion of breast cancer cells to FN-coated substrata, but not collagen (Fig. 3). This is consistent with our prior report that recruitment of Shc to integrin α2β1, a primary collagen receptor for SKBR3 breast cancer cells, occurred in the absence of stimulation and was not further enhanced by GPER-1 activation [44]. In this context, the data presented here support the concept that Shc recruitment is associated with enhanced cellular adhesion to FN, but not collagen. However, this difference may simply reflect synchrony in Shc activation that results from plating unattached cells to their adhesive ligands and does not account for the fact that Shc-integrin recruitment is likely a dynamic process that is associated with assembly and disassembly of focal adhesion structures [67]. It is interesting that Wary et al. [68] further noted that interaction of integrin α5β1 or αvβ3 with FN resulted in cellular proliferation, while engagement of integrin α2β1 or integrin α6β1 with their adhesive ligands results in cell cycle withdrawal, suggesting that differential cellular responses to extracellular matrices of different compositions may depend on the ability of a class of integrins to activate Shc signaling [68].
Shc couples with integrin during matrix engagement [68–70] and localizes to focal adhesions in attached cells [71], adhesive events associated with increased contractility tension at integrin anchorage points [29, 72]. These observations suggest that Shc is able to regulate integrin affinity by its capacity to interpret adhesive interactions with the ECM that is, in turn, determined by its phosphorylation status and interactions with integrin and proteins that accumulate in focal adhesions. The physical association with integrin is determined by an indirect interaction between the SH3 domain of Src-like kinases and the conserved proline-rich CH1 domain encoded within Shc while its conserved PTB domain directs focal adhesion targeting [29]. p66Shc uniquely contains an additional collagen homology domain, CH2, at its N-terminus; however, simple mutational analysis studies indicate that CH2 does not influence targeting to integrin or focal adhesions. Consistent with results reported by others examining the requirements for Shc complex formation with other integrins [64, 65], the primary tyrosyl phosphorylation site on Shc (Tyr317 on p52Shc) is dispensable with regards to integrin association [44], although it serves as an SH2 binding site that promotes the recruitment of Grb2 and a number of proteins that concentrate in focal adhesions, including FAK and Src-like kinases [73, 74]; tyrosine phosphatases, PTPN12, SHP-2 [69]; and tyrosine-phosphorylated proteins such as paxillin, talin, and vinculin [75]. In fact, expression of Shc317Y/F did not block GPER-1-enhanced association of endogenous Shc isoforms with immunopurified integrin α5β1, but rather resulted in its accumulation relative to the endogenous p52 and p46Shc isoforms [44], suggesting a possible role for protein tyrosine phosphatases (PTPs) for the release of Shc from integrin α5β1. This hypothesis is consistent with the concept that PTPs are well known to regulate focal adhesion disassembly, cell spreading, and migration [76–78]. The concept that focal adhesion formation is a dynamic process may suggest that disassembly and reassembly of Shc-integrin complexes is necessary for the formation of more ordered focal adhesions and actin stress cables that may be aligned through SH2 binding proteins [67]. Interestingly, although expression of Shc317Y/F did block GPER-1-mediated FN fibril formation [44], it did not completely impede GPER-1-enhanced recruitment of integrin α5β1 into focal adhesions as integrin α5β1 recruitment occurred in the context of mutant Shc, but resulted in the formation of poorly organized focal adhesions which were not properly aligned with actin stress fibers (Fig. 1). It is noteworthy that SKBR3 317Y/F cells express approximately equivalent amounts of p52Shc317Y/F relative to endogenous wild-type p52Shc and p46Shc isoforms [44]; yet, mutant Shc accumulates on integrin α5β1 relative to wild-type Shc following GPER-1 stimulation. This Shc-mediated defect is also associated with a complete blockade of GPER-1-mediated FN matrix assembly and EGFR transactivation [44], cellular activities that lie downstream of integrin recruitment to focal adhesions. One plausible explanation for this observation is that both mutant and wild-type Shc are equally recruited to integrin α5β1 during integrin clustering and the initial phases of focal adhesion formation but that the presence of Shc317Y/F inhibits its dissociation from integrin α5β1, an event that may be linked to focal contact elongation and disassembly, events that may be required for subsequent centripetal movement of integrin α5β1 to fibrillar adhesions and release of membrane-associated proHB-EGF. Anchorage-independent growth by tumor cells is the best-known predictor of experimental metastasis in mice and the best measure of resident stem cells in human tumors [14, 15]. In our study, we shown that estrogen action via GPER-1 leads to FN-dependent, anchorage-independent growth by human SKBR3 and murine 4 T1 breast cancer cells (Figs. 5 and 6). Prior work investigating the capacity of mammary adenocarcinoma cells to undergo anchorage-independent growth demonstrated a correlation between their capacity to convert soluble FN into fibrils and increased responsiveness to growth factor [14, 15]. As discussed above, the p66Shc isoform appears to play a unique role in sensing cell adhesion as p66Shc promotes anoikis via RhoA activation in detached cells [29]. These findings are consistent with studies which have shown that lung cancer cells lacking p66Shc display traits associated with advanced cancer, including anchorage-independent growth [34]. However, GPER-1-enhanced adhesivity to FN does not simply appear to be a result of preferential recruitment of the p52Shc or p46Shc isoforms to integrin α5β1 as human MDA-MB-231 breast cancer cell lines that express GPER-1 and p66Shc remain competent with regards to their capacity to promote integrin α5β1-dependent EGFR transactivation and enhanced FN adhesivity and anchorage-independent growth [Quinn, Magruder, and Filardo, unpublished results]. Interestingly, a direct linear increase in tumor stem cell activity of human SKBR3 cells has been reported as measured by their capacity to form mammospheroids in low adhesive media following serial passage through immunocompromised mice treated with epirubicin [79]. This finding suggests that chemotherapeutic drugs indirectly provide an environment that favors FN matrix assembly and anchorage-independent growth. It is tempting to speculate that in this harsh environment in which GPER-1-positive breast cancer cells that were presented with estrogenic hormones may show enhanced tumor stem cell activity and anchorage-independent growth.
Taken together, the data presented here support the hypothesis that GPER-1 signals via integrin α 5β1 and Shc to coordinate growth factor responsiveness and anchorage-independent growth, events critical for breast cancer progression. GPER-1 expression in primary breast tumors directly varies with tumor size and metastasis, markers of advanced disease, a relationship that is diametrically opposed to that shared by ER and these same prognostic variables [38]. The more recent finding that GPER-1 expression in triple negative breast cancer directly varies with disease progression underscores a potential role for this newly appreciated estrogen receptor in advanced disease [41]. Our work here, defining the cell biological influence of GPER-1 stimulation on FN matrix assembly and adhesion to this ECM protein, provides insight into the cellular mechanisms by which estrogens may promote the survival of metastatic breast cancer cells.
References
Stoffels JM, Zhao C, Baron W (2013) Fibronectin in tissue regeneration: timely disassembly of the scaffold is necessary to complete the build. Cell Mol Life Sci 70:4243–4253
Astrof S, Hynes RO (2009) Fibronectins in vascular morphogenesis. Angiol 12:165–175
Francis SE, Goh KL, Hodivala-Dilke K, Bader BL, Stark M, Davidson D, Hynes RO (2002) Central roles of α5β1 integrin and fibronectin in vascular development in mouse embryos and embryoid bodies. Arterioscler Thromb Vasc Biol 22:927–933
Faraldo MM, Deugnier MA, Thiery JP, Glukhova MA (2001) Growth defects induced by perturbation of B1-integrin function in the mammary gland epithelium result from a lack of MAPK activation via the Shc and Akt pathways. EMBO Rep 2:431–437
Williams CM, Engler AJ, Slone RD, Galante LL, Schwarzbauer JE (2008) Fibronectin expression modulates mammary epithelial cell proliferation during acinar differentiation. Cancer Res 68:3185–3192
Faull RJ, Kovach NL, Harlan JM, Ginsberg MH (1993) Affinity modulation of integrin α5β1: regulation of the functional response by soluble fibronectin. J Cell Biol 121:155–162
Hocking DC, Sottile J, Langenbach KJ (2000) Stimulation of integrin-mediated cell contractility by fibronectin polymerization. J Biol Chem 275:10673–10682
Ingham KC, Brew SA, Huff S, Litvinovich SV (1997) Cryptic self-association sites in type III modules of fibronectin. J Biol Chem 272:1718–1724
Klotzsch E, Smith ML, Kubow KE, Muntwyler S, Little WC, Beyeler F, Gourdon D, Nelson BJ, Vogel V (2009) Fibronectin forms the most extensible biological fibers displaying switchable force-exposed cryptic binding sites. Proc Natl Acad Sci U S A 106:18267–18272
Valenick LV, Hsia HC, Schwarzbauer JE (2005) Fibronectin fragmentation promotes alpha4beta1 integrin-mediated contraction of a fibrin-fibronectin provisional matrix. Exp Cell Res 309:48–55
Woodward TL, Mienaltowski AS, Modi RR, Bennett JM, Haslam SZ (2001) Fibronectin and the α5β1 integrin are under developmental and ovarian steroid regulation in the normal mouse mammary gland. Endocrinol 142:3214–3222
Faraldo MM, Deugnier MA, Tlouzeau S, Thiery JP, Glukhova MA (2002) Perturbation of beta 1-integrin function in involuting mammary gland results in premature dedifferentiation of secretory epithelial cells. Mol Biol Cell 13(10):3521–3531
Price JE (1996) Metastasis from human breast cancer cell lines. Breast Cancer Res Treat 39:93–102
Saulnier R, Bhardwaj B, Klassen J, Leopold D, Rahimi N, Tremblay E, Mosher D, Elliott B (1996) Fibronectin fibrils and growth factors stimulate anchorage-independent growth of murine mammary carcinoma. Exp Cell Res 222:360–369
Qiao H, Saulnier R, Patryzkat A, Rahimi N, Raptis L, Rossiter J, Tremblay E, Elliott B (2000) Cooperative effect of hepatocyte growth factor and fibronectin in anchorage-independent survival of mammary carcinoma cells: requirement for phosphatidulinositol 3-kinase activity. Cell Growth Differ 11:123–133
Discher DM, Noy D, Strazalka J, Ye S, Moser CC, Lear JD, Blasie JK, Dutton PL (2005) Design of amphiphilic protein maquettes: controlling assembly, membrane insertion, and cofactor interactions. Biochemistry 44(37):12329–12343
Frisch SM, Francis H (1994) Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol 124(4):619–626
Frisch SM, Vuori K, Ruoslahti E, Chan-Hui PY (1996) Control of adhesion-dependent cell survival by focal adhesion kinase. J Cell Biol 134:793–799
Martin SW, Butcher AJ, Berrow NS, Richards MW, Paddon RE, Turner DJ, Dolphin AC, Sihra TS, Fitzgerald EM (2006) Phosphorylation sites on calcium channel alpha 1 and beta subunits regulate ERK-dependent modulation of neuronal N-type calcium channels. Call Calcium 39(3):275–292
Green JA, Berrier AL, Pankov R, Yamada KM (2009) beta1 integrin cytoplasmic domain residues selectively modulate fibronectin matrix assembly and cell spreading through talin and Akt-1. J Biol Chem 284:8148–8159
Ridley AJ, Hall A (1992) The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70:389–399
Sottile J, Hocking DC, Swiatek PJ (1998) Fibronectin matrix assembly enhances adhesion-dependent cell growth. J Cell Sci 111:2933–2943
Wierzbicka-Patynowski I, Schwarzbauer JE (2002) Regulatory role for SRC and phosphatidylinositol 3-kinase in initiation of fibronectin matrix assembly. J Biol Chem 277:19703–19708
Wierzbicka-Patynowski I, Mao Y, Schwarzbauer JE (2007) Continuous requirement for pp 60-Src and phospho-paxillin during fibronectin matrix assembly by transformed cells. J Cell Physiol 210:750–756
Mao Y, Schwarzbauer JE (2005) Stimulatory effects of a three-dimensional microenvironment on cell mediated fibronectin fibrillogenesis. J Cell Sci 118:4427–4436
Accornero P, Miretti S, Cucuzza LS, Martignani E, Baratta M (2010) Epidermal growth factor and hepatocyte growth factor cooperate to enhance cell proliferation, scatter, and invasion in murine mammary epithelial cells. J Mol Endocrinol 44:115–125
Ravichandran KS (2001) Signaling via Shc family adapter proteins. Oncogene 20:6322–6330
Wary KK, Mariotti A, Zurzolo C, Gianeotti FG (1998) A requirement of caveolin-1 and associated kinase Fyn in intergrin signaling and anchorage-dependent cell growth. Cell 94(5):625–634
Ma Z, Myers DP, Wu RF, Nwariaku FE, Terada LS (2007) p66Shc mediates anoikis through RhoA. J Cell Biol 179:23–31
Migliaccio E, Mele S, Salcini AE, Pelicci G, Lai KM, Superti-Furga G, Pawson T, Di Fiore PP, Lanfrancone L, Pelicci PG (1997) Opposite effects of the p52shc/p46shc and p66shc splicing isoforms on the EGF receptor-MAP kinase-fos signalling pathway. EMBO J 16:706–716
Bonati A, Carlo-Stella C, Lunghi P, Albertini R, Pinelli S, Migliaccio E, Sammarelli G, Savoldo B, Tabilio A, Dall'Aglio PP, Pelicci PG (2000) Selective expression and constitutive phosphorylation of SHC proteins [corrected] in the CD34+ fraction of chronic myelogenous leukemias. Cancer Res 60:728–732
Zhang L, Lorenz U, Ravichandran KS (2003) Role of Shc in T-cell development and function. Immunol Rev 191:183–195
Giles AJ, Bender TP, Ravichandran KS (2009) The adaptor protein Shc plays a key role during early B cell development. J Immunol 183:5468–5476
Ma Z, Liu Z, Wu RF, Terada LS (2010) p66(Shc) restrains Ras hyperactivation and suppresses metastatic behavior. Oncogene 29:5559–5567
Frackelton AR Jr, Lu L, Davol PA, Bagdasaryan R, Hafer LJ, Sgroi DC (2006) p66 Shc and tyrosine-phosphorylated Shc in primary breast tumors identify patients likely to relapse despite tamoxifen therapy. Breast Cancer Res 8:R73
Grossman SR, Lyle S, Resnick MB, Sabo E, Lis RT, Rosinha E, Liu Q, Hsieh CC, Bhat G, Frackelton AR Jr, Hafer LJ (2007) p66 Shc tumor levels show a strong prognostic correlation with disease outcome in stage IIA colon cancer. Clin Cancer Res 13:5798–5804
Lee CS, Hall RE, Alexander IE, Koga M, Shine J, Sutherland RL (1990) Inverse relationship between estrogen receptor and epidermal growth factor receptor mRNA levels in human breast cancer cell lines. Growth Factors 3(2):97–103
Filardo EJ, Graeber CT, Quinn JA, Resnick MB, Giri D, DeLellis RA, Steinhoff MM, Sabo E (2006) Distribution of GPR30, a seven-membrane-spanning estrogen receptor, in primary breast cancer and its association with clinicopathological determinants of tumor progression. Clin Cancer Res 12:6359–6366
Arias-Pulido H, Royce M, Gong Y, Joste N, Lomo L, Lee SJ, Chaher N, Verschraegen C, Lara J, Prossnitz ER, Cristofanilli M (2010) GPR30 and estrogen receptor expression: new insights into hormone dependence of inflammatory breast cancer. Breast Cancer Res Treat 123:51–58
Ignatov A, Ignatov T, Weissenborn C, Eggemann H, Bischoff J, Semczuk A, Roessner A, Costa SD, Kalinski T (2011) G-protein-coupled estrogen receptor GPR30 and tamoxifen resistance in breast cancer. Breast Cancer Res Treat 128:457–466
Steiman J, Peralta EA, Louis S, Kamel O (2013) Biology of the estrogen receptor, GPR30, in triple negative breast cancer. Am J Surg 206:698–703
Filardo EJ, Quinn JA, Frackelton AR, Bland KI (2002) Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol 16:70–84
Filardo EJ, Quinn JA, Bland KI, Frackelton AR (2000) Estrogen-induced activation of Erk-1 and Erk-2 requires the G-protein coupled receptor homolog, GPR30, and occurs via the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol 14:1649–1660
Quinn JA, Graeber T, Frackelton R, Kim M, Schwarzbauer JE, Filardo EJ (2009) Coordinate regulation of estrogen-mediated fibronectin matrix assembly and epidermal growth factor receptor transactivation by the G protein-coupled receptor, GPR30. Mol Endocrinol 23:1052–1064
Sechler JL, Takada Y, Schwarzbauer JE (1996) Altered rate of fibronectin matrix assembly by deletion of the first type III repeats. J Cell Biol 134:573–583
Filardo EJ, Brooks PC, Deming SL, Damsky C, Cheresh DA (1995) Requirement of the NPXY motif in the integrin β3 subunit cytoplasmic tail for melanoma cell migration in vitro and in vivo. J Cell Biol 130:441–450
Pierschbacher MD, Hayman EG, Ruoslahti E (1981) Location of the cell-attachment site in fibronectin with monoclonal antibodies and proteolytic fragments of the molecule. Cell 26(2):259–267
Foty R (2011) A simple hanging drop cell culture protocol for generation of 3D spheroids. J Vis Exp 6(51)
Bartosh TJ, Ylostalo JH (2014) Preparation of anti-inflammatory mesenchymal stem/precursor cells (MSCs) through sphere formation using hanging-drop culture technique. Curr Protoc Stem Cell Biol 6(28)
Archacka K, Pozzobon M, Repele A, Rossi CA, Campanella M, De Coppi P (2014) Culturing muscle fibres in hanging drop: a novel approach to solve an old problem. Biol Cell 106(2):72–82
Tsai C, Wu H, Lin C, Lin Y, Chao A, Wang T, Hsueh S, Lai C, Wang H (2013) Estradiol and tamoxifen induce cell migration through GPR30 and activation of focal adhesion kinase (FAK) in endometrial cancers with low or without nuclear estrogen receptor α (ERα). PLoS ONE 8(9):e72999
Pupo M, Pisano A, Lappano R, Santolla MF, De Francesco EM, Abonante S, Rosano C, Maggiolini M (2012) Bisphenol A induces gene expression changes and proliferative effects through GPER in breast cancer cells and cancer-associated fibroblasts. Enviorn Health Perspect 120(8):1177–1182
Ginsberg MH, Partridge A, Shattil SJ (2005) Integrin regulation. Curr Opin Cell Biol 17(5):509–516
Luo BH, Carman CV, Springer TA (2007) Structural basis of integrin regulation and signaling. Annu Rev Immunol 25:619–647
O’Toole TE, Loftus JC, Plow EF, Glass AA, Harper JR, Ginsberg MH (1989) Efficient surface expressin of platelet GPIIb-IIIa requires both subunits. Blood 74(1):14–18
Price J (1996) Metastasis from human breast cancer cell lines. Breast Cancer Res Treat 39(1):93–102
Acconcia F, Barnes CJ, Kumar R (2006) Estrogen and tamoxifen induce cytoskeletal remodeling and migration in endometrial cells. Endocrinol 147:1203–1212
Azios NG, Krishnamoorthy L, Harris M, Cubano LA, Cammer M, Dharmawardhane SF (2007) Estrogen and resveratrol regulate Rac and Cdc42 signaling to the actin cytoskeleton of metastatic breast cancer cells. Neoplasia 9(2):147–158
Sapino A, Pietribiasi F, Bussolati G, Marchisio PC (1986) Estrogen- and tamoxifen-induced rearrangement of cytoskeletal and adhesion structures in breast cancer MCF-7 cells. Cancer Res 46:2526–2531
Ehlers EM, Schubert C (1999) Differences in morphology and cytoskeleton of MCF-7 and MX-1 cells after therapy with OH-tamoxifen and the pure estrogen antagonist ZM 182780. An immunofluorescence and scanning electron microscopic study. Ann Anat 181:231–236
Dong S, Terasaka S, Kiyama R (2011) Bisphenol A induces a rapid activation of Erk1/2 through GPR30 in human breast cancer cells. Environ Pollut 159(1):212–218
Cosemans JMEM, Iserbyt BF, Deckmyn H, Heemskerk JMW (2008) Multiple ways to switch platelet integrins on and off. J Thromb Haemost 6(8):1253–1261
Oberyszyn T, Conti C, Ross M, Oberyszyn A, Tober K, Rackoff A, Robertson F (1998) β2 integrin/ICAM-1 adhesion molecule interactions in cutaneous inflammation and tumor promotion. Carcinogenesis 19(3):445–455
Philips DR, Prasad KS, Manganello J, Bao M, Nannizzi-Alaimo L (2001) Integrin tyrosine phosphorylation in platelet signaling. Curr Opin Cell Biol 13(5):541–554
Cowan KJ, Law DA, Phillips DR (2000) Identification of Shc as the primary protein binding to the tyrosine-phosphorylated beta3 subunit of alpha iibbeta3 during outside-in integrin platelet signaling. J Biol Chem 275:36423–36429
Sweet DT, Chen Z, Wiley DM, Bautch VL, Tzima E (2012) The adaptor protein Shc integrates growth factor and ECM signaling during postnatal angiogenesis. Blood 119:1946–1955
Barberis L, Wary KK, Fiucci G, Liu F, Hirsch E, Brancaccio M, Altruda F, Tarone G, Giancotti FG (2000) Distinct roles of adaptor protein Shc and focal adhesion kinase in integrin signaling to ERK. J Biol Chem 275:36532–36540
Wary K, Mainiero F, Isakoff SJ, Marcantonio EE, Giancotti FG (1996) The adaptor protein Shc couples a class of integrins to the control of cell cycle progression. Cell 87:733–743
Habib T, Herrera R, Decker SJ (1994) Activators of protein kinase C stimulate association of Shc and the PEST tyrosine phosphatase. J Biol Chem 269(41):25243–25246
Deshmukh L, Gorbatyuk V, Vinogradova O (2010) Integrin β3 phosphoylation dictates its complex with the Sch phosphotyrosine-binding (PTB) domain. J Biol Chem 285:34875–34884
Angers-Loustau A, Cote JF, Charest A, Dowbenko D, Spencer S, Laskv LA, Tremblay (1999) Protein tyrosine phosphatase-PEST regulates focal adhesion disassembly, migration, and cytokinesis in fibroblasts. J Cell Biol 144(5):1019–1031
Debnath J (2010) p66Shc and Ras: controlling anoikis from the inside-out. Oncogene 29:5556–5558
Charest A, Wagner J, Jacob S, McGlade CJ, Tremblay ML (1996) Phosphotyrosine-independent binding of Shc to the NPLH sequence of murine protein-tyrosine phosphatase-PEST. Evidence for extended phosphotyrosine binding/phosphotyrosine interaction domain recognition specificity. J Biol Chem 271(14):8424–2429
Sachdev S, Bu Y, Gelman IH (2009) Paxillin-Y118 phosphorylation contributes to the control of Src-induced anchorage-independent growth by FAK and adhesion. BMC Cancer 9(12)
Petita B, Thierya JP (2000) Focal adhesions: structure and dynamics. Biol Cell 92:477–494
Arregui CO, Balsamo J, Lilien J (1998) Impaired integrin-mediated adhesion and signaling in fibroblasts expressing a dominant-negative mutant PTP1B. J Cell Biol 143(3):861–873
Yu DH, Qu CK, Henegariu O, Lu X, Feng GS (1998) Protein-tyrosine phosphatase Shp-2 regulates cell spreading, migration, and focal adhesion. J Biol Chem 273(33):21125–21131
Zheng Y, Lu Z (2013) Regulation of tumor cell migration by protein tyrosine phosphatase (PTP)-proline-, glutamate-, serine-, and threonine-rich sequence (PEST). Chin J Cancer 32(2):75–83
Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, Huang Y, Hu X, Su F, Lieberman J, Song E (2007) Let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 131:1109–1123
Acknowledgments
This work was supported, in part, by a Research Scholar Award from the American Cancer Society (RSG-02-194-01) any by National Institutes of Health Award CA119165-01A2. We would also like to thank the Hixon Lab at Brown University for their kind gift of Bisphenol A for our studies.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Magruder, H.T., Quinn, J.A., Schwartzbauer, J.E. et al. The G Protein-Coupled Estrogen Receptor-1, GPER-1, Promotes Fibrillogenesis via a Shc-Dependent Pathway Resulting in Anchorage-Independent Growth. HORM CANC 5, 390–404 (2014). https://doi.org/10.1007/s12672-014-0195-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-014-0195-9