
Abstract
Neurodegenerative disorders are chronic brain diseases that affect humans worldwide. Although many different factors are thought to be involved in the pathogenesis of these disorders, alterations in several key elements such as the ubiquitin–proteasome system (UPS), the nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway, and the endocannabinoid system (ECS or endocannabinoidome) have been implicated in their etiology. Impairment of these elements has been linked to the origin and progression of neurodegenerative disorders, while their potentiation is thought to promote neuronal survival and overall neuroprotection, as proved with several experimental models. These key neuroprotective pathways can interact and indirectly activate each other. In this review, we summarize the neuroprotective potential of the UPS, ECS, and Nrf2 signaling, both separately and combined, pinpointing their role as a potential therapeutic approach against several hallmarks of neurodegeneration.
Graphical Abstract

Similar content being viewed by others

Introduction
Neurodegenerative disorders (NDs) include a group of chronic brain diseases characterized by progressive neuronal death within specific regions of the nervous system. Human pathologies like Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS) are included in this group and currently represent a significant public health problem (Jellinger 2003; Fu et al. 2018).
In most cases, the origin of NDs is not fully understood, and it is commonly associated with a complex mixture of genetic and environmental factors (Coppede et al. 2006). Phenomena such as overproduction of reactive oxygen species (ROS) lead to oxidative stress (OS), mitochondrial dysfunction and neuroinflammation (Picca et al. 2020), as well as endoplasmic reticulum stress (ERS) (Xiang et al. 2017), and excitotoxic damage (Doble 1999), which are all linked to NDs’ origin and progression.
One of the main causal factors of NDs is related to misfolded proteins which accumulate within cells, triggering toxic mechanisms that disturb neuronal homeostasis and ultimately provoke cell death. Proteins such as amyloid-β (Aβ, related to AD), α-synuclein (α-Syn, related to PD), huntingtin (Htt, related to HD), and tau (related to AD and other tauopathies) have emerged as potential culprits regarding neurodegeneration. While it is true that the cause of the accumulation and toxic effects of these proteins remains unclear, increasing evidence suggests that an impairment in the mechanisms responsible for maintaining cellular proteostasis, such as the autophagosome-lysosome system and the ubiquitin–proteasome system (UPS), might allow these proteins to aggregate and exert toxicity on neural cells (Takalo et al. 2013; Zheng et al. 2016; Di Meco et al. 2020; Monsalvo-Maraver et al. 2023).
Although many experimental therapeutic approaches have been developed to treat NDs, targeting cannabinoid signaling and redox-related pathways, such as Nrf2, has shown promising results against oxidative stress, neuroinflammation, and other related toxic mechanisms that compromise cell function (van Muiswinkel and Kuiperij 2005; Fagan and Campbell 2014). This review is aimed to describe the interactions between key systems involved in the maintenance of neural cell homeostasis, specifically (1) the ubiquitin–proteasome system (UPS), (2) the endocannabinoidome, and (3) the Nrf2 signaling pathway, also discussing the role that these interactions might play in NDs’ pathology and potential treatment.
Ubiquitin–Proteasome System (UPS)
Overview
In eukaryotes, the proteasome is the major protein-degrading complex within the cytosolic and nuclear compartments (Bard et al. 2018). Structurally, the eukaryotic 26S proteasome is composed of two subcomplexes: the 20S core particle and the 19S regulatory particle (Schweitzer et al. 2016). The core particle contains the proteolytic fraction of the proteasome and it is formed by four heptameric rings organized in two pairs: two outer α-rings and two inner β-rings that together form a hollow chamber, in which protein substrates are degraded (Groll et al. 1997; Kopp et al. 1997). In turn, the 19S particle corresponds to the regulatory fraction responsible for recognizing, displaying, translocating, and deubiquitinating substrates for the core particle (Budenholzer et al. 2017).
The proteasome targeted proteins are identified by cells using ubiquitin, a small protein that is attached to lysine residues of other proteins within the cell, thus modifying their function, leading them to interact with other molecules, or altering their localization within the cell compartments; however, ubiquitin is also attached to proteins targeted for proteasomal degradation through a process that requires ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), and ubiquitin ligases (E3). Given that the 19S subcomplex recognizes ubiquitin-attached proteins, allowing them to enter the 20S core for degradation, the ubiquitination machinery and the 26S proteasome form together a whole proteolytic pathway called the UPS (Komander and Rape 2012; Ehlinger and Walters 2013; Kleiger and Mayor 2014; Watanabe et al. 2020).
Physiological Function of UPS in the Nervous System
Within the nervous system, UPS has been reported to be involved in a number of cell processes. Firstly, as early as the embryonic neural tube is being formed, the UPS-ubiquitin ligase protein F-box protein 30 (FBXO-30) mediates the degradation of the retinoic acid receptor, being retinoic acid a vital molecule for neural tube formation (Cheng et al. 2019). Furthermore, UPS appears to modulate differentiation of several cell types of the nervous system as evidence suggests that (1) in Drosophila melanogaster, E3 proteins Slimb and Ago allow glial progenitors to stop proliferating and differentiate into specific lineages (Ho et al. 2009); (2) UPS is involved in motor neuron differentiation, and ubiquitin-like modifier activating enzyme 1 E1 (UBA1) inhibition results in decreased motor neuron viability and neurite growth (Bax et al. 2019); (3) mRNA levels of several proteasome components within the mesencephalon change during postnatal development in rats, suggesting that UPS regulates neuronal development of structures such as the Substantia nigra pars compacta (El-Khodor et al. 2001); and (4) proteasome inhibition appears to be necessary for oligodendrocyte differentiation (Pasquini et al. 2003).
UPS components have also been shown to be necessary for axon growth and guidance during development in different animal models. Regarding axon growth, Highwire E3 protein has been reported to be related to neuromuscular synapse formation in D. melanogaster (DiAntonio et al. 2001), whereas in Xenopus, neural precursor cell expressed developmentally down-regulated protein 4 (Nedd4; an E3-enzyme) proved to enhance axon branching in retinal ganglion cells (Drinjakovic et al. 2010). In mammals, another E3-enzyme (anaphase-promoting complex or APC) mediated axon growth as shown in rat cerebellar granule neurons (Konishi et al. 2004). On the other hand, E3-enzymes Highwire, RPM-1, and Phr1 are strongly related to axon guidance regulation as studies have shown that (1) Highwire loss prevents sister axons from diverging to target the two lobes of the mushroom body; an important structure for learning in Drosophila sp. brain (Shin and DiAntonio 2011); (2) RPM-1 mutants display axon guidance disturbances within mechanosensory neurons of Caenorhabditis elegans (Schaefer et al. 2000); and (3) mice with Phr-1 deficiency exhibit motor neurons with an altered axon guidance in the spinal cord, while axonal growth cones and axons present aberrant morphologies (Lewcock et al. 2007).
Synaptic plasticity is another phenomenon closely related to UPS function. Firstly, studies have reported that Calmodulin-kinase II (CaMKII) enhances proteasome migration towards dendritic spines upon glutamatergic receptor-dependent synaptic stimulation (Bingol and Schuman 2006), and after proteasome migration, CaMKII is able to phosphorylate Rpt-6 proteasome subunit, consequently enhancing dendritic spine growth as shown in hippocampal slice cultures (Bingol et al. 2010; Hamilton et al. 2012). Moreover, UPS is also thought to take part in long-term potentiation (LTP), as evidence suggests that proteasome inhibition upregulates early-phase LTP and facilitates induction of late-phase LTP; however, it also disrupts late-phase LTP maintenance and stability. Noteworthy, other studies show that UPS is necessary for the induction, but not the maintenance, of late-phase LTP, suggesting that available evidence still remains controversial (Fonseca et al. 2006; Dong et al. 2008; Cai et al. 2010; Santos et al. 2015).
It has also been proposed that UPS plays a major role during neurogenesis, as proteasome-assembly chaperone Gankyrin was shown to enhance neurogenesis in neural stem cells and UPS activity inhibition decreases neural stem cell proliferation (Sahu et al. 2019; Singh et al. 2023). Combined, the evidence above described supports the concept that UPS is involved in several processes regarding physiological functions in the nervous system.
Role in Physiopathology of Neurodegenerative Disorders
Since the vast majority of NDs are closely related to abnormally aggregated proteins and their cytotoxic effects, UPS has emerged as a potential therapeutic target to antagonize and inhibit protein-related damage. When the proteasome is experimentally inhibited, aberrant accumulation of Aβ, α-Syn, phosphorylated-Tau (p-Tau), and Htt is exacerbated, as shown with different cell culture models (Zhou et al. 2003; Casarejos et al. 2011; Agholme et al. 2012); notably, proteasome activity reduction was observed in purified proteasomes exposed to Aβ-oligomers (Tseng et al. 2008) and filamentous Htt (Díaz-Hernández et al. 2006), as well as in PC12 cells exposed to mutant α-Syn, and in cultured neurons overexpressing human Tau (Tanaka et al. 2001; Ye et al. 2020).
In contrast, it has been reported that proteasome activity potentiation is able to diminish the toxic effects of ND-related proteins. For instance, (1) the triterpenoid ursolic acid has been used to activate the proteasome, thereby lessening paralysis in C. elegans nematodes exposed to Aβ (Wang et al. 2022a, b, c); (2) Gastrodia elata extracts can activate the proteasome, thus protecting PC12 cells from cell viability reduction induced by Htt (Huang et al. 2011); (3) inducing UPS-mediated degradation of Tau in N2a cells restored normal mitochondrial distribution within cells and rescued viability of Aβ-treated cells (Chu et al. 2016). Combined, this evidence suggests that UPS activity enhancement could be used to target proteostasis impairments in NDs. In addition, other hallmarks of the physiopathology of NDs could be reproduced under experimental conditions by proteasome inhibition. Glial reactivity and motor alterations were reported after an in vivo nigral injection of lactacystin (a proteasome inhibitor) in mice (Deneyer et al. 2019). In several cell culture models, proteasome inhibitors have been shown to reduce neuronal survival and viability, while enhancing ROS production, caspase activity, proapoptotic protein expression and mitochondrial translocation, mitochondrial membrane depolarization, and apoptosis-inducing factor (AIF) and cytochrome c release to the cytosol. Furthermore, UPS inhibitors also decreased the levels of antiapoptotic proteins, diminished (GSH) and oxidized (GSSG) glutathione levels, as well as synapse-related protein levels of βIII-tubulin, drebrin, and synaptophysin (Kikuchi et al. 2003; Papa et al. 2007; Bajic et al. 2012).
A significant amount of evidence is available on the neuroprotective actions derived from UPS upregulation. Table 1 summarizes some of the most relevant reports on this topic.
The Endocannabinoid System (ECS)
Overview
The endocannabinoid system (ECS) is a complex signaling pathway that involves cannabinoid receptors, their endogenous ligands, and the enzymes responsible for ligand biosynthesis and degradation (Lu and Mackie 2021). There are two main ligands belonging to the ECS: anandamide (AEA) and 2-arachidonoyl-glicerol (2-AG), both of which are usually termed endocannabinoids. AEA and 2-AG belong to N-acylethanolamine and 2-acylglycerol families, respectively, and several other molecules from these families share target receptors and metabolic pathways with endocannabinoids, such as oleoylethanolamide and linoleoylethanolamide (N-acylethanolamines), or 2-oleoyl-glycerol and 2-linoleoyl-glycerol (2-acylglycerols). Moreover, other similar molecules like lipoamino acids (N-acyl-serines or N-acyl-taurines) and N-acyl neurotransmitters (N-acyl-dopamines or N-acyl-serotonins) are able to share catabolic enzymes and target receptors with AEA and 2-AG (Cristino et al. 2020).
The two main endocannabinoid receptors are termed cannabinoid receptors 1 (CB1R) and 2 (CB2R), which are conceived as G protein–coupled receptors (GPCR) that signal through Gαi/o G proteins (Hua et al. 2016). Both CB1R and CB2R share a similar tridimensional structure, although they have certain structural differences within their binding sites for cannabinoids, which make them possess different affinities for distinct ligands (Li et al. 2019a, b).
In addition to CB1R/CB2R receptors, further evidence has suggested that endocannabinoids and other related ligands can signal through a number of receptors. Transient receptor potential ion channels of vanilloid type 1–4 (TRPV1-4), transient receptor potential ion channel of melastatin type 8 (TRPM8), transient receptor potential ion channel of ankyrin type 1 (TRPA1) (De Petrocellis et al. 2007, 2012; Raboune et al. 2014; Redmond et al. 2014), T-type calcium channels Cav3.1–3.3 (Ross et al. 2009), peroxisome proliferator-activated receptors α/γ (PPARα/γ) (O'Sullivan 2016), and G protein–coupled receptors GPR18, GPR55, GPR110, and GPR119 (Kohno et al. 2006; Overton et al. 2006; Sharir et al. 2012; Cristino et al. 2020) have all been shown to be modulated by these ligands.
Endocannabinoids and related ligands utilize various anabolic and catabolic pathways, and some of them share key enzymes for their metabolism. The N-acylethanolamine family of cannabinoids is synthesized via N-acylphosphatidylethanolamine-specific phospholipase d-like hydrolase (NAPE-PLD), while diacyl-glycerol lipase (DAGL) synthesizes 2-acylglycerols. In contrast, fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) degrade N-acylethanolamines and 2-acylglycerols, respectively (Basavarajappa 2007; Lu and Mackie 2021). Furthermore, in addition to their basic metabolism, endocannabinoids can be substrates of cyclooxygenase 2 (COX2), lipoxygenase (LOX), and cytochromes P450, which can catalyze the formation of a number of active cannabinoid-derived metabolites (Rouzer and Marnett 2011).
Physiological Significance in the Nervous System
The ECS is critical for the normal function and development of the nervous system. CB1R-expressing cells have been detected in the neural folds, brain primordia, and neural tube of chick embryos. Accordingly, 2-AG and AEA ligands, as well as NAPE-PLD, MAGL, FAAH, and DAGL enzymes, were detected in early chick embryos, pointing at the existence of a functional ECS within the first embryonic developmental stages (Psychoyos et al. 2012).
Nervous system cell differentiation and neural circuitry-building are also partially regulated by the ECS. CB1R-dependent signaling has proven to enhance the postnatal and adult-stage differentiation of neural progenitor cells into astrocytes (Aguado et al. 2006), the fasciculation of corticothalamic and thalamocortical axonal tracts in mice (Wu et al. 2010), the differentiation of neocortex-deep layer pyramidal neurons (Paraíso-Luna et al. 2020), and the radial migration of pyramidal neurons (with AEA and 2-AG acting like chemoattractant agents for cell migration) (Díaz-Alonso et al. 2017).
Several cannabinoid receptors regulate neurogenesis within the adult brain. CB1R and TRPV1 are thought to promote neuronal differentiation in the subventricular zone and the dentate gyrus (Jin et al. 2004). Additionally, other studies in cell cultures showed that dentate gyrus-cell proliferation needs co-stimulation of CB1R and CB2R, while proliferation in the subventricular zone is either stimulated by CB1R or CB2R, separately (Rodrigues et al. 2017). Other cannabinoid receptors that stimulate hippocampal neurogenesis are GPR55 (Hill et al. 2018), PPARγ (Esposito et al. 2011), and Cav3.1 (Yabuki et al. 2021).
Another main function of endocannabinoids within the nervous system involves glutamatergic synapse-retrograde regulation. Endocannabinoids are produced in postsynaptic terminals in response to glutamatergic stimulation, acting like a retrograde messenger that inhibits presynaptic release of glutamate by activating rectifying potassium channels and inhibiting N-type calcium channels (Maejima et al. 2001; Guo and Ikeda 2004).
Beyond the cellular level, ECS receptor signaling mediates complex behaviors, as it has been shown in in vivo rodent models. CB1R is expressed in the hypothalamus, and when it is deleted, animals gain less weight and increase their energy expenditure, revealing the role of CB1R on energy balance (Cardinal et al. 2012). Moreover, CB1R is also expressed in the basolateral amygdala, where it appears to modulate the formation of associative fear memories (Tan et al. 2011). Together, this supports the concept that the physiological role of ECS is crucial for the normal function of the nervous system, and consequently, ECS-function alterations could play important roles in the pathogenesis of neurological diseases, including NDs.
Role in the Physiopathology of Neurodegenerative Disorders
The ECS has emerged as a key factor involved in NDs, both because of its potential as a therapeutic target and its role in ND physiopathology. ECS signaling appears to be disturbed as changes in the expression levels of its components have been reported by several studies. In the hippocampus and entorhinal cortex of postmortem AD brains, CB2R and FAAH were overexpressed in glial cells surrounding Aβ aggregates (Benito et al. 2003). Notably, several studies report CB1R levels unchanged in AD postmortem brains, while others showed a markedly lowered expression, thus demonstrating that the available evidence still remains controversial (Ramírez et al. 2005; Lee et al. 2010). Other reports have shown that, in PD postmortem brains, CB1R levels were increased in the putamen and unchanged in the Substantia nigra (SN) when matched to control brains. In addition, CB2R expression was enhanced in the SN and decreased in the putamen, while MAGL levels were decreased in the SN and increased in the putamen (Navarrete et al. 2018). In contrast, HD patients showed a decrease in CB1R activity in several gray matter regions, and that reduction was inversely correlated with the length of the polyglutamine coding region of the Htt gene (Van Laere et al. 2010). Moreover, a CB1R level decrease was also observed in interneurons of the caudate nucleus of HD patients (Horne et al. 2013).
Although ECS alterations in patients do not exhibit a common pattern, experimental potentiation of cannabinoid signaling has shown common promising results as a neuroprotective approach. Notwithstanding the above, several studies suggest that cannabinoid potentiation, particularly through MAGL-dependent 2-AG degradation, could exert toxic effects via the production of COX2-catalyzed 2-AG-derived metabolites (Valdeolivas et al. 2013). Nonetheless, the vast majority of experimental approaches support the neuroprotective properties of ECS enhancement. In this regard, in a considerable number of studies, cannabinoid signaling potentiation depends on cannabinoid receptors to exert its neuroprotective properties, and neuroprotection tends to involve the inhibition of cell apoptosis/increase of cell viability and reduction of proinflammatory mediators and microglial/astroglial reactivity (Table 2). Altogether, this evidence suggests that ECS might target specific processes within the nervous system to antagonize neural degeneration in multiple NDs.
Nrf2 Signaling Pathway
Overview
The nuclear factor erythroid 2-related factor 2 (Nrf2) is a basic leucine zipper (bZIP) transcription factor that, in humans, is structurally composed of seven Nrf2-ECH homology domains (Neh1-7). Neh1 provides Nrf2 with the bZIP-DNA binding domain, and Neh2 allows Nrf2 to interact with Kelch ECH-associating protein 1 (Keap1) (Canning et al. 2015).
Nrf2 function has been sometimes linked with inflammation (Ahmed et al. 2017), mitochondrial biogenesis (Hayashi et al. 2017), and autophagy (Pajares et al. 2018), as well as with amino acid, glucose, lipid, and purine metabolism (He et al. 2020). Nonetheless, Nrf2 is better known for its role in redox-homeostasis and ROS detoxification. Under basal conditions, Nrf2 is primarily localized within the cytosol, where it interacts with Keap1, which is able to recruit a specific E3 enzymatic complex. These interactions lead to Nrf2 poly-ubiquitinization and ultimately target Nrf2 for proteasomal degradation. In contrast, when ROS are overproduced, certain cysteine residues on Keap 1 become oxidized, preventing Keap1 from interacting with Nrf2 and inhibiting its proteasomal degradation (Ma 2013; Song et al. 2021).
Under oxidative stress conditions, Nrf2 translocates into the cell nucleus and binds to the antioxidant response element (ARE), a DNA sequence within the promoter region of Nrf2 target genes. In the nucleus, Nrf2 interacts with other transcriptional co-activators such as CREB-binding protein (CBP) and small masculoaponeurotic fibrosarcoma proteins (sMaf) (Katoh et al. 2001). Nrf2-ARE target genes are involved in a variety of cell metabolic pathways related to the antioxidant defense system, including (1) the thioredoxin (TRX)-antioxidant system, controlling the expression of TRX, sulfiredoxin (Srx), and thioredoxin reductase (TrxR) (Tanito et al. 2007; Tonelli et al. 2018); (2) the glutathione-antioxidant system, regulating the expression of glutathione peroxidase 2 (Gpx2), glutathione reductase 1 (Gsr1), and both subunits of gamma-glutamylcysteine synthetase (GCS), as well as several glutathione S-transferases (Chan and Kwong 2000; Chanas et al. 2002; Singh et al. 2006; Tonelli et al. 2018); (3) Nrf2 modulates nicotinamide adenine dinucleotide phosphate (NADPH) synthesis, and controls the expression of enzymes that detoxify ROS through NADPH consumption, such as NADPH-quinone dehydrogenase 1 (Nqo1) (Wu et al. 2011; Tonelli et al. 2018); (4) Nrf2 also upregulates the expression of phase I, phase II, and phase III xenobiotic-metabolism enzymes (Lubelska et al. 2016; Tonelli et al. 2018); (5) Nrf2 participates in iron metabolism, particularly upregulating the expression of ferritin, a protein that prevents iron from participating in ROS production via Fenton reaction; (6) finally, but not less important, key antioxidant genes involved in the regulation of redox homeostasis, including superoxide dismutase (SOD), catalase (CAT), and heme oxygenase-1 (HO-1), are also well-known targets for Nrf2 (Zhang et al. 2021a, b). Upregulation of all these proteins neutralizes the overproduction of ROS, thereby limiting oxidative damage and ultimately keeping cell homeostasis (Kerins and Ooi 2018).
Physiological Significance in the Nervous System
Within the nervous system, the transcriptional activity of Nrf2 is involved in a considerable number of physiological processes. Firstly, Nrf2 appears to have a role in neurogenesis and neuronal differentiation as it has been reported that (1) mice lacking Nrf2 show impaired proliferation/differentiation of neural stem cells within the subgranular zone of the hippocampus (Robledinos-Antón et al. 2017); (2) Nrf2 upregulation promoted the migration, proliferation, and differentiation of neural stem cells of the subventricular zone of aged rats (Anandhan et al. 2021); (3) Nrf2 expression mediated the differentiation of neural stem cells into mature neurons or glia in the dentate gyrus of middle age rodents (Ray et al. 2018).
The Nrf2 signaling pathway also crosstalk with neurotrophin signaling in the nervous system. It has been proposed that brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) (neurotrophins) might interact with p75 neurotrophin receptors to stabilize Nrf2 (Ishii et al. 2019). In addition, Nrf2 attenuates the expression of transcriptional repressors of BDNF, while directly enhancing its transcription (Yao et al. 2021). Furthermore, studies in human astrocytes showed that Nrf2 also promotes NGF expression (Mimura et al. 2011). Therefore, neurotrophins and Nrf2 form a positive feedback circuit that might enhance neuronal survival, as well as antioxidant defense-gene expression, in the nervous system.
Notably, Nrf2 is also thought to be involved in synaptic plasticity and memory formation. Recently, it has been reported that Nrf2 is able to enhance the expression of a circular RNA molecule that indirectly enhances CaMKIV expression, thereby promoting hippocampal plasticity and memory formation (Zhang et al. 2022). In agreement, allicin-dependent Nrf2 activation proved to partially rescue the cognitive dysfunction observed in aged mice (Li et al. 2012).
Additional studies reported several other functions of Nrf2. For instance, Nrf2 raised the expression of the mitochondrial biogenesis-related protein PPARγ-coactivator 1α (PGC1α) in the hippocampus, prefrontal cortex, and amygdala (Khalifeh et al. 2017). Several studies reported that Nrf2 activators enhance microglial polarization towards an M2 neuroprotective phenotype (He et al. 2021a, b; Tao et al. 2021; Wang et al. 2023a, b, c). Nrf2 is also activated in astrocytes to counteract Fe2+-induced toxicity (Cui et al. 2016).
Role on the Physiopathology of Neurodegenerative Disorders
As noted above, Nrf2 impacts the physiology of the nervous system in many different manners; however, its primary function involves the direct modulation of redox-homeostasis, a function that is thought to be compromised in many NDs (Heurtaux et al. 2022). In AD postmortem brains, reduced nuclear translocation of Nrf2 was observed within the hippocampus and frontal cortex of AD tissues (Ramsey et al. 2007). In contrast, an efficient Nrf2 nuclear translocation has been reported in PD postmortem brains, although an inconsistent reduction of Nqo1 has been observed in other models, which could suggest that even if Nrf2 is being translocated, the expression of its target genes might remain altered in PD (Ramsey et al. 2007; Cook et al. 2011). Moreover, both PD and AD have been related with certain variants of NFE2L2 (Nrf2 coding gene) that appear to correlate to the onset of these diseases (von Otter et al. 2010, 2014). In HD patient-derived neural stem cell cultures, inhibition of the Nrf2 signaling was noted (Quinti et al. 2017). Regarding ALS patients, reduced mRNA levels of Nrf2 were found in the spinal cord and motor cortex, while increased Keap1 levels were reported only in the motor cortex (Sarlette et al. 2008). Thus, even though Nrf2 signaling impairment varies in different NDs, the alterations of the overall pathway appear to be a constant factor.
Increasing evidence suggests that the enhancement of Nrf2 signaling can result in overall neuroprotection, as shown with a variety of in vivo and in vitro models linked to different NDs (Table 3). The evidence consistently suggests that Nrf2 activation results in neuroprotection, and some studies are even starting to consider the use of Nrf2 activators, such as sulforaphane, for clinical trials (Kim 2021).
Proteasome ↔ Nrf2 Interactions
Both UPS and Nrf2 signaling are interconnected and interfere with the regulation mechanisms of each other. Firstly, Nrf2 enhances the expression of several proteasome-subunits and increases overall proteasomal activity (Kwak et al. 2003; Yang et al. 2007; Arlt et al. 2009; Malhotra et al. 2009; Kapeta et al. 2010; Tsakiri et al. 2013). In corroboration, numerous experimental models have suggested that Nrf2 exerts neuroprotection through proteasome activity improvement. For instance, (1) in Neuro 2A neuroblastoma cells, sulforaphane (Nrf2 activator)-induced neuroprotection against Aβ-induced cytotoxicity was reported to involve the proteasome activity, as proteasome inhibitor MG132 reduced the cytoprotection achieved (Park et al. 2009); (2) a toxic model of adenoviral vector-induced α-Syn-expression in Nrf2-lacking mice showed that Nrf2 deficiency downregulated the expression of several proteasome-subunits, contributing to α-Syn-aggregation, glial reactivity, and overall dopaminergic neuron damage (Lastres-Becker et al. 2012); (3) in SK-N-BE neuroblastoma cell line, 24-hydroxycholesterol treatment indirectly induced Nrf2 activation, consequently activating the proteasome and enhancing Tau protein clearance, an effect that was antagonized by Nrf2-silencing (Testa et al. 2023); (4) in two different α-Syn-overexpression PD models, it was reported that the upregulation of Nrf2/proteasome maturation protein (POMP) pathway enhanced immunoproteasome assembly, thereby leading to α-Syn degradation (Bi et al. 2021); (5) sulforaphane treatment, a well-known Nrf2 enhancer, has shown to protect N2a neuroblastoma-cell cultures from hydrogen peroxide toxicity. This cytoprotection proved to be UPS-dependent, as proteasome inhibition markedly reduced the neuroprotective effects of sulforaphane, which increased the mRNA levels of a number of Nrf2 target genes, including CAT, Nqo1 and the regulatory subunit of GCS (Kwak et al. 2007).
In a heart failure model, Nrf2 enhancement upregulated proteasomal activity, reducing the accumulation of damaged proteins and resulting in overall cardioprotection. In addition, in an insulinoma β-cell line, Nrf2 cytoprotection against tunicamycin was achieved throughout proteasome activity potentiation (Lee et al. 2012; Shimizu et al. 2016). Notwithstanding the above, other studies in different cell lines suggested that DJ-1 protein-mediated Nrf2 activation could both activate and inhibit the proteasome through different pathways (Moscovitz et al. 2015).
As previously reviewed, the UPS also regulates Nrf2 stability and degradation (Ma 2013; Song et al. 2021) and conversely to the previous idea, most evidence suggests that proteasomal inhibition could enhance Nrf2 signaling and consequently achieve neuroprotection. For instance, (1) in 6-hydroxydopamine (6-OHDA)-treated PC12 cells, proteasome inhibitor lactacystin has been shown to reduce oxidative stress, enhanced Nrf2 translocation/Nrf2 target gene expression, and upregulated glutathione levels (Yamamoto et al. 2007; Izumi 2013), (3) paraquat treatment toxicity was attenuated by lactacystin and MG132 (proteasome inhibitors), and consistently, both inhibitors increased the transcriptional activity of Nrf2 (Izumi et al. 2015). Nevertheless, fewer studies have contradictory conclusions, as a genetic-induced inhibition of the 26S proteasome proved to diminish Nrf2, Nqo1, and heme oxygenase 1 (HO-1) protein levels (Ugun-Klusek et al. 2017).
Therefore, whether proteasomal inhibition enhances or decreases Nrf2 signaling remains unclear. However, given that UPS impairment appears to be a pathological hallmark of multiple NDs (Thibaudeau et al. 2018), therapeutic inhibition of the proteasome might not be the best approach to reach Nrf2-signaling potentiation, considering that Nrf2 stabilization can be achieved with absent proteasomal inhibitors, as proven with tert-butylhydroquinone in human neural stem cells (Li et al. 2005).
Nrf2 ↔ Endocannabinoid System Interactions
As reviewed in previous sections, Nrf2/ECS potentiation has been considered a promising therapeutic approach against NDs; however, minor attention has been focused on the interaction between these two major signaling pathways. Increasing evidence suggests that several ECS-related receptors such as CB1R, CB2R, and TRPV1 regulate Nrf2 activity as shown with different tissues and cell models (Li et al. 2016; Lv et al. 2021; Baradaran Rahimi and Askari 2022; Mageed et al. 2022).
Many studies have used cannabinoid signaling as an indirect pathway to achieve Nrf2-dependent neuroprotection, for example, (1) primary neuron cultures were exposed to a toxic treatment of Aβ-oligomers and high glucose; afterwards, the cells were tested with different cannabinoid-related agents including endogenous/synthetic agonists as well as a FAAH inhibitor. All agents upregulated Nrf2, and differentially protected cells by increasing their viability, while reducing Aβ levels and inflammation/oxidative damage-related markers (Elmazoglu et al. 2020); (2) TRPV1 inhibition by a peptide-antagonist resulted in Nrf2 activation, reduction of Aβ processing, accumulation and Aβ precursor protein levels in amyloid precursor protein-over expressing N2a cells (Wang et al. 2022a, b, c); (3) in turn, SOD, CAT, GPx, and Gsr might be regulated by cannabinoids (Elmazoglu et al. 2020), hence highlighting the interaction between these systems.
Further evidence is based on ischemic damage models on neurons, which share some cell damage markers (e.g., oxidative stress and apoptosis) with NDs. For instance, MAGL inhibition rescued oxygen-deprived hippocampal neurons by diminishing apoptosis, as well as oxidative and pro-inflammatory markers, while improving Nrf2 signaling and cell viability. Both the changes observed in cell viability and oxidative markers were partially blocked when silencing Nrf2, thereby demonstrating that cannabinoid upregulation was acting through the Nrf2 pathway to block oxygen deprivation effects (Xu et al. 2021),
Although Nrf2 activation through cannabinoid signaling potentiation has been observed in many cell types, it appears to be particularly important for microglia (Li et al. 2013; Tadijan et al. 2022; Wang et al. 2022a, b, c). Microglia is known to express CB2R receptors and CB2R activation is thought to regulate microglial reactivity, therefore preventing inflammatory damage in several models of NDs (Ashton and Glass 2007; Komorowska-Müller and Schmöle 2020; Young and Denovan-Wright 2022). Outstandingly, it appears that this process might be regulated via Nrf2 signaling, as CB2R agonist JWH133 proved to prevent 1-methyl-4-phenylpyridinium (MPP +)-stimulated microglia from differentiating into a M1 proinflammatory/neurotoxic phenotype, while enhancing their differentiation into a M2 anti-inflammatory/neuroprotective phenotype, an effect that was reverted by Nrf2 inhibition (Wang et al. 2023a, b, c). Additionally, a study in macrophages found similar results, as TRPV1-dependent ion fluxes resulted in an increased Nrf2 signaling and consequently a decreased macrophage differentiation into a M1 phenotype (Lv et al. 2021).
Further evidence based on BV2 microglia-like cells suggested that phytocannabinoids such as cannabidiol (CBD) and Δ9-tetrahydrocannabinol (Δ9-THC) inhibit NF-kB and interferon β/Signal transducer and activator of transcription (STAT) proinflammatory signaling pathways, which might complement the concept that Nrf2 is indirectly able to inhibit NF-kB (Kozela et al. 2010; Wardyn et al. 2015). This previous study also reported that Δ9-THC and CBD act independently from CB1R/CB2R receptors, suggesting that cannabinoids might achieve neuroprotection in this regard through different mechanisms (Kozela et al. 2010). In addition, a microarray analysis of lipopolysaccharide-stimulated BV2 cells showed that mainly CBD is able to upregulate Nrf2, which supports the previous argument (Juknat et al. 2013).
To our current knowledge, little evidence has been found regarding the Nrf2-dependent regulation of the ECS components. In 2020, it was found an ARE sequence within the promoter region of the CB2R-coding gene, where Nrf2 was only reported to regulate CB2R expression in microglia (Galán-Ganga et al. 2020). This report opens up the debate of whether Nrf2 upregulation could enhance cannabinoid signaling via CB2R, boosting the neuroprotective effects of this cannabinoid receptor regarding microglial reactivity and polarization, as previously mentioned.
Finally, cannabinoid-responsive PPARγ receptors are thought to enhance Nrf2 expression as PPARγ response elements in the DNA have been found in the promoter region of NFE2L2. On the other hand, PPARγ is also upregulated by Nrf2, thereby shaping a positive feedback loop between these two transcription factors (Cho et al. 2010; Huang et al. 2010; Lee 2017; Lin et al. 2018).
Evidence regarding PPARγ/Nrf2 interactions is predominantly derived from ischemic/hemorrhagic stroke studies. For instance, (1) PPARγ-agonist pioglitazone was tested in different models of middle cerebral artery occlusion and reperfusion (MCAO); PPARγ activation reduced apoptosis while improving neurological score of rats in vivo, besides it alleviated oxidative damage/excitotoxicity in 6-OHDA/glutamate-treated cells, respectively (Zhao et al. 2021), (2) MCAO-treated rats were exposed to luteoloside, a compound that proved to increase PPARγ and Nrf2 expression, possibly leading to the inhibition of nuclear factor-kB (NF-kB) and the expression of its inflammation-related target genes (Li et al. 2019a, b), (3) sirtuin 1 (Sirt1) protein also proved to exert neuroprotective effects in an ischemia model through the PGC1α/PPARγ/Nrf2 pathway to reduce oxidative stress and enhance cell viability (Zhou et al. 2022). All these models show that ND-related markers such as oxidative damage, neuroinflammation, and excitotoxicity can decrease through the PPARγ/Nrf2 pathway, potentially via PPARγ agonists such as cannabinoid-like compounds.
Ferroptosis is an iron-dependent cell death pathway that has been related to AD, PD, and HD pathologies (Tang et al. 2021); however, the existing evidence is again based on hemorrhagic stroke studies. In intracerebral hemorrhage models, pioglitazone was used to induce the PPARγ/Nrf2 axis, consequently inhibiting ferroptosis in erastin-treated neuron cultures and rats with an intracerebral blood injection (Duan et al. 2022). Additionally, in subarachnoid hemorrhage models, it was proven that the activation of the PPARγ/Nrf2 axis via netrin-1 reduced ferroptotic cell death through the enhancement of Gpx4 and coenzyme-Q10/ferroptosis suppressor protein-1 pathways (Chen et al. 2023). Neuroprotective effects of the PPARγ/Nrf2/Gpx4 pathway against ferroptosis have also been described in an epilepsy model tested on rats (Wang et al. 2023a, b, c). Taken together, these results suggest that the PPARγ/Nrf2 axis might aid in targeting ferroptosis in several NDs.
In addition to stroke models, PPARγ signaling has been tested in inflammation-related studies. NLRP3-inflammasome activation has been shown to be toxic in AD, PD, HD, and ALS models, and several of them suggest that NLRP3 inhibition could exert neuroprotective effects in these NDs (Deora et al. 2020; Hanslik and Ulland 2020; Haque et al. 2020; Chen et al. 2022). Indirect evidence from an alcoholic liver damage model has found that phyto-derivative magnolol is able to increase the levels of PPARγ and Nrf2, while reducing NLRP3 proinflammatory-signaling cascade and the damage associated with it (Liu et al. 2019). In corroboration, both PPARγ and Nrf2 are reportedly able to inhibit NLRP3 (Hou et al. 2018; Yang et al. 2021). Although to our current knowledge, it is still unproved, the PPARγ/Nrf2 axis could reduce ND-related neuroinflammation/cell death through the inhibition of NLRP3.
Finally, a PD-related neuroinflammation model induced by lipopolysaccharide demonstrated that a pioglitazone treatment could improve the behavioral impairment in vivo, while increasing dopaminergic neuron survival within the SN through the activation of the Nrf2/HO-1 pathway and the inhibition of NF-kB signaling (Zakaria et al. 2019).
Proteasome ↔ Endocannabinoid System Interactions
In contrast to the interactions reviewed in previous sections, limited evidence supports a regulatory linkage between the ECS and the UPS pathways. Regarding these interactions, it has been proposed that the phytocannabinoid Δ9-THC is able to inhibit the proteolytic activity of the 20S core particle in the hippocampus (Salgado-Mendialdúa et al. 2018). Additionally, further studies on human astrocytes suggested that Δ9-THC modulated the expression of several genes related to the ubiquitin pathway (Bindukumar et al. 2008). However, the available evidence remains controversial.
There are two main reports that suggest a possible cooperation between the UPS and the ECS to achieve neuroprotection in ND-related models. The first one reported that in N2a cells transfected with mutant Htt, activation of the cannabinoid-related receptor PPARγ by the agonist rosiglitazone rescued cell viability and diminished several markers of cell damage; however, rosiglitazone also enhanced the proteolytic activity of the proteasome. This result is supported by the fact that rosiglitazone also reduced the aggregation of mutant Htt and that one of the PPARγ target genes is part of the proteasome activator complex (Chiang et al. 2015). The second report concluded that in C. elegans nematodes of different strains, CBD treatment increased dopaminergic neuron survival and the lifespan of the worms, while reducing oxidative damage markers. In the strain OW13 (that expresses α-Syn in muscular cells), CBD treatment proved to increase the activity of the proteasome and consistently decreased α-Syn accumulation (Muhammad et al. 2022). These results suggest that cannabinoid potentiation might enhance the UPS activity, potentially exerting neuroprotection against proteostasis impairments.
It is noteworthy that ECS potentiation has been linked with proteasomal UPS activity in other studies less related to NDs. (1) It has been reported that genetic inactivation of CB1R alters myelination and disrupts the differentiation of oligodendrocytes, leading to motor and cognitive deficiencies in vivo, and additionally, it was found that CB1R promoted oligodendrocyte-precursor cell differentiation via the enhancement of the proteasome-dependent degradation of Ras homolog family member A. These previous observations offer a potential therapeutic approach in experimental models of ALS, where myelination and oligodendrocyte differentiation appear to be disrupted (Raffaele et al. 2021; Sánchez-de la Torre et al. 2022), (2) CB1R receptor activation was also linked with neurite outgrowth in N2a cells, a process that is thought to involve the targeting of Rap1-GTPase activating protein II to ubiquitination and proteasome-dependent degradation. These results are of great relevance, given that neurotoxic proteins such as Aβ and α-Syn are able to inhibit neurite outgrowth, a phenomenon that might be targeted through the CB1R/proteasome pathway (Takenouchi et al. 2001; Jordan et al. 2005; Calkins and Reddy et al. 2011).
Concluding Remarks
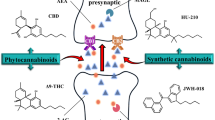
In summary, as NDs are multifactorial, this review provides evidence in support of the involvement of UPS, ECS, and Nrf2 signaling impairment in the origin and progression of several NDs. Experimental manipulation of the UPS, ECS, and Nrf2 has shown promising results as potential therapeutic approaches for NDs. However, particular attention should be focused on the interactions between these pathways, as evidence shows that synergic neuroprotective effects might be achieved when enhancing the activity of Nrf2, ECS, or UPS. Despite the potential of therapeutically targeting these in NDs, the available data are limited as the available evidence is not directly derived from NDs models but rather from ischemic neuronal damage or other non-neuronal cytoprotection models; therefore, further research will be needed to decipher the actual neuroprotective efficacies of the synergic enhancement of Nrf2, ECS, or UPS. A graphical representation summarizing the interaction of the Nrf2, ECS, and UPS systems is shown in Fig. 1.

Summary of the reported interactions and effects between ECS, UPS, and Nrf2 systems. Green arrows represent stimulatory effects, while red arrows represent inhibitory effects
Finally, by recommendation of one reviewer of the manuscript, we include this following section with suggested reviews on the overviews developed herein that may be of support in certain aspects. https://www.sciencedirect.com/science/article/pii/S2213231722002610. https://www.sciencedirect.com/science/article/pii/S0753332223016037. https://www.mdpi.com/2076-3921/12/7/1371. https://www.mdpi.com/1422-0067/24/13/11003. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC9710321/. https://www.annualreviews.org/doi/full/10.1146/annurev-pharmtox-052220-103416. https://molecularneurodegeneration.biomedcentral.com/articles/10.1186/s13024-015-0012-0.
Availability of Data and Materials
Not applicable
References
Agholme L, Hallbeck M, Benedikz E, Marcusson J, Kågedal K (2012) Amyloid-β secretion, generation, and lysosomal sequestration in response to proteasome inhibition: involvement of autophagy. J Alzheimer Dis 31(2):343–358. https://doi.org/10.3233/JAD-2012-120001
Aguado T, Palazuelos J, Monory K, Stella N, Cravatt B, Lutz B, Mariscano G, Kokaia Z, Guzmán M, Galve-Roperh I (2006) The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. J Neurosci 26(5):1551–1561. https://doi.org/10.1523/JNEUROSCI.3101-05.2006
Ahmed SMU, Luo L, Namani A, Wang XJ (1863) Tang X (2017) Nrf2 signaling pathway: pivotal roles in inflammation. Biochim Biophys Acta Mol Basis Dis 2:585–597. https://doi.org/10.1016/j.bbadis.2016.11.005
Anandhan A, Kirwan KR, Corenblum MJ, Madhavan L (2021) Enhanced NRF2 expression mitigates the decline in neural stem cell function during aging. Aging Cell 20(6):e13385. https://doi.org/10.1111/acel.13385
Arlt A, Bauer I, Schafmayer C, Tepel J, Müerköster SS, Brosch M, Röder C, Kalthoff H, Hampe J, Moyer MP, Fölsch UR, Schäfer H (2009) Increased proteasome subunit protein expression and proteasome activity in colon cancer relate to an enhanced activation of nuclear factor E2-related factor 2 (Nrf2). Oncogene 28(45):3983–3996. https://doi.org/10.1038/onc.2009.264
Ashton JC, Glass M (2007) The cannabinoid CB2 receptor as a target for inflammation-dependent neurodegeneration. Curr Neuropharmacol 5(2):73–80. https://doi.org/10.2174/157015907780866884
Bahn G, Park J-S, Yun UJ, Lee YJ, Choi Y, Park JS, Baek SH, Choi BY, Cho YS, Kim HK, Han J, Sul JH, Baik S-H, Lim J, Wakabayashi N, Bae SH, Han J-W, Arumugam TV, Mattson MP, Jo D-G (2019) NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer’s models. Proc Natl Acad Sci 116(25):12516–12523. https://doi.org/10.1073/pnas.1819541116
Bajic N, Jenner P, Ballard CG, Francis PT (2012) Proteasome inhibition leads to early loss of synaptic proteins in neuronal culture. J Neural Transm 119:1467–1476. https://doi.org/10.1007/s00702-012-0816-9
Baradaran Rahimi V, Askari VR (2022) A mechanistic review on immunomodulatory effects of selective type two cannabinoid receptor β-caryophyllene. BioFactors 48(4):857–882. https://doi.org/10.1002/biof.1869
Bard JAM, Goodall EA, Greene ER, Jonsson E, Dong KC, Martin A (2018) Structure and function of the 26S proteasome. Annu Rev Biochem 87:697–724. https://doi.org/10.1146/annurev-biochem-062917-011931
Basavarajappa BS (2007) Critical enzymes involved in endocannabinoid metabolism. Protein Pept Lett 14(3):237–246. https://doi.org/10.2174/092986607780090829
Bax M, McKenna J, Do-Ha D, Stevens CH, Higginbottom S, Balez R, Cabral-da-Silva MEC, Farrawell NE, Engel M, Poronnik P, Yerbury JJ, Saunders DN, Ooi L (2019) The ubiquitin proteasome system is a key regulator of pluripotent stem cell survival and motor neuron differentiation. Cells 8(6):581. https://doi.org/10.3390/cells8060581
Benito C, Núñez E, Tolón RM, Carrier EJ, Rábano A, Hillard CJ, Romero J (2003) Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J Neurosci 23(35):11136–11141. https://doi.org/10.1523/JNEUROSCI.23-35-11136.2003
Bi M, Du X, Xiao X, Dai Y, Jiao Q, Chen X, Zhang L, Jiang H (2021) Deficient immunoproteasome assembly drives gain of α-synuclein pathology in Parkinson’s disease. Redox Biol 47:102167. https://doi.org/10.1016/j.redox.2021.102167
Bindukumar B, Mahajan SD, Reynolds JL, Hu Z, Sykes DE, Aalinkeel R, Schwartz SA (2008) Genomic and proteomic analysis of the effects of cannabinoids on normal human astrocytes. Brain Res 1191:1–11. https://doi.org/10.1016/j.brainres.2007.10.062
Bingol B, Schuman EM (2006) Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature 441(7097):1144–1148. https://doi.org/10.1038/nature04769
Bingol B, Wang C-F, Arnott D, Cheng D, Peng J, Sheng M (2010) Autophosphorylated CaMKIIα acts as a scaffold to recruit proteasomes to dendritic spines. Cell 140(4):567–578. https://doi.org/10.1016/j.cell.2010.01.024
Brandes MS, Zweig JA, Tang A, Gray NE (2021) NRF2 activation ameliorates oxidative stress and improves mitochondrial function and synaptic plasticity, and in A53T α-synuclein hippocampal neurons. Antioxidants 11(1):26. https://doi.org/10.3390/antiox11010026
Budenholzer L, Cheng CL, Li Y, Hochstrasser M (2017) Proteasome structure and assembly. J Mol Biol 429(22):3500–3524. https://doi.org/10.1016/j.jmb.2017.05.027
Cai F, Frey JU, Sanna PP, Behnisch T (2010) Protein degradation by the proteasome is required for synaptic tagging and the heterosynaptic stabilization of hippocampal late-phase long-term potentiation. Neuroscience 169(4):1520–1526. https://doi.org/10.1016/j.neuroscience.2010.06.032
Cai CZ, Zhou HF, Yuan NN, Wu MY, Lee SMY, Ren JY, Lu JH (2019) Natural alkaloid harmine promotes degradation of alpha-synuclein via PKA-mediated ubiquitin-proteasome system activation. Phytomedicine 61:152842. https://doi.org/10.1016/j.phymed.2019.152842
Calkins MJ, Reddy PH (2011) Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim Biophys Acta 4:507–513. https://doi.org/10.1016/j.bbadis.2011.01.007
Canning P, Sorrell FJ, Bullock AN (2015) Structural basis of Keap1 interactions with Nrf2. Free Radic Biol Med 88(Pt B):101–107. https://doi.org/10.1016/j.freeradbiomed.2015.05.034
Cardinal P, Bellocchio L, Clark S, Cannich A, Klugmann M, Lutz B, Marsicano G, Cota D (2012) Hypothalamic CB1 cannabinoid receptors regulate energy balance in mice. Endocrinology 153(9):4136–4143. https://doi.org/10.1210/en.2012-1405
Casarejos MJ, Solano RM, Gomez A, Perucho J, de Yébenes JG, Mena MA (2011) The accumulation of neurotoxic proteins, induced by proteasome inhibition, is reverted by trehalose, an enhancer of autophagy, in human neuroblastoma cells. Neurochem Int 58(4):512–520. https://doi.org/10.1016/j.neuint.2011.01.008
Chan JY, Kwong M (2000) Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim Biophys Acta 1517(1):19–26. https://doi.org/10.1016/S0167-4781(00)00238-4
Chanas SA, Jiang Q, McMahon M, McWalter GK, McLellan LI, Elcombe CR, Henderson CJ, Wolf CR, Moffat GM, Itoh K, Yamamoto M, Hayes JD (2002) Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem J 365(Pt 2):405–416. https://doi.org/10.1042/bj20020320
Chen J, Wang Y, Li M, Zhu X, Liu Z, Chen Q, Xiong K (2023) Netrin-1 alleviates early brain injury by regulating ferroptosis via the PPARγ/Nrf2/GPX4 signaling pathway following subarachnoid hemorrhage. Transl Stroke Res Online ahead of print. https://doi.org/10.1007/s12975-022-01122-4
Chen K-P, Hua K-F, Tsai F-T, Lin T-Y, Cheng C-Y, Yang D-I, Hsu H-T, Ju T-C (2022) A selective inhibitor of the NLRP3 inflammasome as a potential therapeutic approach for neuroprotection in a transgenic mouse model of Huntington’s disease. J Neuroinflammation 19(1):56. https://doi.org/10.1186/s12974-022-02419-9
Cheng X, Pei P, Yu J, Zhang Q, Li D, Xie X, Wu J, Wang S, Zhang T (2019) F-box protein FBXO30 mediates retinoic acid receptor γ ubiquitination and regulates BMP signaling in neural tube defects. Cell Death Dis 10(8):551. https://doi.org/10.1038/s41419-019-1783-y
Cheng YF, Zhu GQ, Wang M, Cheng H, Zhou A, Wang N, Li QL (2009) Involvement of ubiquitin proteasome system in protective mechanisms of puerarin to MPP+-elicited apoptosis. Neurosci Res 63(1):52–58. https://doi.org/10.1016/j.neures.2008.10.009
Chiang M-C, Cheng Y-C, Nicol CJ, Lin K-H, Yen C-H, Chen S-J, Huang R-N (2015) Rosiglitazone activation of PPARγ-dependent signaling is neuroprotective in mutant huntingtin expressing cells. Exp Cell Res 338(2):183–193. https://doi.org/10.1016/j.yexcr.2015.09.005
Cho H-Y, Gladwell W, Wang X, Chorley B, Bell D, Reddy SP, Kleeberger SR (2010) Nrf2-regulated PPARγ expression is critical to protection against acute lung injury in mice. Am J Respir Crit Care Med 182(2):170–182. https://doi.org/10.1164/rccm.200907-1047OC
Chu T-T, Gao N, Li Q-Q, Chen P-G, Yang X-F, Chen Y-X, Zhao Y-F, Li YM (2016) Specific knockdown of endogenous tau protein by peptide-directed ubiquitin-proteasome degradation. Cell Chem Biol 23(4):453–461. https://doi.org/10.1016/j.chembiol.2016.02.016
Chung YC, Bok E, Huh SH, Park J-Y, Yoon S-H, Kim SR, Kim Y-S, Maeng S, Park SH, Jin BK (2011) Cannabinoid receptor type 1 protects nigrostriatal dopaminergic neurons against MPTP neurotoxicity by inhibiting microglial activation. J Immunol 187(12):6508–6517. https://doi.org/10.4049/jimmunol.1102435
Cook AL, Vitale AM, Ravishankar S, Matigian N, Sutherland GT, Shan J, Sutharsan R, Perry C, Silburn PA, Mellick GD, Whitelaw ML, Wells CA, Mackay-Sim A, Wood SA (2011) NRF2 activation restores disease related metabolic deficiencies in olfactory neurosphere-derived cells from patients with sporadic Parkinson’s disease. PLoS ONE 6(7):e21907. https://doi.org/10.1371/journal.pone.0021907
Coppedè F, Mancuso M, Siciliano G, Migliore L, Murri L (2006) Genes and the environment in neurodegeneration. Biosci Rep 26(5):341–367. https://doi.org/10.1007/s10540-006-9028-6
Corpas R, Griñán-Ferré C, Rodríguez-Farré E, Pallàs M, Sanfeliu C (2019) Resveratrol induces brain resilience against Alzheimer neurodegeneration through proteostasis enhancement. Mol Neurobiol 56:1502–1516. https://doi.org/10.1007/s12035-018-1157-y
Cristino L, Bisogno T, Di Marzo V (2020) Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat Rev Neurol 16(1):9–29. https://doi.org/10.1038/s41582-019-0284-z
Cui YB, Ma SS, Zhang CY, Li DP, Yang B, Lv PJ, Xing Q, Huang T, Yang GL, Cao W, Guan FX (2018) Pharmacological activation of the Nrf2 pathway by 3H–1, 2-dithiole-3-thione is neuroprotective in a mouse model of Alzheimer disease. Behav Brain Res 336:219–226. https://doi.org/10.1016/j.bbr.2017.09.011
Cui Z, Zhong Z, Yang Y, Wang B, Sun Y, Sun Q, Yang G-Y, Bian L (2016) Ferrous iron induces Nrf2 expression in mouse brain astrocytes to prevent neurotoxicity. J Biochem Mol Toxicol 30(8):396–403. https://doi.org/10.1002/jbt.21803
Dal Vechio FH, Cerqueira F, Augusto O, Lopes R, Demasi M (2014) Peptides that activate the 20S proteasome by gate opening increased oxidized protein removal and reduced protein aggregation. Free Radic Biol Med 67:304–313. https://doi.org/10.1016/j.freeradbiomed.2013.11.017
De Petrocellis L, Moriello AS, Imperatore R, Cristino L, Starowicz K, Di Marzo V (2012) A re-evaluation of 9-HODE activity at TRPV1 channels in comparison with anandamide: enantioselectivity and effects at other TRP channels and in sensory neurons. Br J Pharmacol 167(8):1643–1651. https://doi.org/10.1111/j.1476-5381.2012.02122.x
De Petrocellis L, Starowicz K, Moriello AS, Vivese M, Orlando P, Di Marzo V (2007) Regulation of transient receptor potential channels of melastatin type 8 (TRPM8): effect of cAMP, cannabinoid CB1 receptors and endovanilloids. Exp Cell Res 313(9):1911–1920. https://doi.org/10.1016/j.yexcr.2007.01.008
Deneyer L, Albertini G, Bentea E, Massie A (2019) Systemic LPS-induced neuroinflammation increases the susceptibility for proteasome inhibition-induced degeneration of the nigrostriatal pathway. Parkinsonism Relat Disord 68:26–32. https://doi.org/10.1016/j.parkreldis.2019.09.025
Deora V, Lee JD, Albornoz EA, McAlary L, Jagaraj CJ, Robertson AAB, Atkin JD, Cooper MA, Schroeder K, Yerbury JJ, Gordon R, Woodruff TM (2020) The microglial NLRP3 inflammasome is activated by amyotrophic lateral sclerosis proteins. Glia 68(2):407–421. https://doi.org/10.1002/glia.23728
Di Meco A, Curtis ME, Lauretti E, Praticò D (2020) Autophagy dysfunction in Alzheimer’s disease: mechanistic insights and new therapeutic opportunities. Biol Psychiatry 87(9):797–807. https://doi.org/10.1016/j.biopsych.2019.05.008
DiAntonio A, Haghighi AP, Portman SL, Lee JD, Amaranto AM, Goodman CS (2001) Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature 412(6845):449–452. https://doi.org/10.1038/35086595
Díaz-Alonso J, de Salas-Quiroga A, Paraíso-Luna J, García-Rincón D, Garcez PP, Parsons M, Andradas C, ZSánchez C, Guillemot F, Guzmán M, Galve-Roperh I, (2017) Loss of cannabinoid CB1 receptors induces cortical migration malformations and increases seizure susceptibility. Cereb Cortex 27(11):5303–5317. https://doi.org/10.1093/cercor/bhw309
Díaz-Alonso J, Paraíso-Luna J, Navarrete C, Del Río C, Cantarero I, Palomares B, Aguareles J, Fernández-Ruiz J, Bellido ML, Pollastro F, Appendino G, Calzado MA, Galve-Roperh I, Muñoz E (2016) VCE-003.2, a novel cannabigerol derivative, enhances neuronal progenitor cell survival and alleviates symptomatology in murine models of Huntington’s disease. Sci Rep 6:29789. https://doi.org/10.1038/srep29789
Díaz-Hernández M, Valera AG, Morán MA, Gómez-Ramos P, Alvarez-Castelao B, Castaño JG, Hernández F, Lucas JJ (2006) Inhibition of 26S proteasome activity by huntingtin filaments but not inclusion bodies isolated from mouse and human brain. J Neurochem 98(5):1585–1596. https://doi.org/10.1111/j.1471-4159.2006.03968.x
Djordjevic ANM, Kapetanou M, Loncarevic-Vasiljkovic N, Todorovic S, Athanasopoulou S, Jovic M, Gonos ES (2021) Pharmacological intervention in a transgenic mouse model improves Alzheimer’s-associated pathological phenotype: involvement of proteasome activation. Free Radic Biol Med 162:88–103. https://doi.org/10.1016/j.freeradbiomed.2020.11.038
Doble A (1999) The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol Ther 81(3):163–221. https://doi.org/10.1016/S0163-7258(98)00042-4
Dong C, Upadhya SC, Ding L, Smith TK, Hegde AN (2008) Proteasome inhibition enhances the induction and impairs the maintenance of late-phase long-term potentiation. Learn Mem 15(5):335–347. http://www.learnmem.org/cgi/doi/https://doi.org/10.1101/lm.984508
Drinjakovic J, Jung H, Campbell DS, Strochlic L, Dwivedy A, Holt CE (2010) E3 ligase Nedd4 promotes axon branching by downregulating PTEN. Neuron 65(3):341–357. https://doi.org/10.1016/j.neuron.2010.01.017
Du Y, Fu M, Huang Z, Tian X, Li J, Pang Y, Song W, Wang YT, Dong Z (2020) TRPV1 activation alleviates cognitive and synaptic plasticity impairments through inhibiting AMPAR endocytosis in APP23/PS45 mouse model of Alzheimer’s disease. Aging Cell 19(3):e13113. https://doi.org/10.1111/acel.13113
Duan C, Jiao D, Wang H, Wu Q, Men W, Yan H, Li C (2022) Activation of the PPARγ prevents ferroptosis-induced neuronal loss in response to intracerebral hemorrhage through synergistic actions with the Nrf2. Front Pharmacol 13:869300. https://doi.org/10.3389/fphar.2022.869300
Ehlinger A, Walters KJ (2013) Structural insights into proteasome activation by the 19S regulatory particle. Biochemistry 52(21):3618–3628. https://doi.org/10.1021/bi400417a
El-Khodor BF, Kholodilov NG, Yarygina O, Burke RE (2001) The expression of mRNAs for the proteasome complex is developmentally regulated in the rat mesencephalon. Brain Res Develop Brain Res 129(1):47–56. https://doi.org/10.1016/S0165-3806(01)00181-x
Elmazoglu Z, Rangel-López E, Medina-Campos ON, Pedraza-Chaverri J, Túnez I, Aschner M, Santamaría A, Karasu Ç (2020) Cannabinoid-profiled agents improve cell survival via reduction of oxidative stress and inflammation, and Nrf2 activation in a toxic model combining hyperglycemia+ Aβ1-42 peptide in rat hippocampal neurons. Neurochem Int 140:104817. https://doi.org/10.1016/j.neuint.2020.104817
Espejo-Porras F, García-Toscano L, Rodríguez-Cueto C, Santos-García I, de Lago E, Fernandez-Ruiz J (2019) Targeting glial cannabinoid CB2 receptors to delay the progression of the pathological phenotype in TDP-43 (A315T) transgenic mice, a model of amyotrophic lateral sclerosis. Br J Pharmacol 176(10):1585–1600. https://doi.org/10.1111/bph.14216
Esposito G, Scuderi C, Valenza M, Togna GI, Latina V, De Filippis D, Cipriano M, Carratù RM, Iuvone T, Steardo L (2011) Cannabidiol reduces Aβ-induced neuroinflammation and promotes hippocampal neurogenesis through PPARγ involvement. PLoS ONE 6(12):e28668. https://doi.org/10.1371/journal.pone.0028668
Fagan SG, Campbell VA (2014) The influence of cannabinoids on generic traits of neurodegeneration. Br J Pharmacol 171(6):1347–1360. https://doi.org/10.1111/bph.12492
Fakhfouri G, Ahmadiani A, Rahimian R, Grolla AA, Moradi F, Haeri A (2012) WIN55212-2 attenuates amyloid-beta-induced neuroinflammation in rats through activation of cannabinoid receptors and PPAR-γ pathway. Neuropharmacology 63(4):653–666. https://doi.org/10.1016/j.neuropharm.2012.05.013
Fonseca R, Vabulas RM, Hartl FU, Bonhoeffer T, Nägerl UV (2006) A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron 52(2):239–245. https://doi.org/10.1016/j.neuron.2006.08.015
Fu H, Hardy J, Duff KE (2018) Selective vulnerability in neurodegenerative diseases. Nat Neurosci 21(10):1350–1358. https://doi.org/10.1038/s41593-018-0221-2
Galán-Ganga M, Del Río R, Jiménez-Moreno N, Díaz-Guerra M, Lastres-Becker I (2020) Cannabinoid CB 2 receptor modulation by the transcription factor Nrf2 is specific in microglial cells. Cell Mol Neurobiol 40(1):167–177. https://doi.org/10.1007/s10571-019-00719-y
Groll M, Ditzel L, Löwe J, Stock D, Bochtler M, Bartunik HD, Huber R (1997) Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 386(6624):463–471. https://doi.org/10.1038/386463a0
Guo J, Ikeda SR (2004) Endocannabinoids modulate N-type calcium channels and G-protein-coupled inwardly rectifying potassium channels via CB1 cannabinoid receptors heterologously expressed in mammalian neurons. Mol Pharmacol 65(3):665–674. https://doi.org/10.1124/mol.65.3.665
Hamilton AM, Oh WC, Vega-Ramirez H, Stein IS, Hell JW, Patrick GN, Zito K (2012) Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron 74(6):1023–1030. https://doi.org/10.1016/j.neuron.2012.04.031
Hanslik KL, Ulland TK (2020) The role of microglia and the Nlrp3 inflammasome in Alzheimer’s disease. Front Neurol 11:570711. https://doi.org/10.3389/fneur.2020.570711
Haque ME, Akther M, Jakaria M, Kim I-S, Azam S, Choi D-K (2020) Targeting the microglial NLRP3 inflammasome and its role in Parkinson’s disease. Mov Disord 35(1):20–33. https://doi.org/10.1002/mds.27874
Hashem J, Hu M, Zhang J, Gao F, Chen C (2021) Inhibition of 2-arachidonoylglycerol metabolism alleviates neuropathology and improves cognitive function in a tau mouse model of Alzheimer’s disease. Mol Neurobiol 58(8):4122–4133. https://doi.org/10.1007/s12035-021-02400-2
Hayashi G, Jasoliya M, Sahdeo S, Saccà F, Pane C, Filla A, Marsili A, Puorro G, Lanzillo R, Morra VB, Cortopassi G (2017) Dimethyl fumarate mediates Nrf2-dependent mitochondrial biogenesis in mice and humans. Hum Mol Genet 26(15):2864–2873. https://doi.org/10.1093/hmg/ddx167
He D, Fu S, Zhou A, Su Y, Gao X, Zhang Y, Huang B, Du J, Liu D (2021a) Camptothecin regulates microglia polarization and exerts neuroprotective effects via activating AKT/Nrf2/HO-1 and inhibiting NF-κB pathways in vivo and in vitro. Front Immunol 12
He F, Ru X, Wen T (2020) Nrf2, a transcription factor for stress response and beyond. Int J Mol Sci 21(13):4777. https://doi.org/10.3390/ijms21134777
He Z, Li X, Han S, Ren B, Hu X, Li N, Du X, Ni J, Yankg X, Liu Q (2021) Bis(ethylmaltolato)oxidovanadium (IV) attenuates amyloid-beta-mediated neuroinflammation by inhibiting NF-κB signaling pathway via a PPARγ-dependent mechanism. Metallomics 13(7):mfab036. https://doi.org/10.1093/mtomcs/mfab036
Heurtaux T, Bouvier DS, Benani A, Helgueta Romero S, Frauenknecht KBM, Mittelbronn M, Sinkkonen L (2022) Normal and pathological Nrf2 signalling in the central nervous system. Antioxidants 11(8):1426. https://doi.org/10.3390/antiox11081426
Hill JD, Zuluaga-Ramirez V, Gajghate S, Winfield M, Persidsky Y (2018) Activation of GPR55 increases neural stem cell proliferation and promotes early adult hippocampal neurogenesis. Br J Pharmacol 175(16):3407–3421. https://doi.org/10.1111/bph.14387
Ho MS-C, Chen H, Chen M, Jacques C, Giangrande A, Chien C-T (2009) Gcm protein degradation suppresses proliferation of glial progenitors. Proc Natl Acad Sci 106(16):6778–6783. https://doi.org/10.1073/pnas.0808899106
Horne EA, Coy J, Swinney K, Fung S, Cherry AET, Marrs WR, Naydenov AV, Lin YH, Sun X, Keene CD, Grouzmann E, Muchowski P, Bates GP, Mackie K, Stella N (2013) Downregulation of cannabinoid receptor 1 from neuropeptide Y interneurons in the basal ganglia of patients with Huntington’s disease and mouse models. Eur J Neurosci 37(3):429–440. https://doi.org/10.1111/ejn.12045
Hou Y, Wang Y, He Q, Li L, Xie H, Zhao Y, Zhao J (2018) Nrf2 inhibits NLRP3 inflammasome activation through regulating Trx1/TXNIP complex in cerebral ischemia reperfusion injury. Behav Brain Res 336:32–39. https://doi.org/10.1016/j.bbr.2017.06.027
Hua T, Vemuri K, Pu M, Qu L, Han GW, Wu Y, Zhao S, Shui W, Li S, Korde A, Lapraire RB, Stahl EL, Ho J-H, Zvonok N, Zhou H, Kufareva I, Wu B, Zhao Q, Hanson MA, Bohn LM, Makriyannis A, Stevens RC, Liu Z-J (2016) Crystal structure of the human cannabinoid receptor CB1. Cell 167(3):750–762. https://doi.org/10.1016/j.cell.2016.10.004
Huang C-L, Yang J-M, Wang K-C, Lee Y-C, Lin Y-L, Yang Y-C, Huang N-K (2011) Gastrodia elata prevents huntingtin aggregations through activation of the adenosine A2A receptor and ubiquitin proteasome system. J Ethnopharmacol 138(1):162–168. https://doi.org/10.1016/j.jep.2011.08.075
Huang J, Tabbi-Anneni I, Gunda V, Wang L (2010) Transcription factor Nrf2 regulates SHP and lipogenic gene expression in hepatic lipid metabolism. Am J Physiol Gastrointest Liver Physiol 299(6):G1211–G1221. https://doi.org/10.1152/ajpgi.00322.2010
Hui Y, Chengyong T, Cheng L, Haixia H, Yuanda Z, Weihua Y (2018) Resveratrol attenuates the cytotoxicity induced by amyloid-β 1–42 in PC12 cells by upregulating heme oxygenase-1 via the PI3K/Akt/Nrf2 pathway. Neurochem Res 43(2):297–305. https://doi.org/10.1007/s11064-017-2421-7
Ibrahim WW, Abdel Rasheed NO (2022) Diapocynin neuroprotective effects in 3-nitropropionic acid Huntington’s disease model in rats: emphasis on Sirt1/Nrf2 signaling pathway. Inflammopharmacology 30(5):1745–1758. https://doi.org/10.1007/s10787-022-01004-z
Ishii T, Warabi E, Mann GE (2019) Circadian control of BDNF-mediated Nrf2 activation in astrocytes protects dopaminergic neurons from ferroptosis. Free Radic Biol Med 133:169–178. https://doi.org/10.1016/j.freeradbiomed.2018.09.002
Izumi Y (2013) Dopaminergic neuroprotection via Nrf2-ARE pathway activation: identification of an activator from green perilla leaves. Yakugaku Zasshi 133(9):983–988. https://doi.org/10.1248/yakushi.13-00166
Izumi Y, Yamamoto N, Matsushima S, Yamamoto T, Takada-Takatori Y, Akaike A, Kume T (2015) Compensatory role of the Nrf2-ARE pathway against paraquat toxicity: relevance of 26S proteasome activity. J Pharmacol Sci 129(3):150–159. https://doi.org/10.1016/j.jphs.2015.09.003
Javed H, Azimullah S, Haque ME, Ojha SK (2016) Cannabinoid type 2 (CB2) receptors activation protects against oxidative stress and neuroinflammation associated dopaminergic neurodegeneration in rotenone model of Parkinson’s disease. Front Neurosci 10:321. https://doi.org/10.3389/fnins.2016.00321
Jellinger KA (2003) General aspects of neurodegeneration. J Neural Transm Suppl 2003(65):101–144. https://doi.org/10.1007/978-3-7091-0643-3_7
Jeon J, Kim W, Jang J, Isacson O, Seo H (2016) Gene therapy by proteasome activator, PA28γ, improves motor coordination and proteasome function in Huntington’s disease YAC128 mice. Neuroscience 324:20–28. https://doi.org/10.1016/j.neuroscience.2016.02.054
Jin K, Xie L, Kim SH, Parmentier-Batteur S, Sun Y, Mao XO, Childs J, Greenberg DA (2004) Defective adult neurogenesis in CB1 cannabinoid receptor knockout mice. Mol Pharmacol 66(2):204–208. https://doi.org/10.1124/mol.66.2.204
Jo MG, Ikram M, Jo MH, Yoo L, Chung KC, Nah S-Y, Hwang H, Rhim H, Kim MO (2019) Gintonin mitigates MPTP-induced loss of nigrostriatal dopaminergic neurons and accumulation of α-synuclein via the Nrf2/HO-1 pathway. Mol Neurobiol 56(1):39–55. https://doi.org/10.1007/s12035-018-1020-1
Jordan JD, He JC, Eungdamrong NJ, Gomes I, Ali W, Nguyen T, Bivona TG, Phillips MR, Devi LA, Iyengar R (2005) Cannabinoid receptor-induced neurite outgrowth is mediated by Rap1 activation through Gαo/i-triggered proteasomal degradation of Rap1GAPII. J Biol Chem 280(12):11413–11421. https://doi.org/10.1074/jbc.M411521200
Juknat A, Pietr M, Kozela E, Rimmerman N, Levy R, Gao F, Coppola G, Geschwind D, Vogel Z (2013) Microarray and pathway analysis reveal distinct mechanisms underlying cannabinoid-mediated modulation of LPS-induced activation of BV-2 microglial cells. PLoS ONE 8(4):e61462. https://doi.org/10.1371/journal.pone.0061462
Kapeta S, Chondrogianni N, Gonos ES (2010) Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts. J Biol Chem 285(11):8171–8184. https://doi.org/10.1074/jbc.M109.031575
Katoh Y, Itoh K, Yoshida E, Miyagishi M, Fukamizu A, Yamamoto M (2001) Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 6(10):857–868. https://doi.org/10.1046/j.1365-2443.2001.00469.x
Kerins MJ, Ooi A (2018) The roles of Nrf2 in modulating cellular iron homeostasis. Antiox Redox Signal 29(17):1756–1773. https://doi.org/10.1089/ars.2017.7176
Khalifeh S, Khodagholi F, Shaerzadeh F, Ghazvini H, Zarrindast MR, Azizi V (2017) Brain region specificity of mitochondrial biogenesis and bioenergetics response to Nrf2 knockdown: a comparison among hippocampus, prefrontal cortex and amygdala of male rat brain. Braz Arch Biol Technol 60. https://doi.org/10.1590/1678-4324-2017160744
Kikuchi S, Shinpo K, Tsuji S, Takeuchi M, Yamagishi S, Makita Z, Niino M, Yabe I, Tashiro K (2003) Effect of proteasome inhibitor on cultured mesencephalic dopaminergic neurons. Brain Res 964(2):228–236. https://doi.org/10.1016/S0006-8993(02)04030-1
Kim BH, Kwon J, Lee D, Mar W (2015) Neuroprotective effect of demethylsuberosin, a proteasome activator, against MPP+-induced cell death in human neuroblastoma SH-SY5Y Cells. Planta Med Lett 2(1):e15–e18. https://doi.org/10.1055/s-0035-1545936
Kim J (2021) Pre-clinical neuroprotective evidences and plausible mechanisms of Sulforaphane in Alzheimer’s disease. Int J Mol Sci 22(6):2929. https://doi.org/10.3390/ijms22062929
Kim S, Viswanath ANI, Park J-H, Lee HE, Park AY, Choi JW, Kim HJ, Londhe AM, Jang BK, Lee J, Hwang H, Lim SM, Pae AN, Park KD (2020) Nrf2 activator via interference of Nrf2-Keap1 interaction has antioxidant and anti-inflammatory properties in Parkinson’s disease animal model. Neuropharmacology 167:107989. https://doi.org/10.1016/j.neuropharm.2020.107989
Kim W, Seo H (2014) Baclofen, a GABAB receptor agonist, enhances ubiquitin-proteasome system functioning and neuronal survival in Huntington’s disease model mice. Biochem Biophys Res Commun 443(2):706–711. https://doi.org/10.1016/j.bbrc.2013.12.034
Kleiger G, Mayor T (2014) Perilous journey: a tour of the ubiquitin-proteasome system. Trends Cell Biol 24(6):352–359. https://doi.org/10.1016/j.tcb.2013.12.003
Kohno M, Hasegawa H, Inoue A, Muraoka M, Miyazaki T, Oka K, Yasukawa M (2006) Identification of N-arachidonylglycine as the endogenous ligand for orphan G-protein-coupled receptor GPR18. Biochem Biophys Res Commun 347(3):827–832. https://doi.org/10.1016/j.bbrc.2006.06.175
Komander D, Rape M (2012) The ubiquitin code. Annu Rev Biochem 81:203–229. https://doi.org/10.1146/annurev-biochem-060310-170328
Komorowska-Müller JA, Schmöle AC (2020) CB2 receptor in microglia: the guardian of self-control. Int J Mol Sci 22(1):19. https://doi.org/10.3390/ijms22010019
Konishi Y, Stegmuller J, Matsuda T, Bonni S, Bonni A (2004) Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science 303(5660):1026–1030. https://doi.org/10.1126/science.1093712
Kopp F, Hendil KB, Dahlmann B, Kristensen P, Sobek A, Uerkvitz W (1997) Subunit arrangement in the human 20S proteasome. Proc Natl Acad Sci 94(7):2939–2944. https://doi.org/10.1073/pnas.94.7.2939
Kozela E, Pietr M, Juknat A, Rimmerman N, Levy R, Vogel Z (2010) Cannabinoids Δ9-tetrahydrocannabinol and cannabidiol differentially inhibit the lipopolysaccharide-activated NF-κB and interferon-β/STAT proinflammatory pathways in BV-2 microglial cells. J Biol Chem 285(3):1616–1626. https://doi.org/10.1074/jbc.M109.069294
Kwak MK, Cho JM, Huang B, Shin S, Kensler TW (2007) Role of increased expression of the proteasome in the protective effects of sulforaphane against hydrogen peroxide-mediated cytotoxicity in murine neuroblastoma cells. Free Radic Biol Med 43(5):809–817. https://doi.org/10.1016/j.freeradbiomed.2007.05.029
Kwak M-K, Wakabayashi N, Greenlaw JL, Yamamoto M, Kensler TW (2003) Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol Cell Biol 23(23):8786–8794. https://doi.org/10.1128/MCB.23.23.8786-8794.2003
Lastres-Becker I, Ulusoy A, Innamorato NG, Sahin G, Rábano A, Kirik D, Cuadrado A (2012) α-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson’s disease. Hum Mol Genet 21(14):3173–3192. https://doi.org/10.1093/hmg/dds143
Lee C (2017) Collaborative power of Nrf2 and PPARγ activators against metabolic and drug-induced oxidative injury. Oxid Med Cell Longev 2017:1378175. https://doi.org/10.1155/2017/1378175
Lee JH, Agacinski G, Williams JH, Wilcock GK, Esiri MM, Francis PT, Wong PT-H, Chen CP, Lai MKP (2010) Intact cannabinoid CB1 receptors in the Alzheimer’s disease cortex. Neurochem Int 57(8):985–989. https://doi.org/10.1016/j.neuint.2010.10.010
Lee S, Hur E-G, Ryoo I-G, Jung K-A, Kwak J, Kwak M-K (2012) Involvement of the Nrf2-proteasome pathway in the endoplasmic reticulum stress response in pancreatic β-cells. Toxicol Appl Pharmacol 264(3):431–438. https://doi.org/10.1016/j.taap.2012.08.021
Lewcock JW, Genoud N, Lettieri K, Pfaff SL (2007) The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron 56(4):604–620. https://doi.org/10.1016/j.neuron.2007.09.009
Li H, Wood JT, Whitten KM, Vadivel SK, Seng S, Makriyannis A, Avraham HK (2013) Inhibition of fatty acid amide hydrolase activates Nrf2 signalling and induces heme oxygenase 1 transcription in breast cancer cells. Br J Pharmacol 170(3):489–505. https://doi.org/10.1111/bph.12111
Li J, Johnson D, Calkins M, Wright L, Svendsen C, Johnson J (2005) Stabilization of Nrf2 by tBHQ confers protection against oxidative stress-induced cell death in human neural stem cells. Toxicol Sci 83(2):313–328. https://doi.org/10.1093/toxsci/kfi027
Li L, Li W-J, Zhen X-R, Liu Q-L, Du Q, Lai Y-J, Liu S-Q (2022) Eriodictyol ameliorates cognitive dysfunction in APP/PS1 mice by inhibiting ferroptosis via vitamin D receptor-mediated Nrf2 activation. Mol Med 28(1):11. https://doi.org/10.1186/s10020-022-00442-3
Li Q, Tian Z, Wang M, Kou J, Wang C, Rong X, Li J, Xie X, Pang X (2019a) Luteoloside attenuates neuroinflammation in focal cerebral ischemia in rats via regulation of the PPARγ/Nrf2/NF-κB signaling pathway. Int Immunopharmacol 66:309–316. https://doi.org/10.1016/j.intimp.2018.11.044
Li X, Han D, Tian Z, Gao B, Fan M, Li C, Li X, Wang Y, Ma S, Cao F (2016) Activation of cannabinoid receptor type II by AM1241 ameliorates myocardial fibrosis via Nrf2-mediated inhibition of TGF-β1/Smad3 pathway in myocardial infarction mice. Cell Physiol Biochem 39(4):1521–1536. https://doi.org/10.1159/000447855
Li X, Hua T, Vemuri K, Ho J-H, Wu Y, Wu L, Popov P, Benchama O, Zvonok N, Locke K, Qu L, Han GW, Iyer MR, Cinar R, Coffey NJ, Wang J, Wu M, Katritch V, Zhao S, Kunos G, Bohn LM, Makriyannis A, Stevens RC, Liu Z-J (2019b) Crystal structure of the human cannabinoid receptor CB2. Cell 176(3):459–467. https://doi.org/10.1016/j.cell.2018.12.011
Li X-H, Li C-Y, Lu J-M, Tian R-B, Wei J (2012) Allicin ameliorates cognitive deficits ageing-induced learning and memory deficits through enhancing of Nrf2 antioxidant signaling pathways. Neurosci Lett 514(1):46–50. https://doi.org/10.1016/j.neulet.2012.02.054
Lin C-C, Yang C-C, Chen Y-W, Hsiao L-D, Yang C-M (2018) Arachidonic acid induces ARE/Nrf2-dependent heme oxygenase-1 transcription in rat brain astrocytes. Mol Neurobiol 55(4):3328–3343. https://doi.org/10.1007/s12035-017-0590-7
Liu H, Zheng Q, Yuan J, Gao Y, Wang T, Zhang H, Li Z (2023) Modulating SQSTM1/p62-dependent selective autophagy of neurons by activating Nrf2 with multifunctional nanoparticles to eliminate α-synuclein aggregates and boost therapy of Parkinson’s disease. NanoToday 49:101770. https://doi.org/10.1016/j.nantod.2023.101770
Liu X, Wang Y, Wu D, Li S, Wang C, Han Z, Wang J, Wang K, Yang Z, Wei Z (2019) Magnolol prevents acute alcoholic liver damage by activating PI3K/Nrf2/PPARγ and inhibiting NLRP3 signaling pathway. Front Pharmacol 10:1459. https://doi.org/10.3389/fphar.2019.01459
Lu H-C, Mackie K (2021) Review of the endocannabinoid system. Biol Psychiatry Cogn Neurosci Neuroimaging 6(6):607–615. https://doi.org/10.1016/j.bpsc.2020.07.016
Lubelska K, Wiktorska K, Mielczarek L, Milczarek M, Zbroińska-Bregisz I, Chilmonczyk Z (2016) Sulforaphane regulates NFE2L2/Nrf2-dependent xenobiotic metabolism phase II and phase III enzymes differently in human colorectal cancer and untransformed epithelial colon cells. Nutr Cancer 68(8):1338–1348. https://doi.org/10.1080/01635581.2016.1224369
Lv Z, Xu X, Sun Z, Yang YX, Guo H, Li J, Sun K, Wu R, Xu J, Jiang Q, Ikegawa S, Shi D (2021) TRPV1 alleviates osteoarthritis by inhibiting M1 macrophage polarization via Ca2+/CaMKII/Nrf2 signaling pathway. Cell Death Dis 12(6):504. https://doi.org/10.1038/s41419-021-03792-8
Ma Q (2013) Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol 53:401–426. https://doi.org/10.1146/annurev-pharmtox-011112-140320
Maejima T, Ohno-Shosaku T, Kano M (2001) Endogenous cannabinoid as a retrograde messenger from depolarized postsynaptic neurons to presynaptic terminals. Neurosci Res 40(3):205–210. https://doi.org/10.1016/S0168-0102(01)00241-3
Mageed SSA, Ammar RM, Nassar NN, Moawad H, Kamel AS (2022) Role of PI3K/Akt axis in mitigating hippocampal ischemia-reperfusion injury via CB1 receptor stimulation by paracetamol and FAAH inhibitor in rat. Neuropharmacology 207:108935. https://doi.org/10.1016/j.neuropharm.2021.108935
Malhotra D, Thimmulappa R, Vij N, Navas-Acien A, Sussan T, Merali S, Zhang L, Kelsen SG, Myers A, Wise R, Tuder R, Biswal S (2009) Heightened endoplasmic reticulum stress in the lungs of patients with chronic obstructive pulmonary disease: the role of Nrf2-regulated proteasomal activity. Am J Respir Crit Care Med 180(12):1196–1207. https://doi.org/10.1164/rccm.200903-0324OC
Mead RJ, Higginbottom A, Allen SP, Kirby J, Bennett E, Barber SC, Heath PR, Coluccia A, Patel N, Gardner I, Brancale A, Grierson AJ, Shaw PJ (2013) S[+]Apomorphine is a CNS penetrating activator of the Nrf2-ARE pathway with activity in mouse and patient fibroblast models of amyotrophic lateral sclerosis. Free Radic Biol Med 61:438–452. https://doi.org/10.1016/j.freeradbiomed.2013.04.018
Mimura J, Kosaka K, Maruyama A, Satoh T, Harada N, Yoshida H, Satoh K, Yamamoto M, Itoh K (2011) Nrf2 regulates NGF mRNA induction by carnosic acid in T98G glioblastoma cells and normal human astrocytes. J Biochem 150(2):209–217. https://doi.org/10.1093/jb/mvr065
Mnich K, Finn DP, Dowd E, Gorman AM (2010) Inhibition by anandamide of 6-hydroxydopamine-induced cell death in PC12 cells. Int J Cell Biol 2010:818497. https://doi.org/10.1155/2010/818497
Monsalvo-Maraver LÁ, Maya-López M, Rangel-López E, Túnez I, Tinkov AA, Skalny A, Ferrer B, Aschner M, Santamaría A (2023) Amyloid beta peptide-mediated alterations in mitochondrial dynamics and its implications for Alzheimer’s disease. CNS Neurol Disord Drug Targets 22(7):1039–1056. https://doi.org/10.2174/1871527321666220616094036
Moscovitz O, Ben-Nissan G, Fainer I, Pollack D, Mizrachi L, Sharon M (2015) The Parkinson’s-associated protein DJ-1 regulates the 20S proteasome. Nat Commun 6:6609. https://doi.org/10.1038/ncomms7609
Mounsey RB, Mustafa S, Robinson L, Ross RA, Riedel G, Pertwee RG, Teismann P (2015) Increasing levels of the endocannabinoid 2-AG is neuroprotective in the 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine mouse model of Parkinson’s disease. Exp Neurol 273:36–44. https://doi.org/10.1016/j.expneurol.2015.07.024
Muhammad F, Liu Y, Wang N, Zhao L, Zhou Y, Yang H, Li H (2022) Neuroprotective effects of cannabidiol on dopaminergic neurodegeneration and α-synuclein accumulation in C. elegans models of Parkinson’s disease. Neurotoxicology 93:128–139. https://doi.org/10.1016/j.neuro.2022.09.001
Navarrete F, García-Gutiérrez MS, Aracil-Fernández A, Lanciego JL, Manzanares J (2018) Cannabinoid CB1 and CB2 receptors, and monoacylglycerol lipase gene expression alterations in the basal ganglia of patients with Parkinson’s disease. Neurotherapeutics 15(2):459–469. https://doi.org/10.1007/s13311-018-0603-x
Oliveira AM, Cardoso SM, Ribeiro M, Seixas RSGR, Silva AMS, Rego AC (2015) Protective effects of 3-alkyl luteolin derivatives are mediated by Nrf2 transcriptional activity and decreased oxidative stress in Huntington’s disease mouse striatal cells. Neurochem Int 91:1–12. https://doi.org/10.1016/j.neuint.2015.10.004
Opattova A, Filipcik P, Cente M, Novak M (2013) Intracellular degradation of misfolded tau protein induced by geldanamycin is associated with activation of proteasome. J Alzheimer Dis 33(2):339–348. https://doi.org/10.3233/JAD-2012-121072
O’Sullivan SE (2016) An update on PPAR activation by cannabinoids. Br J Pharmacol 173(12):1899–1910. https://doi.org/10.1111/bph.13497
Overton HA, Babbs AJ, Doel SM, Fyfe MCT, Gardner LS, Griffin G, Jackson HC, Procter MJ, Rasamison CM, Tang-Christensen M, Widdowson PS, Williams GM, Reynet C (2006) Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab 3(3):167–175. https://doi.org)https://doi.org/10.1016/j.cmet.2006.02.004
Pajares M, Rojo AI, Arias E, Díaz-Carretero A, Cuervo AM, Cuadrado A (2018) Transcription factor NFE2L2/NRF2 modulates chaperone-mediated autophagy through the regulation of LAMP2A. Autophagy 14(8):1310–1322. https://doi.org/10.1080/15548627.2018.1474992
Palazuelos J, Aguado T, Pazos MR, Julien B, Carrasco C, Resel E, Sagredo O, Benito C, Romero J, Azcoitia I, Fernández-Ruiz J, Guzmán M, Galve-Roperh I (2009) Microglial CB2 cannabinoid receptors are neuroprotective in Huntington’s disease excitotoxicity. Brain 132(Pt 11):3152–3164. https://doi.org/10.1093/brain/awp239
Papa L, Gomes E, Rockwell P (2007) Reactive oxygen species induced by proteasome inhibition in neuronal cells mediate mitochondrial dysfunction and a caspase-independent cell death. Apoptosis 12(8):1389–1405. https://doi.org/10.1007/s10495-007-0069-5
Papaevgeniou N, Sakellari M, Jha S, Tavernarakis N, Holmberg CI, Gonos ES, Chondrogianni N (2016) 18α-Glycyrrhetinic acid proteasome activator decelerates aging and Alzheimer’s disease progression in Caenorhabditis elegans and neuronal cultures. Antioxidants Redox Signaling 25(16):855–869. https://doi.org/10.1089/ars.2015.6494
Paraíso-Luna J, Aguareles J, Martín R, Ayo-Martín AC, Simón-Sánchez S, García-Rincón D, Costas-Insua C, García-Taboada E, de Salas-Quiroga A, Díaz-Alonso J, Liste I, Sánchez-Prieto J, Capello S, Guzmán M, Galve-Roperh I (2020) Endocannabinoid signalling in stem cells and cerebral organoids drives differentiation to deep layer projection neurons via CB1 receptors. Development 147(24):dev192161. https://doi.org/10.1242/dev.192161
Park H-M, Kim J-A, Kwak M-K (2009) Protection against amyloid beta cytotoxicity by sulforaphane: role of the proteasome. Arch Pharm Res 32(1):109–115. https://doi.org/10.1007/s12272-009-1124-2
Pasquini LA, Paez PM, Moreno MANB, Pasquini JM, Soto EF (2003) Inhibition of the proteasome by lactacystin enhances oligodendroglial cell differentiation. J Neurosci 23(11):4635–4644. https://doi.org/10.1523/JNEUROSCI.23-11-04635.2003
Picca A, Calvani R, Coelho-Junior HJ, Landi F, Bernabei R, Marzetti E (2020) Mitochondrial dysfunction, oxidative stress, and neuroinflammation: intertwined roads to neurodegeneration. Antioxidants (basel) 9(8):647. https://doi.org/10.3390/antiox9080647
Psychoyos D, Vinod KY, Cao J, Xie S, Hyson RL, Wlodarczyk B, He W, Cooper TB, Hungund BL, Finnell RH (2012) Cannabinoid receptor 1 signaling in embryo neurodevelopment. Birth Defects Res B Dev Reprod Toxicol 95(2):137–150. https://doi.org/10.1002/bdrb.20348
Quinti L, Dayalan Naidu S, Träger U, Chen X, Kegel-Gleason K, Llères D, Connolly C, Chopra V, Low C, Moniot S, Sapp E, Tousley AR, Vodicka P, Van Kanegan MJ, Kaltenbach MS, Crawford LA, Fuszard M, Higgins M, Miller JRC, Farmer RE, Potluri V, Samajdar S, Meisel L, Zhang N, Snyder A, Stein R, Hersch SM, Ellerby LM, Weerapana E, Schwarzschild MA, Steegborn C, Leavitt BR, Degterev A, Trabizi SJ, Lo DC, DiFligia M, Thompson LM, Dinkova-Kostova AT, Kazantsev AG (2017) KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc Natl Acad Sci USA 114(23):E4676–E4685. https://doi.org/10.1073/pnas.161494311
Raboune S, Stuart JM, Leishman E, Takacs SM, Rhodes B, Basnet A, Jameyfield E, McHugh D, Widlanski T, Bradshaw HB (2014) Novel endogenous N-acyl amides activate TRPV1-4 receptors, BV-2 microglia, and are regulated in brain in an acute model of inflammation. Front Cell Neurosci 8:195. https://doi.org/10.3389/fncel.2014.00195
Raffaele S, Boccazzi M, Fumagalli M (2021) Oligodendrocyte dysfunction in amyotrophic lateral sclerosis: mechanisms and therapeutic perspectives. Cells 10(3):565. https://doi.org/10.3390/cells10030565
Ramírez BG, Blázquez C, Del Pulgar TG, Guzmán M, de Ceballos ML (2005) Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci 25(8):1904–1913. https://doi.org/10.1523/JNEUROSCI.4540-04.2005
Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, Hamilton RL, Chu CT, Jordan-Sciutto KL (2007) Expression of Nrf2 in neurodegenerative diseases. J Neuropathol Exp Neurol 66(1):75–85. https://doi.org/10.1097/nen.0b013e31802d6da9
Ray S, Corenblum MJ, Anandhan A, Reed A, Ortiz FO, Zhang DD, Barnes CA, Madhavan L (2018) A role for Nrf2 expression in defining the aging of hippocampal neural stem cells. Cell Transplant 27(4):589–606. https://doi.org/10.1177/0963689718774030
Redmond WJ, Gu L, Camo M, McIntyre P, Connor M (2014) Ligand determinants of fatty acid activation of the pronociceptive ion channel TRPA1. Peer J 2:e248. https://doi.org/10.7717/peerj.248
Robledinos-Antón N, Rojo AI, Ferreiro E, Núñez Á, Krause K-H, Jaquet V, Cuadrado A (2017) Transcription factor NRF2 controls the fate of neural stem cells in the subgranular zone of the hippocampus. Redox Biol 13:393–401. https://doi.org/10.1016/j.redox.2017.06.010
Rodrigues RS, Ribeiro FF, Ferreira F, Vaz SH, Sebastião AM, Xapelli S (2017) Interaction between cannabinoid type 1 and type 2 receptors in the modulation of subventricular zone and dentate gyrus neurogenesis. Front Pharmacol 8:516. https://doi.org/10.3389/fphar.2017.00516
Rodríguez-Cueto C, Santos-García I, García-Toscano L, Espejo-Porras F, Bellido ML, Fernández-Ruiz J, Muñoz E, de Lago E (2018) Neuroprotective effects of the cannabigerol quinone derivative VCE-003.2 in SOD1G93A transgenic mice, an experimental model of amyotrophic lateral sclerosis. Biochem Pharmacol 157:217–226. https://doi.org/10.1016/j.bcp.2018.07.049
Ross HR, Gilmore AJ, Connor M (2009) Inhibition of human recombinant T-type calcium channels by the endocannabinoid N-arachidonoyl dopamine. Br J Pharmacol 156(5):740–750. https://doi.org/10.1111/j.1476-5381.2008.00072.x
Rouzer CA, Marnett LJ (2011) Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem Rev 111(10):5899–5921. https://doi.org/10.1021/cr2002799
Ruiz-Pérez G, de Martín R, Esteban S, Marqués S, Aparicio N, Grande MT, Benito-Cuesta I, Martínez-Relimpio AM, Arnanz MA, Tolón RM, Posada-Ayala M, Cravatt BF, Esteban JA, Romero J, Palenzuela R (2021) Potentiation of amyloid beta phagocytosis and amelioration of synaptic dysfunction upon FAAH deletion in a mouse model of Alzheimer’s disease. J Neuroinflammation 18(1):223. https://doi.org/10.1186/s12974-021-02276-y
Sagredo O, González S, Aroyo I, Pazos MR, Benito C, Lastres-Becker I, Romero JP, Tolón RM, Mechoulam R, Brouillet E, Romero J, Fernández-Ruiz J (2009) Cannabinoid CB2 receptor agonists protect the striatum against malonate toxicity: relevance for Huntington’s disease. Glia 57(11):1154–1167. https://doi.org/10.1002/glia.20838
Sahu I, Nanaware P, Mane M, Mulla SW, Roy S, Venkatraman P (2019) Role of a 19S proteasome subunit-PSMD10Gankyrin in neurogenesis of human neural progenitor cells. Int J Stem Cells 12(3):463–473. https://doi.org/10.15283/ijsc19007
Salgado-Mendialdúa V, Aguirre-Plans J, Guney E, Reig-Viader R, Maldonado R, Bayés À, Oliva B, Ozaita A (2018) Δ9-tetrahydrocannabinol modulates the proteasome system in the brain. Biochem Pharmacol 157:159–168. https://doi.org/10.1016/j.bcp.2018.08.026
Sánchez-de la Torre A, Aguado T, Huerga-Gómez A, Santamaría S, Gentile A, Chara JC, Matute C, Monory K, Mato S, Guzmán M, Lutz B, Galve-Roperh I, Palazuelos J (2022) Cannabinoid CB1 receptor gene inactivation in oligodendrocyte precursors disrupts oligodendrogenesis and myelination in mice. Cell Death Dis 13(7):585. https://doi.org/10.1038/s41419-022-05032-z
Santos AR, Mele M, Vaz SH, Kellermayer B, Grimaldi M, Colino-Oliveira M, Rombo DM, Comprido D, Sebastião AM, Duarte CB (2015) Differential role of the proteasome in the early and late phases of BDNF-induced facilitation of LTP. J Neurosci 35(8):3319–3329. https://doi.org/10.1523/JNEUROSCI.4521-14.2015
Sarlette A, Krampfl K, Grothe C, von Neuhoff N, Dengler R, Petri S (2008) Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 67(11):1055–1062. https://doi.org/10.1097/NEN.0b013e31818b4906
Schaefer AM, Hadwiger GD, Nonet ML (2000) rpm-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron 26(2):345–356. https://doi.org/10.1016/S0896-6273(00)81168-x
Schweitzer A, Aufderheide A, Rudack T, Beck F, Pfeifer G, Plitzko JM, Sakata E, Schulten K, Förster F, Baumeister W (2016) Structure of the human 26S proteasome at a resolution of 3.9 Å. Proc Natl Acad Sci USA 113(28):7816–7821. https://doi.org/10.1073/pnas.1608050113
Seo H, Sonntag KC, Kim W, Cattaneo E, Isacson O (2007) Proteasome activator enhances survival of Huntington’s disease neuronal model cells. PLoS ONE 2(2):e238. https://doi.org/10.1371/journal.pone.0000238
Sharir H, Console-Bram L, Mundy C, Popoff SN, Kapur A, Abood ME (2012) The endocannabinoids anandamide and virodhamine modulate the activity of the candidate cannabinoid receptor GPR55. J Neuroimmune Pharmacol 7(4):856–865. https://doi.org/10.1007/s11481-012-9351-6
Shimizu Y, Nicholson CK, Lambert JP, Barr LA, Kuek N, Herszenhaut D, Tan L, Murohara T, Hansen JM, Husain A, Naqvi N, Calvert JW (2016) Sodium sulfide attenuates ischemic-induced heart failure by enhancing proteasomal function in an Nrf2-dependent manner. Circ Heart Fail 9(4):e002368. https://doi.org/10.1161/CIRCHEARTFAILURE.115.002368
Shin JE, DiAntonio A (2011) Highwire regulates guidance of sister axons in the Drosophila mushroom body. J Neurosci 31(48):17689–17700. https://doi.org/10.1523/JNEUROSCI.3902-11.2011
Singh A, Rangasamy T, Thimmulappa RK, Lee H, Osburn WO, Brigelius-Flohé R, Kensler TW, Yamamoto M, Biswal S (2006) Glutathione peroxidase 2, the major cigarette smoke–inducible isoform of GPX in lungs, is regulated by Nrf2. Am J Respir Cell Mol Biol 35(6):639–650. https://doi.org/10.1165/rcmb.2005-0325OC
Singh SJ, Tandon A, Phoolmala ST, Singh N, Goyal S, Priya S, Chaturvedi RK (2023) Bisphenol-A (BPA) impairs hippocampal neurogenesis via inhibiting regulation of the ubiquitin proteasomal system. Mol Neurobiol 60(6):3277–3298. https://doi.org/10.1007/s12035-023-03249-3
Song M-Y, Lee D-Y, Chun K-S, Kim E-H (2021) The role of NRF2/KEAP1 signaling pathway in cancer metabolism. Int J Mol Sci 22(9):4376. https://doi.org/10.3390/ijms22094376
Tadijan A, Vlašić I, Vlainić J, Đikić D, Oršolić N, Jazvinšćak Jembrek M (2022) Intracellular molecular targets and signaling pathways involved in antioxidative and neuroprotective effects of cannabinoids in neurodegenerative conditions. Antioxidants (basel) 11(10):2049. https://doi.org/10.3390/antiox11102049
Takalo M, Salminen A, Soininen H, Hiltunen M, Haapasalo A (2013) Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am J Neurodegener Dis 2(1):1–14. PMID: PMC3601466
Takenouchi T, Hashimoto M, Hsu LJ, Mackowski B, Rockenstein E, Mallory M, Masliah E (2001) Reduced neuritic outgrowth and cell adhesion in neuronal cells transfected with human alpha-synuclein. Mol Cell Neurosci 17(1):141–150. https://doi.org/10.1006/mcne.2000.0923
Tan H, Lauzon NM, Bishop SF, Chi N, Bechard M, Laviolette SR (2011) Cannabinoid transmission in the basolateral amygdala modulates fear memory formation via functional inputs to the prelimbic cortex. J Neurosci 31(14):5300–5312. https://doi.org/10.1523/JNEUROSCI.4718-10.2011
Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa A, Dawson VL, Dawson TM, Ross CA (2001) Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet 10(9):919–926. https://doi.org/10.1093/hmg/10.9.919
Tang D, Chen X, Kang R, Kroemer G (2021) Ferroptosis: molecular mechanisms and health implications. Cell Res 31(2):107–125. https://doi.org/10.1038/s41422-020-00441-1
Tanito M, Agbaga M-P, Anderson RE (2007) Upregulation of thioredoxin system via Nrf2-antioxidant responsive element pathway in adaptive-retinal neuroprotection in vivo and in vitro. Free Radic Biol Med 42(12):1838–1850. https://doi.org/10.1016/j.freeradbiomed.2007.03.018
Tao W, Hu Y, Chen Z, Dai Y, Hu Y, Qi M (2021) Magnolol attenuates depressive-like behaviors by polarizing microglia towards the M2 phenotype through the regulation of Nrf2/HO-1/NLRP3 signaling pathway. Phytomedicine 91:153692. https://doi.org/10.1016/j.phymed.2021.153692
Testa G, Giannelli S, Sottero B, Staurenghi E, Giaccone G, Caroppo P, Gamba P, Leonarduzzi G (2023) 24-Hydroxycholesterol induces Tau proteasome-dependent degradation via the SIRT1/PGC1α/Nrf2 pathway: a potential mechanism to counteract Alzheimer’s disease. Antioxidants (basel) 12(3):631. https://doi.org/10.3390/antiox12030631
Thibaudeau TA, Anderson RT, Smith DM (2018) A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat Commun 9(1):1097. https://doi.org/10.1038/s41467-018-03509-0
Tonelli C, Chio IIC, Tuveson DA (2018) Transcriptional regulation by Nrf2. Antioxid Redox Signal 29(17):1727–1745. https://doi.org/10.1089/ars.2017.7342
Tsakiri EN, Sykiotis GP, Papassideri IS, Terpos E, Dimopoulos MA, Gorgoulis VG, Bohmann D, Trougakos IP (2013) Proteasome dysfunction in Drosophila signals to an Nrf2-dependent regulatory circuit aiming to restore proteostasis and prevent premature aging. Aging Cell 12(5):802–813. https://doi.org/10.1111/acel.12111
Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM (2008) Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging 29(11):1607–1618. https://doi.org/10.1016/j.neurobiolaging.2007.04.014
Ugun-Klusek A, Tatham MH, Elkharaz J, Constantin-Teodosiu D, Lawler K, Mohamed H, Paine SML, Anderson G, Mayer RH, Lower J, Billet EE, Bedford L (2017) Continued 26S proteasome dysfunction in mouse brain cortical neurons impairs autophagy and the Keap1-Nrf2 oxidative defence pathway. Cell Death Dis 8(1):e2531–e2531. https://doi.org/10.1038/cddis.2016.443
Valdeolivas S, Pazos MR, Bisogno T, Piscitelli F, Iannotti FA, Allarà M, Sagredo O, Di Marzo V, Fernández-Ruiz J (2013) The inhibition of 2-arachidonoyl-glycerol (2-AG) biosynthesis, rather than enhancing striatal damage, protects striatal neurons from malonate-induced death: a potential role of cyclooxygenase-2-dependent metabolism of 2-AG. Cell Death Dis 4(10):e862–e862. https://doi.org/10.1038/cddis.2013.387
Van Laere K, Casteels C, Dhollander I, Goffin K, Grachev I, Bormans G, Vandenberghe W (2010) Widespread decrease of type 1 cannabinoid receptor availability in Huntington disease in vivo. J Nucl Med 51(9):1413–1417. https://doi.org/10.2967/jnumed.110.077156
van Muiswinkel FL, Kuiperij HB (2005) The Nrf2-ARE signalling pathway: promising drug target to combat oxidative stress in neurodegenerative disorders. Curr Drug Targets CNS Neurol Disord 4(3):267–281. https://doi.org/10.2174/1568007054038238
Vasilopoulou MA, Gioran A, Theodoropoulou M, Koutsaviti A, Roussis V, Ioannou E, Chondrogianni N (2022) Healthspan improvement and anti-aggregation effects induced by a marine-derived structural proteasome activator. Redox Biol 56:102462. https://doi.org/10.1016/j.redox.2022.102462
von Otter M, Bergström P, Quattrone A, De Marco EV, Annesi G, Söderkvist P, Wettinger SB, Drodzik M, Bialecka M, Nissbrandt H, Klein C, Nilsson M, Hammarsten O, Nilsson S, Zetterberg H (2014) Genetic associations of Nrf2-encoding NFE2L2 variants with Parkinson’s disease-a multicenter study. BMC Med Genet 15:131. https://doi.org/10.1186/s12881-014-0131-4
von Otter M, Landgren S, Nilsson S, Zetterberg M, Celojevic D, Bergström P, Minthon L, Bogdanovich N, Andreasen N, Gustafson DR, Skoog I, Wallin A, Tasa G, Blennow K, Nilsson M, Hammarsten O, Zetterberg H (2010) Nrf2-encoding NFE2L2 haplotypes influence disease progression but not risk in Alzheimer’s disease and age-related cataract. Mech Ageing Dev 131(2):105–110. https://doi.org/10.1016/j.mad.2009.12.007
Wang D-P, Kang K, Sun J, Lin Q, Lv Q-L, Hai J (2022a) URB597 and andrographolide improve brain microvascular endothelial cell permeability and apoptosis by reducing oxidative stress and inflammation associated with activation of Nrf2 signaling in oxygen-glucose deprivation. Oxid Med Cell Longev 2022:4139330. https://doi.org/10.1155/2022/4139330
Wang M-J, Liu X, Chi S, Wang F-Y, Zhang L, Yao M-Y, Liu T, Gao J (2023a) Activation of PPARγ prevents ferroptosis-induced damage through Nrf2/Gpx4 signaling pathway in rat hippocampus following epilepsy. NeuroReport 34(6):368–374. https://doi.org/10.1097/WNR.0000000000001892
Wang M, Liu M, Ma Z (2023b) Cannabinoid type 2 receptor activation inhibits MPP+-induced M1 differentiation of microglia through activating PI3K/Akt/Nrf2 signal pathway. Mol Biol Rep 50(5):4423–4433. https://doi.org/10.1007/s11033-023-08395-4
Wang N, Wang E, Wang R, Muhammad F, Li T, Yue J, Zhou Y, Zhi D, Li H (2022b) Ursolic acid ameliorates amyloid β-induced pathological symptoms in Caenorhabditis elegans by activating the proteasome. Neurotoxicology 88:231–240. https://doi.org/10.1016/j.neuro.2021.12.004
Wang X, Bian Y, Wong CTT, Lu J-H, Lee SM-Y (2022) TRPV1 modulator ameliorates Alzheimer-like amyloid-β neuropathology via Akt/Gsk3β-mediated Nrf2 activation in the Neuro-2a/APP cell model. Oxid Med Cell Longev 2022. https://doi.org/10.1155/2022/1544244
Wang Y, Cai Z, Zhan G, Li X, Li S, Wang X, Li S, Luo A (2023c) Caffeic acid phenethyl ester suppresses oxidative stress and regulates M1/M2 microglia polarization via Sirt6/Nrf2 pathway to mitigate cognitive impairment in aged mice following anesthesia and surgery. Antioxidants (basel) 12(3):714. https://doi.org/10.3390/antiox12030714
Wardyn JD, Ponsford AH, Sanderson CM (2015) Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem Soc Trans 43(4):621–626. https://doi.org/10.1042/BST20150014
Watanabe Y, Taguchi K, Tanaka M (2020) Ubiquitin, autophagy and neurodegenerative diseases. Cells 9(9):2022. https://doi.org/10.3390/cells9092022
Wen J, Li S, Zheng C, Wang F, Luo Y, Wu L, Cao J, Guo B, Yu P, Zhang G, Li S, Sun Y, Yang X, Zhang Z, Wang Y (2021) Tetramethylpyrazine nitrone improves motor dysfunction and pathological manifestations by activating the PGC-1α/Nrf2/HO-1 pathway in ALS mice. Neuropharmacology 182:108380. https://doi.org/10.1016/j.neuropharm.2020.108380
Wu C-S, Zhu J, Wager-Miller J, Wang S, O’Leary D, Monory K, Lutz B, Mackie K, Lu H-C (2010) Requirement of cannabinoid CB(1) receptors in cortical pyramidal neurons for appropriate development of corticothalamic and thalamocortical projections. Eur J Neurosci 32(5):693–706. https://doi.org/10.1111/j.1460-9568.2010.07337.x
Wu KC, Cui JY, Klaassen CD (2011) Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol Sci 123(2):590–600. https://doi.org/10.1093/toxsci/kfr183
Xiang C, Wang Y, Zhang H, Han F (2017) The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis 22:1–26. https://doi.org/10.1007/s10495-016-1296-4
Xu J, Guo Q, Huo K, Song Y, Li N, Du J (2021) JZL184 protects hippocampal neurons from oxygen-glucose deprivation-induced injury via activating Nrf2/ARE signaling pathway. Hum Exp Toxicol 40(7):1084–1094. https://doi.org/10.1177/0960327120984220
Yabuki Y, Matsuo K, Yu M, Xu J, Sakimura K, Shioda N, Fukunaga K (2021) Cav3.1 t-type calcium channel is critical for cell proliferation and survival in newly generated cells of the adult hippocampus. Acta Physiol 232(1):e13613. https://doi.org/10.1111/apha.13613
Yamamoto N, Sawada H, Izumi Y, Kume T, Katsuki H, Shimohama S, Akaike A (2007) Proteasome inhibition induces glutathione synthesis and protects cells from oxidative stress: relevance to Parkinson disease. J Biol Chem 282(7):4364–4372. https://doi.org/10.1074/jbc.M603712200
Yang C-C, Wu C-H, Lin T-C, Cheng Y-N, Chang C-S, Lee K-T, Tsai P-J, Tsai Y-S (2021) Inhibitory effect of PPARγ on NLRP3 inflammasome activation. Theranostics 11(5):2424. https://doi.org/10.7150/thno.46873
Yang W, Chen L, Ding Y, Zhuang X, Kang UJ (2007) Paraquat induces dopaminergic dysfunction and proteasome impairment in DJ-1-deficient mice. Hum Mol Genet 16(23):2900–2910. https://doi.org/10.1093/hmg/ddm249
Yao W, Lin S, Su J, Cao Q, Chen Y, Chen J, Zhang Z, Hashimoto K, Qi Q, Zhang J-C (2021) Activation of BDNF by transcription factor Nrf2 contributes to antidepressant-like actions in rodents. Transl Psychiatry 11(1):140. https://doi.org/10.1038/s41398-021-01261-6
Ye J, Yin Y, Liu H, Fang L, Tao X, Wei L, Zuo Y, Yin Y, Ke D, Wang J-Z (2020) Tau inhibits PKA by nuclear proteasome-dependent PKAR2α elevation with suppressed CREB/GluA1 phosphorylation. Aging Cell 19(1):e13055. https://doi.org/10.1111/acel.13055
Young AP, Denovan-Wright EM (2022) The dynamic role of microglia and the endocannabinoid system in neuroinflammation. Front Pharmacol 12:806417. https://doi.org/10.3389/fphar.2021.806417
Yu H, Liu X, Chen B, Vickstrom CR, Friedman V, Kelly TJ, Bai X, Zhao L, Hillard CJ, Liu Q-S (2021) The neuroprotective effects of the CB2 agonist GW842166x in the 6-OHDA mouse model of Parkinson’s disease. Cells 10(12):3548. https://doi.org/10.3390/cells10123548
Zakaria A, Rady M, Mahran L, Abou-Aisha K (2019) Pioglitazone attenuates lipopolysaccharide-induced oxidative stress, dopaminergic neuronal loss and neurobehavioral impairment by activating Nrf2/ARE/HO-1. Neurochem Res 44:2856–2868. https://doi.org/10.1007/s11064-019-02907-0
Zhang C, Zhao M, Wang B, Su Z, Guo B, Qin L, Zhang W, Zheng R (2021a) The Nrf2-NLRP3-caspase-1 axis mediates the neuroprotective effects of Celastrol in Parkinson’s disease. Redox Biol 47:102134. https://doi.org/10.1016/j.redox.2021.102134
Zhang R, Gao Y, Li Y, Geng D, Liang Y, He Q, Wang L, Cui H (2022) Nrf2 improves hippocampal synaptic plasticity, learning and memory through the circ-Vps41/miR-26a-5p/CaMKIV regulatory network. Exp Neurol 351:113998. https://doi.org/10.1016/j.expneurol.2022.113998
Zhang W, Feng C, Jiang H (2021b) Novel target for treating Alzheimer’s diseases: crosstalk between the Nrf2 pathway and autophagy. Ageing Res Rev 65:101207. https://doi.org/10.1016/j.arr.2020.101207
Zhao Y, Lützen U, Gohlke P, Jiang P, Herdegen T, Culman J (2021) Neuroprotective and antioxidative effects of pioglitazone in brain tissue adjacent to the ischemic core are mediated by PI3K/Akt and Nrf2/ARE pathways. J Mol Med (berl) 99(8):1073–1083. https://doi.org/10.1007/s00109-021-02065-3
Zheng Q, Huang T, Zhang L, Zhou Y, Luo H, Xu H, Wang X (2016) Dysregulation of ubiquitin-proteasome system in neurodegenerative diseases. Front Aging Neurosci 8:303. https://doi.org/10.3389/fnagi.2016.00303
Zhou H, Cao F, Wang Z, Yu Z-X, Nguyen H-P, Evans J, Li S-H, Li X-J (2003) Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J Cell Biol 163(1):109–118. https://doi.org/10.1083/jcb.200306038
Zhou H, Shao M, Guo B, Li C, Lu Y, Yang X, Lee SMY (2019) Tetramethylpyrazine analogue T-006 promotes the clearance of alpha-synuclein by enhancing proteasome activity in Parkinson’s disease models. Neurotherapeutics 16(4):1225–1236. https://doi.org/10.1007/s13311-019-00759-8
Zhou Y, Peng L, Li Y, Zhao Y (2022) Silent information regulator 1 ameliorates oxidative stress injury via PGC-1α/PPARγ-Nrf2 pathway after ischemic stroke in rat. Brain Res Bull 178:37–48. https://doi.org/10.1016/j.brainresbull.2021.11.001
Acknowledgements
Students from Programa Delfín 2022 Diana C. Medrano-Cruz (UAS), Uriel García-Flores (BUAP), Sara I. Castañeda-Roque (BUAP), Dan L. Romero-Méndez (BUAP), Elizabeth Varillas-Galicia (BUAP), Daniela Rojas-Lobato (BUAP), Santiago Navas-Escobar (UNAD-Colombia), Karen Rentería-Pérez (BUAP).
Funding
MA was supported by a grant from the National Institute of Environmental Health Sciences (NIEHS) R01ES10563 and R01ES07331.
Author information
Authors and Affiliations
Consortia
Contributions
LAMM, AS: conceptualization, writing of original draft. EAON, JNO, GPD2022: information compilation. MML, ERL: selection of compiled information. IT, AAT, YT: critical contributions to the manuscript and supervision. LAAM, MA, AS: editing the final draft.
Corresponding authors
Ethics declarations
Ethics Approval
Not applicable
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Monsalvo-Maraver, L.A., Ovalle-Noguez, E.A., Nava-Osorio, J. et al. Interactions Between the Ubiquitin–Proteasome System, Nrf2, and the Cannabinoidome as Protective Strategies to Combat Neurodegeneration: Review on Experimental Evidence. Neurotox Res 42, 18 (2024). https://doi.org/10.1007/s12640-024-00694-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12640-024-00694-3