
Abstract
Purpose of Review
Brain metastasis is a significant cause of mortality in breast cancer patients and treatment options are limited. This article will focus on the dynamic intercellular communication between metastatic cancer cells, the tumor microenvironment, and neighboring brain cells to breach the brain’s defenses and promote tumor progression.
Recent Findings
Recent advances have further elucidated how tumor cells traverse through the blood–brain barrier and implicated the blood–cerebrospinal fluid barrier as a potential entry point. Tumor cells once within the brain milieu utilize factors secreted by and dynamically reprogram neurons, glial cells, and infiltrating leukocytes for their own colonization and survival.
Summary
Current research has illuminated key mechanisms metastatic breast cancer cells utilize to infiltrate the brain and assimilate in their new environment. New multimodal treatments are investigating not only chemotherapy and radiation that can target metastatic tumor cells, but also immune checkpoint inhibitor–based therapies to sensitize the immune microenvironment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The brain is a common site for metastasis from primary cancers, with lung accounting for 20–56%, breast cancer for 5–20% and melanoma for 7–16% [1]. Lung cancer is the most common source of brain metastasis in males, whereas breast cancer is the most common source in females. Breast cancer is a genetically and histologically heterogenous disease which is categorized by the expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor (HER2/neu) or basal-like triple negative (TN), which lacks expression of ER, PR, and HER2 receptors [2, 3•]. Triple-negative breast cancer (TNBC) is considered comparatively more aggressive and has a poorer prognosis than other types of breast cancer. Breast-to-brain metastasis (BBM) is diagnosed more frequently in TNBC (25–27%) and HER2+ (11–20%) while incidence of metastasis in luminal A (8–15%), ER+PR+HER2−, and luminal B (8–11%), ER+PR+HER2±, is comparatively lower [2]. The prognosis for breast cancer patients with brain metastases is generally poor, with a median survival of 2 to 27 months in absence of treatment [4]. Moreover, brain metastases often lead to neurocognitive impairments, functional decline, and reduced quality of life.
Metastatic cancer cell dissemination to the brain is a complex process. The steps involve invasion through the stromal environment, intravasation into the vasculature directly or via lymphatic system generating circulatory tumor cells (CTCs), and adhesion and extravasation into the metastatic site [5]. The immunologic landscape and local microenvironment discretely shape tumor cell repertoires to either rapidly proliferate and metastasize or remain dormant until a favorable environment is achieved to colonize. The brain microenvironment is particularly unique and is comprised of two primary environments separating the CNS from the circulatory system: (1) the brain parenchyma, housing neurons and glial cells, is protected by the blood–brain barrier (BBB) and (2) the ventricles, bathed in cerebrospinal fluid (CSF), is protected by the blood–CSF barrier (BCSFB). Each of these local environments differs in their barrier defense against invading pathogens and tumor cells, anatomy, and nutrient availability which markedly alters brain metastatic spread [6].
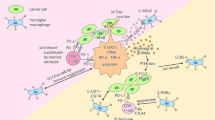
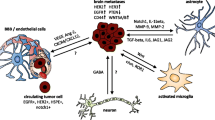
This review aims to summarize recent findings regulating breast-to-brain metastasis, with a particular emphasis on the dynamic interplay between metastatic cells and the CNS barriers, glial cells, and the invading and local immune microenvironment (Fig. 1). We believe, by comprehending the factors and agents responsible for regulating communication between the brain and the invading breast cancer cells we can develop innovative therapeutic approaches to combat this deadly condition.

Schematic of breast cancer cell interactions in the brain. An extensive direct and indirect crosstalk between the blood–brain barrier and blood–cerebrospinal fluid barrier needs to occur for circulating tumor cells to breach the CNS defenses and colonize the brain. Once within the brain parenchyma, the tumor cells interact with neurons, astrocytes, metabolic factors, microglia/infiltrating myeloids, and lymphocytes
Barriers of Tumor Cell Entry: BBB AND BCSFB
The central nervous system maintains its homeostasis through the blood–brain barrier (BBB) which tightly regulates the exchange of substances between the blood and the brain by preventing passive diffusion of blood-borne solutes. The BBB actively transports essential nutrients to the brain while the blood–cerebrospinal fluid barrier (BCSFB) filters nutrients from the blood and removes waste products from the brain [7]. Both barriers are unique anatomical structures defined by tight and adherent junctions between brain endothelial or epithelial cells, strictly regulating the entry of cells into the brain. Successful brain metastatic outgrowth requires tumor cells to breach one or both of these barriers, and in this section, we examine the factors and cells regulating this process.
Blood–Brain Barrier (BBB)
The BBB consists of a complex network of highly specialized structures that interact with each other. These structures include vascular brain endothelial cells (BECs), which are connected by an ensheathed basement membrane containing pericytes, as well as astrocytic foot processes that project outward. Unlike peripheral endothelial cells, BECs do not have fenestrations and instead use a network of tight junctions (TJs) to prevent the uncontrolled paracellular diffusion of water-soluble molecules [8, 9].
Disruption of the BBB by CTCs is a crucial step for metastatic tumor cell extravasation. Proteases secreted by tumor cells such as cysteine cathepsin S have been reported as a regulator of breast-to-brain metastasis which mediate BBB transmigration by proteolytic processing of (JAM)-B, a junctional adhesion molecule [10]. In a clinical study of 21 matrix metalloproteinases, only matrix metalloproteinase-1 (MMP1) was found to be significantly associated with brain metastasis by degrading claudin and occludin but not zonula occludens-1 (ZO-1) of the BBB. This process is regulated by an upregulation of cyclooxygenase-2 (COX2) in these brain metastatic cells which induced prostaglandins to upregulate MMP1 production. Furthermore, COX2 and prostaglandins were able to activate astrocytes to release chemokine C-C motif ligand 7 (CCL7) to direct tumor cell entry and promote their self-renewal in the brain [11]. Another study utilizing an in vivo model for TNBC brain metastasis demonstrated that tumor cells upregulated vascular growth factor angiopoietin 2 (ANG2) expression in brain endothelial cells which led to disruption of ZO-1 and claudin-5 tight junction protein structures [12].
In addition to disrupting the BBB, cancer cells interact with endothelial cells and undergo transcriptional changes to promote their migration and extravasation into the brain. Recent reports identified the role of reader YTH N6-methyladenosine RNA binding protein 3 (YTHDF3) overexpression in promoting breast cancer cell metastasis through interactions with endothelial cells and astrocytes [13]. The YTHDF3 protein family directly binds and reads modified regulatory N6-methyladenosine (m6A) sites on mRNAs, affecting RNA stability and translation [14, 15]. Chang et al. showed that knockdown of YTHDF3 reader in TNBC cells (4T1Br and MDA-MB-231Br) significantly reduced endothelial adhesion, implicating a critical crosstalk between tumor and endothelial cells [13]. Tavora et al. demonstrated that breast cancer cells co-opt innate RNA sensing and induce a chemotactic signaling pathway in the brain endothelium which drives tumor extravasation and metastasis. Their deep sequencing results showed that metastatic tumors induced high expression of axon guidance gene slit guidance ligand 2 (SLIT2) in endothelium compartments which along with its receptor, roundabout homolog 1 (ROBO1), promoted cancer cell migration in vivo. They identified an upstream signal of double-stranded RNA derived from tumor cells which induced expression of endothelial SLIT2 by acting on the RNA sensing Toll-like receptor 3 (TLR3) [16••]. Furthermore, genes involved in an organ-specific metastatic interaction such as the expression of ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 5 (ST6GALNAC5) in breast cancer cells were reported to enhance adhesion to brain endothelial cells and their passage through BBB by cell surface glycosylation [17]. In vitro co-culture experiments of endothelial cells with TNBC MDA-MB-231 significantly upregulated the gene expression of ANG2 and vascular endothelial growth factor (VEGF) in the tumor cells, promoting angiogenesis and interaction [18]. Similarly, breast cancer cells have shown to “educate” endothelial cells to upregulate VEGFR2 and subsequently signal transducer and activator of transcription 3 (STAT3) to promote tumor extravasation into the brain [19, 20]. In their study, Zhang and colleagues observed an intriguing intercellular communication between endothelial cells and TNBC cells via signaling involving plasminogen activator inhibitor-1 (PAI-1) and chemokine ligand-5 (CCL5). The researchers induced an epithelial to mesenchymal transition (EMT) in TNBC cells using exogenous transforming growth factor beta (TGF-β), which caused the cells to produce PAI-1. This in turn stimulated the expression and secretion of CCL5 from endothelial cells. The CCL5 acted in a paracrine manner on the TNBC cells, enhancing their migration [21]. However, this study was not conducted utilizing brain endothelial cells and will need to be validated in that context.
In addition to endothelial cells, pericytes dynamically interact with tumor cells to promote entry through the BBB. Pericytes serve as immediate responders capable of interacting with metastasized tumor cells to mediate tumor growth and angiogenesis by regulating focal adhesion kinase protein (FAK) expression, as reported in mouse models of melanoma, lung carcinoma, and pancreatic B-cell insulinoma [22]. In TNBC and melanoma, pericytes were also shown to secrete high levels of extracellular matrix proteins and insulin-like growth factor 2 (IGF2) to promote cell proliferation, illustrating its multifaceted pro-metastatic role [23]. Pericyte expression of a transmembrane glycoprotein receptor endosialin (CD248) regulated mesenchymal cell proliferation by platelet-derived growth factor receptor beta (PDGFRβ) signaling and promoted metastatic dissemination in 4T1 murine breast cancer cells [24]. These pericytes in the brain preserved BBB stability by controlling expression of extracellular matrix generation, transcytosis, and junctional components [25]. The two subpopulations viz. CD13+ pericytes and desmin+ pericytes were reported to alter BBB permeability and promote breast cancer metastasis [26].
Blood–Cerebrospinal Fluid Barrier (BCSFB)
Similar to the endothelial barrier in the BBB, the BCSFB is characterized by apical tight junctions that are located within choroid plexus epithelial cells (CPEs). These tight junctions act as a physical barrier, restricting the unregulated movement of water-soluble molecules. Although there are minimal reports of breast-to-brain metastasis through the BCSFB, the presence of fenestrated endothelial cells and “leakier” choroid plexus epithelial cells as compared to “tighter” brain endothelial cells may enable easier tumor cell traversal. MDA-MB-231 breast cancer cells were found to disrupt tight junctions in the choroid plexus epithelium through activation of complement component c3 which allowed plasma components and other growth factors to enter the CSF and promote cancer cell growth [27]. In an in vitro study in our laboratory, we demonstrated that increased cortisol levels can facilitate BBM through the BCSFB by enhancing the migratory capacity of breast cancer cells. This study cautions the use of glucocorticoids to treat breast cancer patients; however, further in vivo investigation is required to accurately establish cortisol’s role [28]. The current limited work on BBM entry through the BCSFB will need to be expanded upon in the years to come, perhaps lending insight from the plethora of recent findings examining BCSFB entry in other cancers such as neuroblastoma and leukemia [29].
Effects of the Microenvironment on Colonization of Breast Cancer to the Brain
After breaching the CNS barriers, tumor colonization in the brain is dependent on interaction with the neuronal microenvironment which is coordinated by various molecular pathways involving the surrounding glial cells, neurons, and immune cells [7]. In this section, we review different cell types within the brain and their interaction with colonizing metastatic breast cancer cells.
Astrocytes
Astrocytes comprise roughly half of the cell population in the brain and have a major role in maintaining BBB integrity and tissue homeostasis. Moreover, they serve as the principle stromal constituents of primary and metastatic brain tumors and can aid in both tumor apoptosis and tumor promotion [30]. Reactive astrocyte subtypes A1 and A2 have been reported to be associated with ischemia and neuroinflammation, where A1 is proinflammatory and A2 promotes tissue repair through the production of neurotrophic factors [31, 32]. Future studies are needed to elucidate the exact mechanism regulating reactive astrocyte formation upon encountering danger associated molecular patters (DAMP) signals and interconversion within the astrocyte subtypes based on the brain tumor microenvironment.
Utilizing human brain metastasis biopsies and rat models of breast-to-brain metastasis, Sarmiento et al. demonstrated that during micrometastasis, astrocytes became activated in a STAT3 dependent pathway, impaired cerebrovascular responses to stimuli, and disrupted traditional astrocyte–endothelial interactions, thereby further promoting brain metastatic progression [33]. In another model of TNBC brain metastases, astrocytes released growth factors such as transforming growth factor beta2 (TGF-β2) [34], which upregulate angiopoietin-4 (ANG4, a secreted glycoprotein) in tumor cells and contribute to BBM development [35]. Furthermore, it has been reported that astrocyte derived insulin-like growth factor (IGF) binding protein 2 (IGFBP2) and chitinase-3-like protein-1 (CHI3LI) prime the CSF as a favorable niche for TNBC cells. IGFBP2 promotes proliferation through activation of IGF signaling and CHI3LI facilitates anchorage of the metastatic lesions [36]. The same group previously demonstrated that the transactivation of Reelin by astrocytes in HER2+ breast cancer was markedly increased, resulting in enhanced proliferation and survival ability. Notably, heightened Reelin expression was only detected in HER2+ metastases but not in TN primary tumors or in TN breast-to-brain metastatic cells that were in direct contact with astrocytes [37]. In primary gliomas, a high degree of bilateral communication has been reported between astrocytes and gliomas mediated through extracellular vesicles (EVs). The EVs released from glioblastoma cells taken up by astrocytes increases their oncogenic signaling pathways (receptor tyrosine kinase and mitogen-activated protein kinase) and migratory capabilities, converting them to a reactive astrocyte subtype [38]. Astrocytes in turn release miRNA containing EVs which downregulate phosphatase and tensin homolog (PTEN), a tumor suppressor gene [39]. This unique finding remains to be studied in a breast-to-brain metastatic model.
The overwhelming current literature in BBMs sheds light on astrocyte involvement primarily through pro-tumor mechanisms, but reports in other metastatic cancers have implicated an anti-tumor role which need to be examined in a BBM context. Additionally, the function of another glial cell type, oligodendrocytes, on metastasizing breast cancer cells has yet to be reported. However, extensive studies on the formation of oligodendrogliomas can help guide this inspection.
Neurons and Neurotransmitters
It has been previously established that cancer cells can grow around neurons and invade their niche by chemokine mediated attachment, a phenomenon referred to as perineural invasion [40]. Later, in a landmark finding, it was shown that prostate to brain cancer metastases were infiltrated by newly generated neurons, analogous to angiogenesis and regulated tumor development [41•]. In gliomas, interaction with neurons has been reported to promote glioma growth through alpha amino-3-hydroxy-5 methyl-4 isoxalolepropionic acid (AMPA) receptor dependent neuron glioma synapses, forming an electrically coupled network [42]. Although the studies conducted thus far have unveiled an association and axis of crosstalk between neurons and tumors, there is still a significant lack of exploration regarding this interplay in metastatic breast cancer cells. However, some of the critical findings are discussed below.
We recently showed that exposure of tumor cells to the neuronal environment upregulates neurotransmitter receptor and synaptic mediators mRNAs (CNR1, HTR3A, BDNF, CEBPD, TIMP1) in both brain and non-brain metastatic breast and lung cancer cells. Consequently, our findings indicated that neurotransmitter treatment to dormant tumor cells led to a dose dependent increase in expression of Reelin, an extracellular matrix glycoprotein that plays a crucial role in neuronal synaptic communication, and in regulating tumor migration and invasiveness in metastatic cells [43].
Another recent study demonstrated that breast cancer cells can functionally interact with neurons to promote their proliferation and colonization by co-opting a neuronal signaling pathway activated by glutamate ligands of aspartate receptor (NMDAR) [44]. A number of reports also suggest the role of neurotransmitters in progression and growth of breast cancer [45]. Involvement of acetylcholine through nicotinic acetylcholine receptor (nAChR) signaling cascades such as ERK/MAPK, JAK/PI3K, and STAT3/TWIST1 were shown to induce proliferation, apoptosis resistance, and growth in BBMs, implicating a unique interplay with the neuronal environment [46,47,48,49]. Interestingly, dopamine receptor-1 (D1R) was found to act as a tumor suppressor in breast cancer cells [50]. New findings have hinted at the role of VEGF and NGF in promoting axonogenesis of breast cancer, and additional findings may open new avenues to target the tumor-neuron microenvironment [51].
Metabolic Plasticity of Tumor Cells in the Brain
Regardless of the metastatic potential of tumor cells, they largely fail to colonize a metastatic site. Only cancer cells with the appropriate adaptions successfully colonize their niche, and one technique to create a permissive microenvironment is by establishing a pre-metastatic niche conducive to survival. Tumor cells secrete chemokines and extracellular vesicles [38] to alter the dynamic map of metabolites at the metastatic site and adapt to their new environment [52]. The human brain exclusively relies on glucose; however, under certain conditions it is capable of utilizing alternative fuels such as ketone bodies, fatty acids, and acetate [53]. For instance, astrocytes pre-utilize glucose, resulting in low glucose levels and high levels of glutamate and branched chain alpha-keto acids in the interstitial compartment of the brain [54]. Cancers such as glioblastoma are critically dependent on oxidative phosphorylation and metabolize acetate by upregulating acetyl-coenzyme A synthetase short-chain family member 2 (ACSS2), an acetyl-coA converting enzyme [55, 56]. The brain metastatic breast cancer line MDA-MB-231Br3 was found to evolve from glucose-dependent to glucose-independent proliferation in order to survive in a low glucose CNS environment. These cells possibly upregulated gluconeogenesis and utilized the oxidation of glutamine and branched chain amino acids for purine synthesis as an alternate energy source [57].
The role of γ-amino butyric acid (GABA) as a metabolite exploited for metastatic tumor growth has not garnered enough attention. GABA is traditionally an inhibitory neurotransmitter highly expressed throughout the CNS, but has been shown to contribute to cancer cell proliferation [58]. Neman et al. showed that HER2+ and TN breast-to-brain metastatic cells overexpressed key regulatory proteins in the GABA shunt pathway and parvalbumin, a calcium binding GABAergic neuronal synaptic plasticity protein, and were able to metabolize extracellular GABA to promote increased tumor cell proliferation [59]. In a recent report, brain-colonizing breast cancer cells upregulated epithelial to mesenchymal transcription factor lymphoid enhancer–binding factor-1 (LEF1) to help resist glutathione depletion and improve antioxidant capacity of the breast cancer cells [60]. Furthermore, it was also shown that LEF1 mediated a survival benefit to the metastasized breast cancer cells in the brain by conferring reactive oxygen synthase (ROS) resistance and boosting glutathione levels [60].
Immune Crosstalk in the BBM Microenvironment
Successful breast cancer metastatic spread into the CNS niche is supported by several mechanisms regulated by resident and infiltrating immune cells. While once heralded as an immune sanctuary, the metastatic brain microenvironment is now known to have a diverse blood-borne immune presence including a lymphocyte population comprised of CD3+, CD4+, CD8+, FoxP3+, NK cells, and myeloid-derived cells in addition to brain resident CNS microglia [61]. In this section, we segment the role of microglia/infiltrating myeloids from infiltrating lymphocytes to examine how understanding of these cells in the context of their support or elimination of metastases can provide avenues for improved patient outcomes.
Microglia/Infiltrating Myeloids
Infiltrating tumor cells have conventionally been observed to commandeer various mechanisms, inducing microglia and infiltrating myeloids to adopt an M2 macrophage phenotype. This particular subtype of macrophages is widely recognized as pro-tumorigenic, leading to the extended survival and proliferation of cancer cells [62, 63]. However, recent advancements in the study of breast cancer brain metastases demonstrate that the involvement of microglia/macrophages is heterogenous and subject to high levels of plasticity conferring a diverse functional contribution to tumor progression [64]. Utilizing a TNBC immunocompetent mouse model, Andreou et al. examined the function of microglia/peripheral macrophages during the initial phases of metastatic development (first 28 days) [65]. They identified groups of proinflammatory (iNOS+, COX2+) and anti-inflammatory (MRC1+ and Arg1+) microglia/macrophages in the metastatic tumor whose levels remained steady across the time-course. By using mannosylated clodronate liposomes to specifically remove the anti-inflammatory microglia/macrophage population, there was a significant decrease in the metastatic tumor burden. This reduction occurred by inducing apoptosis of TNBC cells. Further examination of microglial permeation demonstrated that these macrophages infiltrated into the breast cancer tumor and established contacts with tumor cells after successful extravasation and stimulated a five-fold increase in the proliferation of metastatic cells [66].
Additional studies examining the mechanisms of microglia/macrophage tumor growth implicate neutrophin-3, Wnt, and P13K dependent pathways. Louie et al. demonstrated that breast cancer cells which metastasized to the brain secrete high levels of neurotrophin-3 which function to diminish the number of fully activated cytotoxic microglia recruited to the tumor site and allows for a “permissive” microenvironment for brain metastatic growth [67]. According to Blazquez et al., P13K was recognized as a crucial regulator of macrophages/microglia that promote metastasis during BBM. By administering a pan-P13K class 1 inhibitor, the metastasis-promoting characteristics of these cells were decreased, and they were redirected towards a classically activated M1 phenotype that is known for its anti-tumor properties [68]. Pukrop et al. illustrated that microglia promote invasion and colonization of brain tissue by breast cancer cells through a Wnt-dependent pathway, which was reversed by Wnt-inhibitor Dickkopf-2 [69]. While it may appear enticing to formulate therapies to modulate macrophage/microglia polarity towards a M1 phenotype through toll-like receptor 4 (TLR4) and P13K mediated pathways to inhibit BBM outgrowth, it is pertinent to recognize the neurotoxic effects of M1 polarized microglia may be irreparable [70].
The distinct roles of microglia and infiltrating myeloids in the brain tumor microenvironment has been controversial, but increased understanding of markers denoting varied cell states within this plastic population and new tools such as mass cytometry, digital spatial profiling, and multiplex immunohistochemistry have allowed us to tackle this contentious topic. Recently, Guldner et al. demonstrated through multimodal single cell analysis that infiltrating myeloid cells minimally influenced BBMs while microglia promoted BBM outgrowth through upregulation of C-X-C motif chemokine 10 (Cxcl10) to recruit immune-suppressive microglia to BBMs [71••]. There is now an abundance of literature characterizing myeloid infiltration of breast tumors and their association with poor prognosis and decreased survival in patients with breast cancer. As such, there is an increasing focus on the role of targeting myeloid suppression in breast cancer as a strategy to sensitize the tumor microenvironment to therapies such as immune checkpoint inhibition. In particular, recent studies examining MDSCs have implicated them as an attractive partner in breast cancer metastasis because of their plasticity, immunosuppression, and tumor-promoting role throughout the entire metastatic cascade [72]. Unfortunately, much remains to be studied about their direct involvement in promoting brain metastases and adaptation to the CNS microenvironment.
Infiltrating Lymphocytes and Immunotherapies
Use of immune checkpoint inhibition (ICI) is becoming increasingly common among many solid tumor malignancies. Patients with TNBC have shown the most promising responses to these therapies; however, understanding of their efficacy or lack thereof, in breast-to-brain metastases remains to be determined [73, 74]. The healthy human brain contains almost no lymphocytes, but recent studies have illustrated that few B- and T-lymphocytes are present within BBMs due to disruption of the BBB [61]. Further examination reveals a decrease of lymphocytes within BBMs as compared to either matched primary breast tumors or other metastatic sites [75]. The researchers suggested that this could be attributed to immune evasion mechanisms employed by the tumor, as well as the unique immune environment of the brain. Despite decreased lymphocytes, there was no significant difference in programmed cell death ligand-1 (PD-L1) positivity between primary breast tumors and associated brain metastases. PD-L1 has been studied as a predictive biomarker and is currently used in some countries including the USA, to determine eligibility for use of ICI in breast and other solid tumor malignancies given it is often associated with response to therapy [76]. Similar expression of PD-L1 in brain metastases suggests response to checkpoint inhibition in brain metastases should match that seen in the primary tumor, however it often does not [73, 74]. Further research done by Chongsathidkiet et al. showed that in experimental models of BBMs, T cells are sequestered in the bone marrow because of tumor-imposed loss of sphingosine 1-phosphate receptor (S1P1) from the T-cell surface [77]. Among the few cytotoxic T cells that have breached the BBM environment, breast cancer cells have shown to downregulate or modify major histocompability complex-1 (MHC-I) on their surface to escape immunosurveillance [78]. Tumor cells may also recruit MDSCs and Tregs to deplete T cells in the BBM microenvironment, although this remains to be studied.
Despite these limitations, because BBMs commonly express PD-L1 and PD-L2 they appear to be an untapped target for therapy with immune checkpoint inhibitors. Furthermore, patients with low levels of tumor-infiltrating lymphocytes in brain metastases have worse prognoses than those with higher counts [75]. As a result, there have been recent clinical trials to investigate the role of immunotherapy for BBM patients as they can cross through the BBB. One phase 2 trial examined the effect of MEDI4736, a novel PD-L1 inhibitory monoclonal antibody but was unfortunately terminated due to a withdrawal of funding and change in clinical practices [NCT02669914]. Novel studies have demonstrated that immune checkpoint inhibition fails to induce responses without sufficient T-cell infiltration in tumor cells, a key issue with BBMs [79]. To remedy this, researchers are examining combination therapy with chemotherapy, radiation, or epigenetic modulation which can stimulate the immune system to activate CD4 and CD8 positive T cells, upregulate PD-L1 on tumor cells and alter other immune cells in the microenvironment to improve response with subsequent treatment with immune checkpoint inhibitors [80]. Trials investigating combinations with chemotherapy are numerous but two notable studies led to recent approvals in the USA by the Federal drug administration (FDA) for TNBC [73, 74]. There are currently three ongoing phase 1/2 clinical trials examining the treatments of BBMs with “priming” stereotactic radiosurgery followed by a PD-L1 inhibitor: atezolizumab (NCT03483012), nivolumab (NCT03807765), and pembrolizumab (NCT03449238). In addition, combining dual immune checkpoint inhibitor therapy with a histone deacetylase inhibitor entinostat has been studied in a phase Ib trial (NCT02453620) and demonstrated preliminary efficacy correlated with decreased suppression of MDSCs and increased CD8+ T cell infiltration in advanced solid tumors, including in patients with breast cancer [81]. While other immune checkpoint therapies targeting CTLA-4 and IDO have shown response in melanoma, non-small-cell lung cancer, and renal cell carcinoma metastasizing to the brain, they have yet to be examined in the context of BBMs [82, 83]. A greater understanding of the function of immune cells in BBMs can open novel avenues for treatment.
Conclusion
For patients, brain metastasis is one of the leading causes of death. Over the past decade, the development of advanced research methods to decipher the signaling pathways and metabolic processes guiding brain metastasis has progressed tremendously, but still faces a formidable challenge in drug delivery. Currently, ongoing clinical trials examining the role of combination radiotherapy and immune checkpoint inhibitors holds considerable promise. We recommend future studies to go beyond the BBB–tumor interaction and closely examine the role of the BCSFB as a potential mode of tumor cell entry, which has been overlooked in the past. Lastly, a more in depth understanding of myeloid and lymphoid populations infiltrating brain metastases will help elucidate potential novel therapeutic combinations which could allow improved efficacy of immune checkpoint inhibition in patients with breast-to-brain metastases. As different scientific disciplines continue to converge and collaborate, we hope new undiscovered methods to understand the pathophysiology of breast-to-brain metastases will emerge.
Abbreviations
- ADAM:
-
A Disintegrin and Metalloproteinase
- Arg1:
-
Arginase 1
- BDNF:
-
Brain-derived Neurotrophic Factor
- CD:
-
Cluster of Differentiation
- CREB:
-
CAMP-Response Element Binding Protein
- ERK-:
-
Extracellular Signal Regulated Kinases
- FOXP3-:
-
Forhead Box P3
- GABA:
-
γ-amino Butyric Acid
- iNOS:
-
inducible Nitric Oxide Synthase
- JAK:
-
janus kinase
- LEF1:
-
Lymphoid Enhancer Binding Factor 1
- MAPK:
-
Mitogen-Activated Protein Kinase
- NGF:
-
Nerve Growth Factor
- MHC:
-
Major Histocompatibility Complex
- MMP:
-
Matrix Metalloproteinase
- MRC1:
-
Mannose Receptor C type-1
- NK:
-
Natural Killer Cells
- NMDAR:
-
N-methyl-D-aspartate Receptor
- PI3K:
-
Phosphoinositide 3-kinase
- RTK:
-
Receptor Tyrosine Kinase
- TCA:
-
Tricarboxylic Acid Cycle
- TWIST1:
-
Twist Family BHLH Transcription Factor-1
- Wnt:
-
Wingless-related Integration Site
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ostrom QT, Wright CH, Barnholtz-Sloan JS. Brain metastases: epidemiology. Handb Clin Neurol. 2018;149:27–42.
Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, Speers CH, et al. Metastatic behavior of breast cancer subtypes. J Clin Oncol. 2010;28(20):3271–7.
Ebright RY, Lee S, Wittner BS, Niederhoffer KL, Nicholson BT, Bardia A, et al. Deregulation of ribosomal protein expression and translation promotes breast cancer metastasis. Science. 2020;367(6485):1468–73. This finding suggests that overexpression of RPL15 gene enhances metastatic growth and selectively modulates translation of ribosomal proteins and cell cycle regulators.
Ogawa K, Yoshii Y, Nishimaki T, Tamaki N, Miyaguni T, Tsuchida Y, et al. Treatment and prognosis of brain metastases from breast cancer. J Neuro-Oncol. 2008;86(2):231–8.
Obenauf AC, Massague J. Surviving at a distance: organ-specific metastasis. Trends Cancer. 2015;1(1):76–91.
Valiente M, Ahluwalia MS, Boire A, Brastianos PK, Goldberg SB, Lee EQ, et al. The evolving landscape of brain metastasis. Trends Cancer. 2018;4(3):176–96.
Hosonaga M, Saya H, Arima Y. Molecular and cellular mechanisms underlying brain metastasis of breast cancer. Cancer Metastasis Rev. 2020;39(3):711–20.
Engelhardt B, Sorokin L. The blood-brain and the blood-cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol. 2009;31(4):497–511.
Metzger F, Mischek D, Stoffers F. The connected steady state model and the interdependence of the CSF proteome and CSF flow characteristics. Front Neurosci. 2017;11:241.
Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol. 2014;16(9):876–88.
Wu K, Fukuda K, Xing F, Zhang Y, Sharma S, Liu Y, et al. Roles of the cyclooxygenase 2 matrix metalloproteinase 1 pathway in brain metastasis of breast cancer. J Biol Chem. 2015;290(15):9842–54.
Avraham HK, Jiang S, Fu Y, Nakshatri H, Ovadia H, Avraham S. Angiopoietin-2 mediates blood-brain barrier impairment and colonization of triple-negative breast cancer cells in brain. J Pathol. 2014;232(3):369–81.
Chang G, Shi L, Ye Y, Shi H, Zeng L, Tiwary S, et al. YTHDF3 Induces the translation of m(6)A-enriched gene transcripts to promote breast cancer brain metastasis. Cancer Cell. 2020;38(6):857–71 e7.
Frye M, Harada BT, Behm M, He C. RNA modifications modulate gene expression during development. Science. 2018;361(6409):1346–9.
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27(3):315–28.
Tavora B, Mederer T, Wessel KJ, Ruffing S, Sadjadi M, Missmahl M, et al. Tumoural activation of TLR3-SLIT2 axis in endothelium drives metastasis. Nature. 2020;586(7828):299–304. This research work suggests that cancer cells expoits the innate RNA sensing mechanism to trigger a chemotactic signaling in the endothelium leading to intravasation and metastasis.
Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459(7249):1005–9.
Buchanan CF, Szot CS, Wilson TD, Akman S, Metheny-Barlow LJ, Robertson JL, et al. Cross-talk between endothelial and breast cancer cells regulates reciprocal expression of angiogenic factors in vitro. J Cell Biochem. 2012;113(4):1142–51.
Brenet M, Martinez S, Perez-Nunez R, Perez LA, Contreras P, Diaz J, et al. Thy-1 (CD90)-induced metastatic cancer cell migration and invasion are beta3 Integrin-dependent and involve a Ca(2+)/P2X7 receptor signaling axis. Front Cell Dev Biol. 2020;8:592442.
Lee HT, Xue J, Chou PC, Zhou A, Yang P, Conrad CA, et al. Stat3 orchestrates interaction between endothelial and tumor cells and inhibition of Stat3 suppresses brain metastasis of breast cancer cells. Oncotarget. 2015;6(12):10016–29.
Zhang W, Xu J, Fang H, Tang L, Chen W, Sun Q, et al. Endothelial cells promote triple-negative breast cancer cell metastasis via PAI-1 and CCL5 signaling. FASEB J. 2018;32(1):276–88.
Lechertier T, Reynolds LE, Kim H, Pedrosa AR, Gomez-Escudero J, Munoz-Felix JM, et al. Pericyte FAK negatively regulates Gas6/Axl signalling to suppress tumour angiogenesis and tumour growth. Nat Commun. 2020;11(1):2810.
Molnar K, Meszaros A, Fazakas C, Kozma M, Gyori F, Reisz Z, et al. Pericyte-secreted IGF2 promotes breast cancer brain metastasis formation. Mol Oncol. 2020;14(9):2040–57.
Viski C, Konig C, Kijewska M, Mogler C, Isacke CM, Augustin HG. Endosialin-expressing pericytes promote metastatic dissemination. Cancer Res. 2016;76(18):5313–25.
Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468(7323):562–6.
Lyle LT, Lockman PR, Adkins CE, Mohammad AS, Sechrest E, Hua E, et al. Alterations in pericyte subpopulations are associated with elevated blood-tumor barrier permeability in experimental brain metastasis of breast cancer. Clin Cancer Res. 2016;22(21):5287–99.
Boire A, Zou Y, Shieh J, Macalinao DG, Pentsova E, Massague J. Complement component 3 adapts the cerebrospinal fluid for leptomeningeal metastasis. Cell. 2017;168(6):1101–13 e13.
Herrera RA, Deshpande K, Martirosian V, Saatian B, Julian A, Eisenbarth R, et al. Cortisol promotes breast-to-brain metastasis through the blood-cerebrospinal fluid barrier. Cancer Rep (Hoboken). 2021;5:e1351.
März M, Meyer S, Erb U, Georgikou C, Horstmann MA, Hetjens S, et al. Pediatric acute lymphoblastic leukemia—conquering the CNS across the choroid plexus. Leuk Res. 2018;71:47–54.
Wasilewski D, Priego N, Fustero-Torre C, Valiente M. Reactive astrocytes in brain metastasis. Front Oncol. 2017;7:298.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–7.
Priego N, Zhu L, Monteiro C, Mulders M, Wasilewski D, Bindeman W, et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat Med. 2018;24(7):1024–35.
Sarmiento Soto M, Larkin JR, Martin C, Khrapitchev AA, Maczka M, Economopoulos V, et al. STAT3-mediated astrocyte reactivity associated with brain metastasis contributes to neurovascular dysfunction. Cancer Res. 2020;80(24):5642–55.
Wang L, Cossette SM, Rarick KR, Gershan J, Dwinell MB, Harder DR, et al. Astrocytes directly influence tumor cell invasion and metastasis in vivo. PLoS One. 2013;8(12):e80933.
Gong X, Hou Z, Endsley MP, Gronseth EI, Rarick KR, Jorns JM, et al. Interaction of tumor cells and astrocytes promotes breast cancer brain metastases through TGF-beta2/ANGPTL4 axes. NPJ Precis Oncol. 2019;3:24.
Ansari KI, Bhan A, Liu X, Chen MY, Jandial R. Astrocytic IGFBP2 and CHI3L1 in cerebrospinal fluid drive cortical metastasis of HER2+breast cancer. Clin Exp Metastasis. 2020;37(3):401–12.
Jandial R, Choy C, Levy DM, Chen MY, Ansari KI. Astrocyte-induced Reelin expression drives proliferation of Her2(+) breast cancer metastases. Clin Exp Metastasis. 2017;34(2):185–96.
Nieland L, Morsett LM, Broekman MLD, Breakefield XO, Abels ER. Extracellular vesicle-mediated bilateral communication between glioblastoma and astrocytes. Trends Neurosci. 2021;44(3):215–26.
O'Brien ER, Howarth C, Sibson NR. The role of astrocytes in CNS tumors: pre-clinical models and novel imaging approaches. Front Cell Neurosci. 2013;7:40.
Marchesi F, Piemonti L, Mantovani A, Allavena P. Molecular mechanisms of perineural invasion, a forgotten pathway of dissemination and metastasis. Cytokine Growth Factor Rev. 2010;21(1):77–82.
Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, et al. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341(6142):1236361.
Venkatesh HS, Morishita W, Geraghty AC, Silverbush D, Gillespie SM, Arzt M, et al. Electrical and synaptic integration of glioma into neural circuits. Nature. 2019;573(7775):539–45.
Deshpande K, Martirosian V, Nakamura BN, Iyer M, Julian A, Eisenbarth R, et al. Neuronal exposure induces neurotransmitter signaling and synaptic mediators in tumors early in brain metastasis. Neuro-Oncology. 2022;24(6):914–24.
Zeng Q, Michael IP, Zhang P, Saghafinia S, Knott G, Jiao W, et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature. 2019;573(7775):526–31.
Drell TL, Joseph J, Lang K, Niggemann B, Zaenker KS, Entschladen F. Effects of neurotransmitters on the chemokinesis and chemotaxis of MDA-MB-468 human breast carcinoma cells. Breast Cancer Res Treat. 2003;80(1):63–70.
Chen ZB, Liu C, Chen FQ, Li SY, Liang Q, Liu LY. Effects of tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) on the activation of ERK1/2 MAP kinases and the proliferation of human mammary epithelial cells. Environ Toxicol Pharmacol. 2006;22(3):283–91.
Guha P, Bandyopadhyaya G, Polumuri SK, Chumsri S, Gade P, Kalvakolanu DV, et al. Nicotine promotes apoptosis resistance of breast cancer cells and enrichment of side population cells with cancer stem cell-like properties via a signaling cascade involving galectin-3, alpha9 nicotinic acetylcholine receptor and STAT3. Breast Cancer Res Treat. 2014;145(1):5–22.
Kalantari-Dehaghi M, Parnell EA, Armand T, Bernard HU, Grando SA. The nicotinic acetylcholine receptor-mediated reciprocal effects of the tobacco nitrosamine NNK and SLURP-1 on human mammary epithelial cells. Int Immunopharmacol. 2015;29(1):99–104.
Nishioka T, Kim HS, Luo LY, Huang Y, Guo J, Chen CY. Sensitization of epithelial growth factor receptors by nicotine exposure to promote breast cancer cell growth. Breast Cancer Res. 2011;13(6):R113.
Borcherding DC, Tong W, Hugo ER, Barnard DF, Fox S, LaSance K, et al. Expression and therapeutic targeting of dopamine receptor-1 (D1R) in breast cancer. Oncogene. 2016;35(24):3103–13.
Han H, Yang C, Zhang Y, Han C, Zhang G. Vascular endothelial growth factor mediates the sprouted axonogenesis of breast cancer in rat. Am J Pathol. 2021;191(3):515–26.
Elia I, Doglioni G, Fendt SM. Metabolic hallmarks of metastasis formation. Trends Cell Biol. 2018;28(8):673–84.
Ebert D, Haller RG, Walton ME. Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J Neurosci. 2003;23(13):5928–35.
Hu Y, Wilson GS. Rapid changes in local extracellular rat brain glucose observed with an in vivo glucose sensor. J Neurochem. 1997;68(4):1745–52.
Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, Clement-Schatlo V, et al. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012;26(17):1926–44.
Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. 2014;159(7):1603–14.
Chen J, Lee HJ, Wu X, Huo L, Kim SJ, Xu L, et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res. 2015;75(3):554–65.
Szczaurska K, Mazurkiewicz M, Opolski A. The role of GABA-ergic system in carcinogenesis. Postepy Hig Med Dosw. 2003;57(5):485–500.
Neman J, Termini J, Wilczynski S, Vaidehi N, Choy C, Kowolik CM, et al. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc Natl Acad Sci U S A. 2014;111(3):984–9.
Blazquez R, Rietkotter E, Wenske B, Wlochowitz D, Sparrer D, Vollmer E, et al. LEF1 supports metastatic brain colonization by regulating glutathione metabolism and increasing ROS resistance in breast cancer. Int J Cancer. 2020;146(11):3170–83.
Berghoff AS, Lassmann H, Preusser M, Höftberger R. Characterization of the inflammatory response to solid cancer metastases in the human brain. Clin Exp Metastasis. 2013;30(1):69–81.
Rippaus N, Taggart D, Williams J, Andreou T, Wurdak H, Wronski K, et al. Metastatic site-specific polarization of macrophages in intracranial breast cancer metastases. Oncotarget. 2106;7(27):41473–87.
Raza M, Prasad P, Gupta P, Kumar N, Sharma T, Rana M, et al. Perspectives on the role of brain cellular players in cancer-associated brain metastasis: translational approach to understand molecular mechanism of tumor progression. Cancer Metastasis Rev. 2018;37(4):791–804.
Simon A, Yang M, Marrison JL, James AD, Hunt MJ, O’Toole PJ, et al. Metastatic breast cancer cells induce altered microglial morphology and electrical excitability in vivo. J Neuroinflammation. 2020;17(1).
Andreou KE, Soto MS, Allen D, Economopoulos V, De Bernardi A, Larkin JR, et al. Anti-inflammatory microglia/macrophages as a potential therapeutic target in brain metastasis. Front Oncol. 2017;7.
Fitzgerald DP, Palmieri D, Hua E, Hargrave E, Herring JM, Qian Y, et al. Reactive glia are recruited by highly proliferative brain metastases of breast cancer and promote tumor cell colonization. Clin Exp Metastasis. 2008;25(7):799–810.
Louie E, Chen XF, Coomes A, Ji K, Tsirka S, Chen EI. Neurotrophin-3 modulates breast cancer cells and the microenvironment to promote the growth of breast cancer brain metastasis. Oncogene. 2013;32(35):4064–77.
Blazquez R, Wlochowitz D, Wolff A, Seitz S, Wachter A, Perera-Bel J, et al. PI3K: A master regulator of brain metastasis-promoting macrophages/microglia. Glia. 2018;66(11):2438–55.
Pukrop T, Dehghani F, Chuang H-N, Lohaus R, Bayanga K, Heermann S, et al. Microglia promote colonization of brain tissue by breast cancer cells in a Wnt-dependent way. Glia. 2010;58(12):1477–89.
Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2016;53(2):1181–94.
Guldner IH, Wang Q, Yang L, Golomb SM, Zhao Z, Lopez JA, et al. CNS-native myeloid cells drive immune suppression in the brain metastatic niche through Cxcl10. Cell. 2020;183(5):1234–48.e25. Findings from this study suggest that an immune-suppressive niche in the brain metastic microenvironment primarily stems from CNS native myeloid and not bone marrow–derived cells.
Trovato R, Canè S, Petrova V, Sartoris S, Ugel S, De Sanctis F. The Engagement Between MDSCs and Metastases: Partners in Crime. Front Oncol. 2020;10.
Cortes J, Cescon DW, Rugo HS, Nowecki Z, Im S-A, Yusof MM, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): a randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet. 2020;396(10265):1817–28.
Schmid P, Cortes J, Pusztai L, McArthur H, Kümmel S, Bergh J, et al. Pembrolizumab for early triple-negative breast cancer. N Engl J Med. 2020;382(9):810–21.
Ogiya R, Niikura N, Kumaki N, Yasojima H, Iwasa T, Kanbayashi C, et al. Comparison of immune microenvironments between primary tumors and brain metastases in patients with breast cancer. Oncotarget. 2017;8(61):103671–81.
Davis AA, Patel VG. The role of PD-L1 expression as a predictive biomarker: an analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J ImmunoTherapy Cancer. 2019;7(1).
Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med. 2018;24(9):1459–68.
Bates JP, Derakhshandeh R, Jones L, Webb TJ. Mechanisms of immune evasion in breast cancer. BMC Cancer. 2018;18(1).
Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, et al. Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell. 2016;29(3):285–96.
Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti–PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Investig. 2014;124(2):687–95.
Roussos Torres ET, Rafie C, Wang C, Lim D, Brufsky A, LoRusso P, et al. Phase I study of entinostat and nivolumab with or without ipilimumab in advanced solid tumors (ETCTN-9844). Clin Cancer Res. 2021;27(21):5828–37.
Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, et al. Combined nivolumab and ipilimumab in melanoma metastatic to the brain. N Engl J Med. 2018;379(8):722–30.
Nitschke NJ, Bjoern J, Iversen TZ, Andersen MH, Svane IM. Indoleamine 2,3-dioxygenase and survivin peptide vaccine combined with temozolomide in metastatic melanoma. Stem Cell Invest. 2017;4(9):77.
Funding
Open access funding provided by SCELC, Statewide California Electronic Library Consortium. DD, MI, JN supported by R01CA223544-01A1. ERT supported by the Concern Founation, METAVivor, USC Norris CCC, and USC Wright Foundation. FC supported by KL2TR001854.
Author information
Authors and Affiliations
Contributions
DD: writing, review, and figure generation; MI: writing, review, and figure generation; FC: editing, reviewing; ERT: writing, edit, review; JN: writing, edit, review, concept.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing interests
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Das, D., Iyer, M., Roussos Torres, E.T. et al. Breast-to-Brain Metastasis: from Microenvironment to Plasticity. Curr Breast Cancer Rep 15, 142–151 (2023). https://doi.org/10.1007/s12609-023-00488-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12609-023-00488-0