
Abstract
The sarcomere is the fundamental structural and functional unit of striated muscle and is directly responsible for most of its mechanical properties. The sarcomere generates active or contractile forces and determines the passive or elastic properties of striated muscle. In the heart, mutations in sarcomeric proteins are responsible for the majority of genetically inherited cardiomyopathies. Here, we review the major determinants of cardiac sarcomere mechanics including the key structural components that contribute to active and passive tension. We dissect the molecular and structural basis of active force generation, including sarcomere composition, structure, activation, and relaxation. We then explore the giant sarcomere-resident protein titin, the major contributor to cardiac passive tension. We discuss sarcomere dynamics exemplified by the regulation of titin-based stiffness and the titin life cycle. Finally, we provide an overview of therapeutic strategies that target the sarcomere to improve cardiac contraction and filling.
Similar content being viewed by others

Introduction
Cardiac diseases are the leading cause of death for both men and women in Western countries (Who 2019) and are often characterized by alterations of active forces(contraction) and/orpassive forces (myocardium passive tension) (van der Velden and Stienen 2019). The main source of both active and passive tension is the cardiomyocyte. Cardiomyocytes represent about 50% of the cells in the human heart ventricles (Litvinukova et al. 2020). They are specialized striated muscle cells that actively generate force, pumping blood into the vascular system, and then relax allowing the passive filling of the ventricles up to the limits provided by the passive tension of the organ. Within the cardiomyocyte, sarcomeres are responsible for the generation of active and passive forces. Sarcomeres are highly ordered multiprotein complexes longitudinally aligned to give the characteristic striated look of cardiomyocytes and skeletal muscle cells (Figure 1A). Active forces are generated by actins and myosins sliding along each other in a process that involves cross-bridge formation, while passive tension in the sarcomere is generated by the giant spring protein titin (Maruyama et al. 1977a; Maruyama et al. 1977b; Horowits and Podolsky 1988). Titin is the largest protein in the human proteome, and its homeostasis represents a major challenge for muscle cells. In addition to actin, myosin, and titin, which make up the thin, thick, and elastic filament system, respectively, more than 200 proteins populate the cardiac sarcomere and are continuously and dynamically assembled, modified, and degraded to maintain and adapt cardiac function (Martin and Kirk 2020). Structural and functional alterations of the sarcomere and its regulators underlie a wide spectrum of different cardiac diseases, and it is therefore of critical importance to understanding cardiac sarcomere mechanics. In this review, we revisit the main determinants of sarcomere mechanics, distinguishing between active and passive forces. We discuss the structure of thin, thick, and titin filaments of the sarcomeres; the cross-bridge formation; and the importance of other sarcomere-associated proteins. We then describe the Frank-Starling mechanism and Ca2+ homeostasis as major determinants for sarcomere activation and the interrelation of diastolic filling and systolic ejection. We explore titin as it is the source of passive tension within the sarcomere and discuss current knowledge about titin life cycle as a paradigm for sarcomere homeostasis. Finally, we discuss current and potential therapeutic strategies targeting the sarcomere with examples for both active and passive forces. Overall, we aim to provide insights into the complexity of sarcomere mechanics and its relevance to cardiac disorders.

Sarcomeric structure-function relations. A Schematic of sarcomere (above) and sarcomere ultrastructure observed by electron microscopy (below). The sarcomere is an elastic scaffold that consists of structural proteins lined out from Z-disk to M-band, including actin (black), myosin (green), and titin (grey) that extends through the half-sarcomere. Scale is 100 nm. BLength-tension relation, a contributor to the Frank-Starling Law of the heart. Adapted from (Gordon et al. 1966). C Sarcomere length changes during systole and diastole. D Myofilament compressions during diastolic filling, Myosin in green and actin in red
Sarcomere structure and protein composition
At its core, the mature cardiac sarcomere is a regular hexagonal lattice of thin, actin-containing filaments attached to the Z-disk and thick, myosin-containing filaments interconnected in the middle of the sarcomere via the M-band (Figure 1A). In addition, the filamentous protein titin spans the half-sarcomere from the Z-disk to M-band and stabilizes contraction, among other functions discussed below. The structural properties of sarcomeric proteins and their arrangement are central to cardiac contraction. Sarcomeres are connected in series at the Z-disk and arranged in parallel to form bundles of myofibrils about 1 μm in diameter. Sliding of thick and thin filaments relative to each other results in muscle contraction.
Thick and thin filaments
The thick filament mainly consists of myosin molecules. The myosin superfamily encodes 18 classes of myosin motors, which are ubiquitous in eukaryotes and participate in several cellular motile processes (Hartman and Spudich 2012). A subset of class II myosins power muscle contraction in striated muscles, MYH6 (α-MyHC) and MYH7 (β-MyHC), are known as the cardiac myosin heavy chain isoforms and, albeit 93% identical in humans, display significantly different functional properties. α-MyHC has a higher ATPase activity but generates less force than β-MyHC (Pope et al. 1980; Aksel et al. 2015). In the adult human ventricle, the cardiac myosin composition is 95% β-MyHC and 5% α-MyHC (Reiser et al. 2001), a ratio that further changes in favor of β-MyHC in cardiac diseases (Bouvagnet et al. 1989; Nadal-Ginard and Mahdavi 1989). There are two functional units in class II myosins, a globular motor domain (myosin head) that contains the catalytic ATPase site and binds actin and an α-helical coiled-coil rod domain that dimerizes and assembles into bipolar thick filaments. In the center of the thick filament, the bare zone is free of myosin heads as a consequence of the bipolar arrangement of the myosin molecules. Three-dimensional studies on tarantula thick filaments showed that two myosin heads pack together to form an interacting-heads motif (IHM) (Woodhead et al. 2005). The IHM forms only in relaxed muscle and is an evolutionarily conserved motif among species and muscle types (Alamo et al. 2016). Relaxed myosin exists in two conformations: disordered relaxation (DRX), with one of the two paired myosin heads folded (blocked head) and super relaxation (SRX), with both heads folded back along the thick-filament backbone. As compared to the DRX conformation, myosins in SRX do not participate in contraction and conserve energy, while providing reserve heads that can be activated in response to increased mechanical need (McNamara et al. 2015). The thick filaments contain cardiac myosin-binding protein-C (cMyBP-C) (Carrier et al. 2015), which resides in the A band and interacts with myosin (Starr and Offer 1978; Alyonycheva et al. 1997) and titin (Labeit et al. 1992; Soteriou et al. 1993) and helps maintain sarcomeric structure and regulates cardiac contraction and relaxation.
The thin filament consists of a double-stranded helix of G-actin also called filamentous actin (F-actin), two tropomyosin (Tm) strands, and the three troponin (Tn) subunits, TnT, TnI, and TnC, which together form the Ca2+-regulatory complex of the thin filament. Tm strands lie within the two grooves of F-actin and provide the thin filament with stability, flexibility, and cooperativity. The adult human heart expresses about 80% α-cardiac actin and 20% α-skeletal actin encoded by ACTC and ACTA1 genes, respectively. Higher expression of α-skeletal actin is associated with increased contractility in mouse (Hewett et al. 1994) and in diseased human hearts (Copeland et al. 2010). Myosin thick filaments and actin thin filaments interdigitate and slide past one another to cause sarcomere shortening (Powers et al. 2021). At the functional level, the myosin head directly interacts with two adjacent actin monomers (Behrmann et al. 2012). This attachment of myosin to actin is a multi-step process (Holmes et al. 2004; Behrmann et al. 2012) that starts with calcium binding to TnC, resulting in reduced affinity of TnI for actin (da Silva and Reinach 1991) and exposing myosin-binding sites on actin (McKillop and Geeves 1993). Myosin binding to actin further displaces tropomyosin and prevents its return into the blocking position (Xu et al. 1999). When calcium dissociates from troponin, myosin heads detach from actin progressively, allowing Tm and the troponin complex to move back to an inhibitory position on the thin filament.
Titin filaments
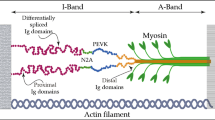
Within the sarcomere, the giant protein titin spans from the Z-disk to the M-band and functions as a scaffold and molecular spring. Titin is encoded by a single gene of 363 exons, and its passive tension can be adjusted in the long term by titin isoform switching via alternative splicing — e.g., peri- and postnatally to meet the increased needs of the developing heart — or more readily by post-translational modifications. The titin region located in the I-band is extensible and consists of immunoglobulin-like (Ig) domains arranged in tandem, the PEVK sequence (rich in proline, glutamate, valine, and lysine residues), and the N2B element. Each functions as a distinct spring element in series. The portion of titin located in the A band is inextensible and composed of regular Ig and fibronectin type 3 (Fn3) domains that form so-called super-repeats. The C-terminal region of titin is located at the M-band and contains a kinase domain, while the N-terminus is in the Z disk of the sarcomere. Titin filaments with opposite polarity overlap and interconnect at both Z-disk and M-band, forming a contiguous filament along the myofibril. Single-molecule studies using laser tweezers and atomic force microscopy (Kellermayer et al. 1997; Li et al. 2002; Watanabe et al. 2002a; Watanabe et al. 2002b) have shown that titin can be compared to a modular polymer of connected elastic segments, each with distinct extensibility, acting as a multistage spring in response to stress applied in the axial direction. Without external force, titin behaves as an entropic spring, where each “module” of the spring is folded and resists stretch. When external force is applied, titin domains unfold and participate in the entropic elasticity of the newly adjusted spring characterized by an increased length and a different spring constant. Upon relaxation, each module folds individually. The unfolding/refolding order is determined by the intrinsic properties of folding pathway for each individual titin domain. Immunolabeling of selected titin domains in rodent left ventricular myocardium revealed that tandem Ig domains in the I-band are extended first, followed by the PEVK segment, and lastly the N2B segment (Linke et al. 1999; Trombitas et al. 1999; Trombitas et al. 2000). As a result, sarcomere passive tension rises slowly at first and then exponentially — limited by the extension of the I-band domains, the rupture of interfilament interactions, and the resistance of non-extensible regions. In fact, Ig and Fn3 domains in the A-band region of titin do not unfold/refold significantly during physiological contraction cycles, because of the strength of interaction between titin and the thick filaments (Wang et al. 1991, 1993). This feature preserves the efficiency of the sarcomere and may function as a molecular ruler, regulating assembly of the thick filament.
Accessory proteins of the sarcomere
A large number of accessory proteins are present within the sarcomere and support the structural, mechanical, signaling, and transport functions. These proteins populate predominantly the Z-disk and the M-band of the sarcomere and are often characterized by a dynamic sarcomeric and/or cellular localization in response to intra- and extra-cellular signals (Lange et al. 2006). For instance, ubiquitin E3 ligases MURF1/2/3, FHL protein family, and CARP are located in the cardiac sarcomere and in the nucleus. MURFs are present at both the M-band and Z-disk suggesting a specialized location-dependent role and can translocate to the nucleus and participate to cardiac transcriptional regulation (Willis et al. 2009; Perera et al. 2011; Willis et al. 2014). FHL2 is highly expressed in the heart (Scholl et al. 2000) and present in two regions of the sarcomere (I-band and M-band), as well as the nucleus, and focal contacts. CARP localizes at the I-band and Z-disk and acts predominantly as a corepressor of transcription through interaction with the ubiquitous transcription factor YB-1 (Jeyaseelan et al. 1997; Zou et al. 1997). Recently, the complexity of the protein networks that connect to the sarcomere Z-disk has been explored using biotin ligase (BioID) inserted at titin’s Z-disk region, providing a census of the sarcomeric proteome in the heart and skeletal muscle in vivo (Rudolph et al. 2020). This approach was used in neonatal and adult heathy mice but could be extended to other sarcomeric regions and pathophysiological states.
Sarcomeric cardiomyopathies
Several genetic cardiac diseases result from mutations in genes that encode for sarcomere proteins. Particularly, hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) are the two predominant types of cardiomyopathy and are highly variable both genetically and phenotypically (Masarone et al. 2018). HCM is characterized by preserved or elevated systolic function but diminished relaxation resulting from asymmetric thickening of the ventricular walls, cardiac fibrosis, and cardiomyocyte disarray (Maron 2010; Harvey and Leinwand 2011). Conversely, in DCM patients, systolic performance is reduced, and the ventricle is dilated rather than hypertrophic (Hershberger et al. 2013; McNally et al. 2013). Both HCM and DCM have been linked to hundreds of sarcomeric gene mutations and different pathogenic mechanisms (Yotti et al. 2019). Generally, sarcomeric proteins with missense mutations are expected to incorporate into the sarcomere and contribute to disease by disrupting normal mechanical function. Conversely, gene mutations as insertions, deletions, premature stop codons, or altered splice sites likely result in unstable proteins that degrade prematurely and cause cardiomyopathies through a haploinsufficiency mechanism (Marston et al. 2009; Kampourakis et al. 2018). HCM mutations reside mainly in MYH7 and MYBPC3, and more rarely in MYL2 or MYL3, which encode the ventricular myosin regulatory light chain (RLC) or myosin essential light chain (ELC), respectively (Richards et al. 2015; Marian and Braunwald 2017). Most HCM mutations in MYH7, MYL2, and MYL3 alter residues that participate in IHM and change the charge of the encoded amino acid, possibly resulting in destabilization of SRX myosin heads and an increased proportion of myosin heads in DRX (Alamo et al. 2017). MYBPC3 mutations associated with HCM have also been linked to changes in the myosin DRX/SRX-ratio in human hearts (McNamara et al. 2017; Toepfer et al. 2019). This is likely the result of intermyofilament interactions between cMyBP-C and myosin that have been recently assessed with unprecedented resolution in the heart (Brunello et al. 2020). A shift in balance favoring DRX over SRX would increase the number of available heads for actin interactions, resulting in hypercontractility, impairment of relaxation, and increased energy consumption, all hallmarks of HCM. A different mechanism is likely responsible for HCM missense mutations occurring in the rod domain of β-MyHC (Blair et al. 2002; Waldmuller et al. 2002; Richard et al. 2003; Karkkainen et al. 2004; Van Driest et al. 2004; Hougs et al. 2005; Perrot et al. 2005). These mutations are located too far from the IHM and the head to affect myosin SRX-DRX ratio or myosin motor and ATPase function but can impair assembly and stability of myofilaments (Buvoli et al. 2008; Armel and Leinwand 2009, 2010; Wolny et al. 2013). Thin filament mutations associated with HCM are predominantly located in TPM1 (α-tropomyosin), TNNT2 (TnT), TNNI3 (TnI), and ACTC1, resulting in a phenotypically distinct class of patients with increased risks for cardiac dysfunction and heart failure (Coppini et al. 2014). These mutations lead to increase calcium sensitivity and altered response to signaling pathways, which enhances contractility but impairs relaxation (Cheng and Regnier 2016; Gangadharan et al. 2017).
As DCM is characterized by the reduced mechanical force generation, pathogenic gene mutations (Kamisago et al. 2000; McNally et al. 2013) result generally in opposite molecular mechanisms as compared to HCM. In fact, DCM mutations in MYH7 reduce myosin ATPase activity and motor function (Schmitt et al. 2006), while DCM mutations in thin-filament proteins decrease myofibril calcium sensitivity, resulting in reduced tension and faster relaxation (Robinson et al. 2007; Gangadharan et al. 2017). Approximately 90% of titin mutations are associated with DCM phenotype, and the remaining to HCM (Greaser 2009). Specifically, mutations that lead to titin truncated variants are highly associated with DCM (Herman et al. 2012; Merlo et al. 2013; Tharp et al. 2019). Integrated analysis of sequencing and transcriptional data from large human cohorts has demonstrated that the effect of titin-truncated variants is dependent on the position of the truncation within the protein (Roberts et al. 2015). The majority of DCM patients carry titin-truncated variants located in the A-band (Herman et al. 2012; Roberts et al. 2015; Akinrinade et al. 2016; Schafer et al. 2017), but in general, truncations occurring in constitutive (highly expressed) exons of titin lead to DCM (Roberts et al. 2015). Mechanisms by which titin-truncating variants cause DCM are probably related to haploinsufficiency rather than dominant negative effects. In fact, truncated titin peptides are not found in DCM hearts (Roberts et al. 2015) likely due to nonsense-mediated decay and rapid turnover of the mutant peptides (Schafer et al. 2017). Accessory proteins of the sarcomere contribute to cardiac contraction and are linked mechanically to thin, thick, and titin filaments as well as additional non-sarcomeric compartments. Mutations of these proteins can also lead to HCM and DCM (Selcen and Carpen 2008; Lange et al. 2020; Wadmore et al. 2021) and have shown to exhibit altered contractility via different mechanisms (Adams et al. 2007; Friedrich et al. 2012; Crocini et al. 2013).
Active forces
The contractile function of the heart is intimately related to the mechanical properties of sarcomeres as their building blocks and is determined by structural parameters of orientation and density of cardiac sarcomeres and by temporal parameters affecting sarcomere activation and relaxation.
The Frank-Starling mechanism
Cardiac contraction and shortening dynamically adjusts on a beat-to-beat basis in response to changes during cardiac filling. This phenomenon is historically referred to as the Frank–Starling Law of the heart (Zimmer 2002; Shiels and White 2008; Sequeira and van der Velden 2015). The law describes the direct relationship between length of the cardiac fibers and the force generated in contraction. This mechanism links changes in cardiac filling to the subsequent ejection of blood into the circulation: an increase in venous return dilates the ventricles, stretching the myocardium that respond by increasing both contractility and stroke volume. In this way, the Frank–Starling mechanism is central to the regulation of cardiac output. For decades, the underlying mechanism for the Frank-Starling Law was considered the degree of overlap between the actin and myosin filaments, or sarcomere length, which would guarantee maximum number of cross-bridges (Gordon et al. 1966). The development of active tension is optimal near the resting sarcomere length, ~ 2 μm, and decreases at higher or lower sarcomere lengths. One can define a descending limb of the isometric length–tension relation, at sarcomere lengths above 2.2 μm, and an ascending limb of the isometric length–tension relation, at sarcomere lengths below ~ 2 μm (Figure 1B). The cardiac sarcomere operates between ~ 2 and 1.8 μm (Spotnitz et al. 1966), i.e., in the ascending limb of the isometric length–tension relation, in which active tension increases with an increase in sarcomere length. Increased diastolic filling results in an increase in sarcomere length and thus an increase in the tension-generating capacity as more myosin heads within the cardiac myocytes are able to bind to actin (Figure 1C). However, a significant increase in active tension occurs over the region 2.0–2.2 μm in mammals (Fabiato and Fabiato 1975; Allen and Kentish 1985), suggesting that myofilament overlap cannot completely account for the length–tension relation in cardiac muscle (Jewell and Wilkie 1960; Hill 1964). Titin has been suggested to play a major role in the length-dependent activation of active force in the heart. Radial forces generated by titin would reduce lateral spacing between thick and thin filaments at higher sarcomere length (Figure 1C, D) that may also influence the affinity of Ca2+ ions to the negatively charged myofilaments (McDonald and Moss 1995; Wang and Fuchs 1995). This would at least in part explain direct relationship between myofilament length and their sensitivity to Ca2+ ions, as indicated in the Frank-Starling Law. Several other structural and molecular mechanisms are likely participating to the Frank-Starling Law and include post-translation modifications of myofilament proteins and sarcomere-independent components (Sequeira and van der Velden 2017). Although a unifying idea that describes the Frank–Starling Law of the heart is still missing, it is an essential component for the cardiac function, and its understanding could help develop therapeutic strategies for human heart disease.
Calcium homeostasis
Cytosolic calcium concentration is fundamental for mechanical activation of the sarcomere. At rest, intracellular calcium concentration is at submicromolar level but rises during the plateau phase of the cardiac action potential, thanks to the cardiac-specific voltage-sensitive L-type Ca2+ channels. This unique cardiac current triggers release of additional calcium from the sarcoplasmic reticulum via the ryanodine receptor (RyR, calcium-induced calcium release) (Bers 2002) that activates contraction. Collectively, this process is called excitation–contraction (E-C) coupling (Fozzard et al. 1992; Bers 2002; Page, 2002). The level of sarcomere activation is not simply proportional to cytosolic calcium concentration, but rather it is the result of a complex and dynamic signaling process that is regulated by numerous factors (de Tombe 2003). However, there is generally a direct relationship between the magnitude of the calcium transient and the sarcomere (ventricle) contraction (Gwathmey and Hajjar 1990; Backx et al. 1995; Bassani et al. 1995), thus making calcium one of the major determinants of heart contractility. Altered intracellular Ca2+ handling underlies numerous cardiac diseases. In heart failure, for example, E-C coupling is affected by functionally defective L-type Ca2+ channels (Piot et al. 1996; Barrere-Lemaire et al. 2000; He et al. 2001) and increased space between L-type Ca2+ channels and the RyR (Gomez et al. 1997), as well as decreased Ca2+ content in the sarcoplasmic reticulum (Lindner et al. 1998; O'Rourke et al. 1999; Hobai and O'Rourke 2001) and altered channel-gating property of RyR (Yamamoto et al. 1999; Ono et al. 2000; Yano et al. 2000; Marx et al. 2001). Sarcomere activation not only depends on the amount of Ca2+ released at each given heartbeat but also on the rate of Ca2+ release across the cell (Yano et al. 2005). To ensure a synchronized activation of all sarcomeres within each cell, mammalian cardiac myocytes are provided with a complex network of membrane invaginations called the transverse-axial tubular system or t-tubules(Ferrantini et al. 2013). T-tubules allow for rapid and homogenous propagation of the cardiac action potential, thus Ca2+ entry, and affect cardiac sarcomere mechanics. Loss of t-tubules by acute formamide-induced osmotic shock (detubulation) induces prolonged contraction kinetics and impaired force–frequency response and can be, at least in part, mitigated by improving Ca2+ synchrony and propagation (Ferrantini et al. 2014). T-tubular structural remodeling reduces Ca2+ synchrony in failing human hearts (Louch et al. 2004) and murine cardiac disease models (Louch et al. 2006; Wei et al. 2010; Heinzel et al. 2011), and also, t-tubule functional remodeling (Crocini et al. 2017) can affect Ca2+ propagation and contraction (Crocini et al. 2014; Crocini et al. 2016a; Crocini et al. 2016b; Scardigli et al. 2018).
Passive forces
When cardiac muscle is stretched beyond its resting sarcomere length, it develops passive tension. Passive tension is essential for the heart as it contributes to the diastolic wall tension that determines the extent of filling of the ventricle and the subsequent stroke volume (for reviews, see Allen and Kentish 1985; Brady,1991a). Passive tension is also important in the activated myocardium because it participates in determining the shortening velocity of cardiac trabeculae (de Tombe and ter Keurs 1992) and cardiac myocytes (Sweitzer and Moss 1993). Over the working range of the heart (sarcomere lengths 1.8-2.2 μm), collagen and titin are the most important contributors to passive tension, with collagen dominating at the longer sarcomere lengths of the working range and titin at the shorter lengths (Linke et al. 1994; Granzier and Irving 1995). Collagen is a fibrous protein of the extracellular matrix packed to form long, thin, and strong fibrils. Notably, cardiac fibrosis with altered collagen expression and composition is a hallmark of many cardiac diseases and can dramatically affect cardiac contraction and relaxation (Travers et al. 2016). Minor contributions are also provided by intermediate filaments, measured as ~ 10-fold less than titin, and even less by microtubules. Here, we focus on the titin protein as it represents the sarcomeric source of passive tension.
Titin-based passive tension
As mentioned, titin-based stiffness can be adjusted both in the long term by titin isoform switching (Granzier and Irving 1995; Opitz et al. 2004) or more rapidly by post-translational modifications (Koser et al. 2019). Abnormal isoform ratios or post-translational modifications can dramatically affect passive tension of cardiomyocytes and have been described in many human cardiac diseases (LeWinter and Granzier 2014). In the heart, there are 3 main types of titin isoforms: fetal cardiac titin, adult N2BA, and adult N2B. They differ in their I-band extensible regions. Fetal cardiac titin isoforms are longer and more compliant than either N2B or N2BA (Lahmers et al. 2004; Opitz et al. 2004) (Figure 2A). These isoforms could be beneficial in fetal-neonatal development because of the low filling pressure of the fetal heart and the structural constraints provided by other tissues that limit cardiac reserve in the fetus (Walker and de Tombe 2004). Fetal isoforms gradually disappear during postnatal development in favor of the mature N2BA and N2B. Titin N2BA isoforms have a longer PEVK sequence and a variable number of additional Ig domains resulting in more compliant isoforms than N2B titin. Both isoforms are co-expressed in the cardiac sarcomere, and their ratio is a determinant of passive stiffness. In adult human left ventricle, the N2BA/N2B ratio is ~0.6 and can change in disease (Neagoe et al. 2002; Makarenko et al. 2004; Nagueh et al. 2004) (Figure 2B). The N2BA:N2B ratio is increased in DCM patients (Nagueh et al. 2004) and would result in reduced passive tension leading to reduced diastolic forces and dilation of the heart, both hallmarks of DCM. Switching to the longer N2BA isoform could represent an initial compensatory mechanism that improves diastolic function; however, long-term reduction of passive tension and diastolic pressure could worsen contractile performance in systole (Makarenko et al. 2004).

Titin-based passive tension. A Developmental changes in titin isoform expression facilitate the transition from fetal to adult force generation. B The relationship of sarcomere length and passive tension changes in disease via posttranslational modifications (protein kinases PKA, PKC, PKG acting on the titin spring region) and changes in titin isoform expression (N2BA to N2B titin isoform ratio)
The RNA-binding motif 20 (RBM20) is a well-characterized regulator of cardiac isoform expression for titin and over 30 additional genes (Guo et al. 2012; Maatz et al. 2014; van den Hoogenhof et al. 2018; Lennermann et al. 2020). RBM20 is regarded as a splicing repressor. In the case of titin pre-mRNA, RBM20 binds and represses splicing of large stretches of exons, allowing alternative splice sites at the 3′ or 5′ end of RBM20-repressed regions to splice together (Li et al. 2013). However, loss of RBM20 function leads to larger titin proteins, with reduced N2B isoform and upregulation of N2BA isoform and another, giant, and even more compliant titin isoform called N2BA-G. As a result, myocardial stiffness is reduced, and resting sarcomere length is increased (Greaser et al. 2008; Guo et al. 2012; Methawasin et al. 2014; Beqqali et al. 2016; Methawasin et al. 2016). RBM20 mutations lead to a severe form of DCM with high rates of heart failure, arrhythmias, and sudden cardiac death (Brauch et al. 2009; Li et al. 2010; Wells et al. 2013; van den Hoogenhof et al. 2018), a complex phenotype resulting from missplicing of several RBM20 target genes (Guo et al. 2012; Maatz et al. 2014). In particular, missplicing of the RBM20-target calcium/calmodulin-dependent protein kinase II (CaMKII), a crucial modulator of E-C coupling in cardiomyocytes (Maier and Bers 2007), is likely responsible for altered cellular calcium handling and cellular calcium overload observed in RBM20 cardiomyopathy (van den Hoogenhof et al. 2018).
In order to meet beat-to-beat requirements of the heart, titin tension can be modulated via post-translational modifications of titin spring elements (Koser et al. 2019), especially N2B and PEVK unique sequences. A few kinases have been implicated in titin phosphorylation, including protein kinase A (PKA), protein kinase G (PKG), and CaMKII (Kruger et al. 2009; Hamdani et al. 2013; Kotter et al. 2013; Koser et al. 2019). PEVK phosphorylation (via CaMKII) increases titin stiffness, while N2B phosphorylation (via PKG and PKA) decreases it. Abnormal regulation of signaling pathways has been found in numerous cardiac diseases and can affect titin passive tension. For example, human failing hearts show increased cardiomyocyte passive tension via hyperphosphorylation of PEVK and reduced phosphorylation of N2B (Hopf et al. 2018). Targeting intracellular pathways holds very promising therapeutic potential and has been already validated in animal models (Eisenberg et al. 2016; Hopf et al. 2018; Leite-Moreira et al. 2018; Slater et al. 2019).
Titin life cycle
Considering the impressive molecular weights of titin and the plethora of possible isoforms, it could be tempting to think that titin protein homeostasis is a static and slow process. However, titin dynamics studies revealed an unexpected level of protein mobility in and out of sarcomeres (da Silva et al. 2011; Rudolph et al. 2019; Cadar et al. 2020). Pulse-chase experiments demonstrated that the protein half-life of titin is approximately 70 h in cultured skeletal muscle cells (Isaacs et al. 1989). Using isolated neonatal cardiomyocytes from a titin-eGFP knock-in mouse model in combination with fluorescent recovery after photobleaching (FRAP) imaging, a later work (da Silva et al. 2011) reported that the exchange half-life for titin in the sarcomere is 2.1 h. Taken together, these two works would suggest the existence of a pool of sarcomeric and non-sarcomeric titin molecules that can rapidly exchange in the sarcomere as they move within hours and are replaced within days. Titin synthesis, motility, and sarcomeric (dis)integration have been further resolved using FRAP and a bifluorescently labeled knock-in mouse to simultaneously visualize both ends of titin molecules (Rudolph et al. 2019). This work demonstrated that there is a pool (> 15%) of soluble titin molecules readily available for integration into the sarcomere. Additionally, this work showed that titin is inserted into the mature sarcomere stochastically from either ends of the protein — starting at the Z-disk or at the M-band — in contrast with myofibrillogenesis predictions (Rhee et al. 1994; Du et al. 2008). Titin dynamics differed considerably between embryonic and mature cardiomyocytes, highlighting the complexity of cardiac sarcomere assembly and roles at various life stages.
The sarcomere as a therapeutic target
There is a remarkable growth of knowledge about sarcomere mechanics and how active and passive forces are regulated in disease states. Translation of these findings to the clinics has been facilitated by both technological advances in model systems and analysis tools, including the generation of small and large animal models (e.g., using CRISPR/Cas9 technology) and the advent of induced pluripotent stem cell (iPSC) technology. The differentiation of iPSCs into cardiomyocytes has seen tremendous advances (Hirt et al. 2014; Ronaldson-Bouchard et al. 2018), and efforts combining iPSC-derived cardiomyocytes with clinically relevant animal models are generating fundamental insights to understanding mechanisms of cardiac pathology and to exploring different therapeutic strategies (Coppini et al. 2014). For instance, a functional readout has been developed to study TnT mutations leading to HCM or DCM (Pettinato et al. 2020), RBM20 therapy has been validated in a DCM model (Briganti et al. 2020), and disruption of SRX state has been linked to hypercontractility in cells harboring a MYH7 mutation (Vander Roest et al. 2021). These new experimental tools confirm that the sarcomere is an excellent therapeutic target (Table 1) — due to both the number of mutations in sarcomeric proteins causing human disease and because of its unique role in determining systolic and diastolic properties. Therapeutic approaches that directly target the primary cause of inherited cardiomyopathies, i.e., genetic mutations of sarcomere proteins, are limited, but targeting the sarcomere has shown promising results in ameliorating a wide variety of cardiomyopathies. We discuss here sarcomere targets and potential therapies distinguishing between active and passive forces.
Targeting active forces
Genetic approaches to revert sarcomere mutations have been explored in animal models and iPSC-derived cardiomyocytes (Gedicke-Hornung et al. 2013; Jiang et al. 2013; Mearini et al. 2013; Prondzynski et al. 2017; van Kampen and van Rooij 2019), but translation to the patient could be challenging due to possible off-target effects of gene-editing, invasiveness, and poor efficiency of delivery methods. To date, cardiac gene therapy in adult heart failure patients has been attempted with an adeno-associated viral vector encoding the non-sarcomeric gene SERCA2a under control of a cytomegalovirus promoter and intracoronary infusion (Jessup et al. 2011; Zsebo et al. 2014; Greenberg et al. 2016; Hulot et al. 2017; Lyon et al. 2020) with relatively disappointing outcomes. Considering their central role in sarcomere mechanics, Ca2+ handling and myofilament Ca2+ sensitivity stand out as targets to treat cardiac disease. L-type Ca2+ channel blockers are well established to treat intracellular calcium overload in cardiovascular disease (Rosing et al. 1979; Akhtar et al. 1989; Udelson et al. 1989; Gistri et al. 1994; Ho et al. 2015). Promising results to reduce myofilament Ca2+ sensitivity have been obtained with catechins, contained in green tea, in an animal model of genetic HCM (Adhikari and Wang 2004). Furthermore, molecules developed to inhibit calmodulin (Hidaka et al. 1980; Silver et al. 1986; Osawa et al. 1998) hold promise to exert a similar inhibitory effect on myofilaments due to the structural homology between calmodulin and cTnC. In the heart, the positive inotropic effect of β-adrenergic signaling is accompanied by reduced myofilament Ca2+ sensitivity via PKA phosphorylation of cTnI (Ser23/24) (Zhang et al. 1995; Kentish et al. 2001). β-Adrenergic receptor antagonists (β-blockers) have been used for decades to treat a variety of cardiac diseases, including hypertension, heart failure, cardiac arrhythmias, and myocardial infarction. Among the β-blockers used in clinical therapy, nebivolol has been reported to additionally desensitize cardiac myofilaments (Zeitz et al. 2000). Clinical trials using small molecules to change sarcomere mechanics are promising and include the selective cardiac myosin activator omecamtiv mecarbil to treat systolic heart failure (Teerlink et al. 2021) and the selective myosin inhibitor mavacamtem to treat obstructive HCM (Olivotto et al. 2020). Mavacamtem acts on myosin and stabilizes the SRX configuration thus reducing hypercontractility (Kawas et al. 2017; Anderson et al. 2018). Cardiac myosin isoform ratio is known to change in favor of β-MyHC in disease; thus, strategies to counteract this isoform shift could be of benefit. The RNA transcript of the ancient myosin 7b (MYH7b) — non-translated in mammalian hearts (Rossi et al. 2010; Warkman et al. 2012; Lee et al. 2019; Peter et al. 2019) — includes a long non-coding RNA that has been demonstrated capable of regulating the expression ratio of α-MyHC and β-MyHC in human iPSC-derived cardiomyocytes, and thus may represent a novel therapeutic target (Broadwell et al. 2021).
Targeting passive forces
Titin is an excellent target to address cardiac diseases related to altered passive tension. High sarcomere passive stiffness due to low PKG titin phosphorylation is found in some patients with heart failure. PKG activity depends on the levels of cyclic guanosine monophosphate and may be increased via inhibition of phosphodiesterases. However, treatments of heart failure patients with the phosphodiesterase-5 inhibitor, sildenafil (Redfield et al. 2013; Hoendermis et al. 2015) did not improve outcomes. Promising results have been obtained by inhibiting phosphodiesterase-9A in diseased human tissues (Lee et al. 2015), although its expression in failing human cardiomyocytes remains uncertain (Li et al. 2019). Other studies have shown that the specific stimulation of β3 adrenergic receptors may exert beneficial effects incardiomyocytes by increasing PKG activity (Gauthier et al. 1998; Hammond and Balligand 2012). PKG phosphorylates the same PKA site in cTnI (Ser23/24) reducing myofilament Ca2+ sensitivity (Lee et al. 2010). Thus, activation of β3 adrenergic receptor would conveniently decrease myofilament Ca2+ sensitivity while improving sarcomere relaxation.
In a mouse model of heart failure with preserved ejection fraction, administration of metformin, used in patients to treat type 2 diabetes, improved diastolic function based on the phosphorylation of a PKA site on N2B domain of titin (Slater et al. 2019). An alternative strategy to change passive tension builds on RBM20 to target titin isoform expression and has been explored in different experimental models of diastolic dysfunction (Methawasin et al. 2014; Hinze et al. 2016; Methawasin et al. 2016). Improved ventricular filling and exercise capacity, reduced end-diastolic pressure, and increased expression of hypertrophic genes were associated with negative effects on the Frank–Starling mechanism, force generation, and slower cross-bridge kinetics (Pulcastro et al. 2016), highlighting the tight connection between passive and active forces in the sarcomere. Together with titin, the extracellular matrix represents a major contributor of cardiac passive tension. Fibrosis, the excessive deposition of collagen and altered composition of the extracellular matrix, is a hallmark of several cardiac diseases. Although we focused this review on forces originating from the sarcomere, combating cardiac fibrosis remains an essential clinical intervention (Travers et al. 2016). Notably, fibrosis can induce a switch to the more compliant N2BA titin isoform to partially counteract increased collagen-based stiffness (Neagoe et al. 2002; Makarenko et al. 2004; Nagueh et al. 2004; Hamdani et al. 2010). Although non-sarcomeric in nature, antifibrotic therapy could affect titin isoform composition. Advances in biomaterial sciences to control the extracellular stiffness (Walker et al. 2021; Jian et al. 2014; Crocini et al. 2020; Shimkunas et al. 2021), could help dissecting the crosstalk between the cardiac sarcomere and the extracellular matrix and their relative contribution to cardiac passive tension.
Concluding remarks
Cardiac diseases remain an enormous burden and unresolved medical problem worldwide. The need for novel therapeutic approaches and interventions is obvious, and targeting cardiac sarcomeres is a powerful and effective strategy to combat cardiac disorders. Significant progress has been made toward understanding cardiac sarcomere structure and function, but a comprehensive and detailed picture is still missing — starting with the complete survey of the dynamic sarcomeric proteome, its interactions and adaptation in health and disease.
Change history
01 July 2023
A Correction to this paper has been published: https://doi.org/10.1007/s12551-023-01086-1
References
Adams V, Linke A, Wisloff U, Doring C, Erbs S, Krankel N, Witt CC, Labeit S, Muller-Werdan U, Schuler G, Hambrecht R (2007) Myocardial expression of Murf-1 and MAFbx after induction of chronic heart failure: Effect on myocardial contractility. Cardiovasc Res 73:120–129
Adhikari BB, Wang K (2004) Interplay of troponin- and myosin-based pathways of calcium activation in skeletal and cardiac muscle: the use of W7 as an inhibitor of thin filament activation. Biophys Rev 86:359–370
Akhtar M, Tchou P, Jazayeri M (1989) Use of calcium channel entry blockers in the treatment of cardiac arrhythmias. Circulation 80:IV31–IV39
Akinrinade O, Alastalo TP, Koskenvuo JW (2016) Relevance of truncating titin mutations in dilated cardiomyopathy. Clin Genet 90:49–54
Aksel T, Choe Yu E, Sutton S, Ruppel KM, Spudich JA (2015) Ensemble force changes that result from human cardiac myosin mutations and a small-molecule effector. Cell rep 11:910–920
Alamo L, Qi D, Wriggers W, Pinto A, Zhu J, Bilbao A, Gillilan RE, Hu S, Padron R (2016) Conserved intramolecular interactions maintain myosin interacting-heads motifs explaining tarantula muscle super-relaxed state structural basis. J Mol Biol 428:1142–1164
Alamo L, Ware JS, Pinto A, Gillilan RE, Seidman JG, Seidman CE, Padron R (2017) Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. eLife 13(6):e24634
Allen DG, Kentish JC (1985) The cellular basis of the length-tension relation in cardiac muscle. J Mol Cell Cardiol 17:821–840
Alyonycheva TN, Mikawa T, Reinach FC, Fischman DA (1997) Isoform-specific interaction of the myosin-binding proteins (MyBPs) with skeletal and cardiac myosin is a property of the C-terminal immunoglobulin domain. J Biol Chem 272:20866–20872
Anderson RL, Trivedi DV, Sarkar SS, Henze M, Ma W, Gong H, Rogers CS, Gorham JM, Wong FL, Morck MM, Seidman JG, Ruppel KM, Irving TC, Cooke R, Green EM, Spudich JA (2018) Deciphering the super relaxed state of human beta-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc Natl Acad Sci U.S.A 115:E8143–E8152
Armel TZ, Leinwand LA (2009) Mutations in the beta-myosin rod cause myosin storage myopathy via multiple mechanisms. Proc Natl Acad Sci U.S.A 106:6291–6296
Armel TZ, Leinwand LA (2010) A mutation in the beta-myosin rod associated with hypertrophic cardiomyopathy has an unexpected molecular phenotype. Biochem Biophys Res Commun 391:352–356
Backx PH, Gao WD, Azan-Backx MD, Marban E (1995) The relationship between contractile force and intracellular [Ca2+] in intact rat cardiac trabeculae. J Gen Physiol 105:1–19
Barrere-Lemaire S, Piot C, Leclercq F, Nargeot J, Richard S (2000) Facilitation of L-type calcium currents by diastolic depolarization in cardiac cells: impairment in heart failure. Cardiovasc Res 47:336–349
Bassani JW, Yuan W, Bers DM (1995) Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am J Physiol 268:C1313–C1319
Behrmann E, Muller M, Penczek PA, Mannherz HG, Manstein DJ, Raunser S (2012) Structure of the rigor actin-tropomyosin-myosin complex. Cell 150:327–338
Beqqali A, Bollen IA, Rasmussen TB, van den Hoogenhof MM, van Deutekom HW, Schafer S, Haas J, Meder B, Sorensen KE, van Oort RJ, Mogensen J, Hubner N, Creemers EE, van der Velden J, Pinto YM (2016) A mutation in the glutamate-rich region of RNA-binding motif protein 20 causes dilated cardiomyopathy through missplicing of titin and impaired Frank-Starling mechanism. Cardiovasc Res 112:452–463
Bers DM (2002) Cardiac excitation-contraction coupling. Nature 415:198–205
Blair E, Redwood C, de Jesus OM, Moolman-Smook JC, Brink P, Corfield VA, Ostman-Smith I, Watkins H (2002) Mutations of the light meromyosin domain of the beta-myosin heavy chain rod in hypertrophic cardiomyopathy. Circ Res 90:263–269
Bouvagnet P, Mairhofer H, Leger JO, Puech P, Leger JJ (1989) Distribution pattern of alpha and beta myosin in normal and diseased human ventricular myocardium. Basic Res Cardiol 84:91–102
Brauch KM, Karst ML, Herron KJ, de Andrade M, Pellikka PA, Rodeheffer RJ, Michels VV, Olson TM (2009) Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol 54:930–941
Briganti F, Sun H, Wei W, Wu J, Zhu C, Liss M, Karakikes I, Rego S, Cipriano A, Snyder M, Meder B, Xu Z, Millat G, Gotthardt M, Mercola M, Steinmetz LM (2020) iPSC modeling of RBM20-Deficient DCM identifies upregulation of RBM20 as a therapeutic strategy. Cell Rep 32:108117
Broadwell LJ, Smallegan MJ, Rigby KM, Navarro-Arriola JS, Montgomery RL, Rinn JL, Leinwand LA (2021) Myosin 7b is a regulatory long noncoding RNA (lncMYH7b) in the human heart. J Biol Chem 296:100694
Brunello E, Fusi L, Ghisleni A, Park-Holohan SJ, Ovejero JG, Narayanan T, Irving M (2020) Myosin filament-based regulation of the dynamics of contraction in heart muscle. Proc Natl Acad Sci U S A 117:8177–8186
Buvoli M, Hamady M, Leinwand LA, Knight R (2008) Bioinformatics assessment of beta-myosin mutations reveals myosin's high sensitivity to mutations. Trends Cardiovasc Med 18:141–149
Cadar AG, Feaster TK, Bersell KR, Wang L, Hong T, Balsamo JA, Zhang Z, Chun YW, Nam YJ, Gotthardt M, Knollmann BC, Roden DM, Lim CC, Hong CC (2020) Real-time visualization of titin dynamics reveals extensive reversible photobleaching in human induced pluripotent stem cell-derived cardiomyocytes. Am J Physiol Cell physiol 318:C163–C173
Carrier L, Mearini G, Stathopoulou K, Cuello F (2015) Cardiac myosin-binding protein C (MYBPC3) in cardiac pathophysiology. Gene 573:188–197
Cheng Y, Regnier M (2016) Cardiac troponin structure-function and the influence of hypertrophic cardiomyopathy associated mutations on modulation of contractility. Arch Biochem Biophys 601:11–21
Copeland O, Nowak KJ, Laing NG, Ravenscroft G, Messer AE, Bayliss CR, Marston SB (2010) Investigation of changes in skeletal muscle alpha-actin expression in normal and pathological human and mouse hearts. J Muscle Res Cell Motil 31:207–214
Coppini R, Ho CY, Ashley E, Day S, Ferrantini C, Girolami F, Tomberli B, Bardi S, Torricelli F, Cecchi F, Mugelli A, Poggesi C, Tardiff J, Olivotto I (2014) Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin-filament gene mutations. J Am Coll Cardiol 64:2589–2600
Crocini C, Arimura T, Reischmann S, Eder A, Braren I, Hansen A, Eschenhagen T, Kimura A, Carrier L (2013) Impact of ANKRD1 mutations associated with hypertrophic cardiomyopathy on contraction parameters of engineered heart tissue. Basic Res Cardiol 108:349
Crocini C, Coppini R, Ferrantini C, Yan P, Loew LM, Tesi C, Cerbai E, Poggesi C, Pavone FS, Sacconi L (2014) Defects in T-tubular electrical activity underlie local alterations of calcium release in heart failure. Proc Natl Acad Sci U S A 111:15196–15201
Crocini C, Coppini R, Ferrantini C, Yan P, Loew LM, Poggesi C, Cerbai E, Pavone FS, Sacconi L (2016a) T-Tubular Electrical Defects Contribute to Blunted beta-Adrenergic Response in Heart Failure. Int J Mol Sci 17(9):1471
Crocini C, Ferrantini C, Scardigli M, Coppini R, Mazzoni L, Lazzeri E, Pioner JM, Scellini B, Guo A, Song LS, Yan P, Loew LM, Tardiff J, Tesi C, Vanzi F, Cerbai E, Pavone FS, Sacconi L, Poggesi C (2016b) Novel insights on the relationship between T-tubular defects and contractile dysfunction in a mouse model of hypertrophic cardiomyopathy. J Mol Cell Cardiol 91:42–51
Crocini C, Ferrantini C, Coppini R, Sacconi L (2017) Electrical defects of the transverse-axial tubular system in cardiac diseases. J Physiol 595:3815–3822
Crocini C, Walker CJ, Anseth KS, Leinwand LA (2020) Three-dimensional encapsulation of adult mouse cardiomyocytes in hydrogels with tunable stiffness. Prog Biophys Mol Biol 154:71–79
da Silva AC, Reinach FC (1991) Calcium binding induces conformational changes in muscle regulatory proteins. Trends Biochem Sci 16:53–57
da Silva LK, Pietas A, Radke MH, Gotthardt M (2011) Titin visualization in real time reveals an unexpected level of mobility within and between sarcomeres. J Cell Biol 193:785–798
de Tombe PP (2003) Cardiac myofilaments: mechanics and regulation. J Biomech 36:721–730
de Tombe PP, ter Keurs HE (1992) An internal viscous element limits unloaded velocity of sarcomere shortening in rat myocardium. J Physiol 454:619–642
Du A, Sanger JM, Sanger JW (2008) Cardiac myofibrillogenesis inside intact embryonic hearts. Dev Biol 318:236–246
Eisenberg T et al (2016) Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med 22:1428–1438
Fabiato A, Fabiato F (1975) Dependence of the contractile activation of skinned cardiac cells on the sarcomere length. Nature 256:54–56
Ferrantini C, Crocini C, Coppini R, Vanzi F, Tesi C, Cerbai E, Poggesi C, Pavone FS, Sacconi L (2013) The transverse-axial tubular system of cardiomyocytes. Cell Mol Life Sci: CMLS 70:4695–4710
Ferrantini C, Coppini R, Sacconi L, Tosi B, Zhang ML, Wang GL, de Vries E, Hoppenbrouwers E, Pavone F, Cerbai E, Tesi C, Poggesi C, ter Keurs HE (2014) Impact of detubulation on force and kinetics of cardiac muscle contraction. J Gen Physiol 143:783–797
Fozzard HA, Haber E, Jennings RB, Katz AM (1992) The heart and cardiovascular system. Raven Press, New York
Friedrich FW et al (2012) Evidence for FHL1 as a novel disease gene for isolated hypertrophic cardiomyopathy. Hum Mol Genet 21:3237–3254
Gangadharan B, Sunitha MS, Mukherjee S, Chowdhury RR, Haque F, Sekar N, Sowdhamini R, Spudich JA, Mercer JA (2017) Molecular mechanisms and structural features of cardiomyopathy-causing troponin T mutants in the tropomyosin overlap region. Proc Natl Acad Sci U S A 114:11115–11120
Gauthier C, Leblais V, Kobzik L, Trochu JN, Khandoudi N, Bril A, Balligand JL, Le Marec H (1998) The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J Clin Investig 102:1377–1384
Gedicke-Hornung C, Behrens-Gawlik V, Reischmann S, Geertz B, Stimpel D, Weinberger F, Schlossarek S, Precigout G, Braren I, Eschenhagen T, Mearini G, Lorain S, Voit T, Dreyfus PA, Garcia L, Carrier L (2013) Rescue of cardiomyopathy through U7snRNA-mediated exon skipping in Mybpc3-targeted knock-in mice. EMBO Mol Med 5:1128–1145
Gistri R, Cecchi F, Choudhury L, Montereggi A, Sorace O, Salvadori PA, Camici PG (1994) Effect of verapamil on absolute myocardial blood flow in hypertrophic cardiomyopathy. Am J Cardiol 74:363–368
Gomez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA, Lederer WJ (1997) Defective excitation-contraction coupling in experimental cardiac hypertrophy and heart failure. Science 276:800–806
Gordon AM, Huxley AF, Julian FJ (1966) The variation in isometric tension with sarcomere length in vertebrate muscle fibres. J Physiol 184:170–192
Granzier HL, Irving TC (1995) Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J 68:1027–1044
Greaser ML (2009) Stressing the giant: a new approach to understanding dilated cardiomyopathy. J Mol Cell Cardiol 47:347–349
Greaser ML, Warren CM, Esbona K, Guo W, Duan Y, Parrish AM, Krzesinski PR, Norman HS, Dunning S, Fitzsimons DP, Moss RL (2008) Mutation that dramatically alters rat titin isoform expression and cardiomyocyte passive tension. J Mol Cell Cardiol 44:983–991
Greenberg B, Butler J, Felker GM, Ponikowski P, Voors AA, Desai AS, Barnard D, Bouchard A, Jaski B, Lyon AR, Pogoda JM, Rudy JJ, Zsebo KM (2016) Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet 387:1178–1186
Guo W et al (2012) RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med 18:766–773
Gwathmey JK, Hajjar RJ (1990) Intracellular calcium related to force development in twitch contraction of mammalian myocardium. Cell Calcium 11:531–538
Hamdani N, Borbely A, Veenstra SP, Kooij V, Vrydag W, Zaremba R, Dos Remedios C, Niessen HW, Michel MC, Paulus WJ, Stienen GJ, van der Velden J (2010) More severe cellular phenotype in human idiopathic dilated cardiomyopathy compared to ischemic heart disease. J Muscle Res Cell Motil 31:289–301
Hamdani N, Krysiak J, Kreusser MM, Neef S, Dos Remedios CG, Maier LS, Kruger M, Backs J, Linke WA (2013) Crucial role for Ca2(+)/calmodulin-dependent protein kinase-II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ Res 112:664–674
Hammond J, Balligand JL (2012) Nitric oxide synthase and cyclic GMP signaling in cardiac myocytes: from contractility to remodeling. J Mol Cell Cardiol 52:330–340
Hartman MA, Spudich JA (2012) The myosin superfamily at a glance. J Cell Sci 125:1627–1632
Harvey PA, Leinwand LA (2011) The cell biology of disease: cellular mechanisms of cardiomyopathy. J Cell Biol 194:355–365
He J, Conklin MW, Foell JD, Wolff MR, Haworth RA, Coronado R, Kamp TJ (2001) Reduction in density of transverse tubules and L-type Ca(2+) channels in canine tachycardia-induced heart failure. Cardiovasc Res 49:298–307
Heinzel FR, MacQuaide N, Biesmans L, Sipido K (2011) Dyssynchrony of Ca2+ release from the sarcoplasmic reticulum as subcellular mechanism of cardiac contractile dysfunction. J Mol Cell Cardiol 50:390–400
Herman DS et al (2012) Truncations of titin causing dilated cardiomyopathy. N Eng J Med 366:619–628
Hershberger RE, Hedges DJ, Morales A (2013) Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol 10:531–547
Hewett TE, Grupp IL, Grupp G, Robbins J (1994) Alpha-skeletal actin is associated with increased contractility in the mouse heart. Circ Res 74:740–746
Hidaka H, Yamaki T, Naka M, Tanaka T, Hayashi H, Kobayashi R (1980) Calcium-regulated modulator protein interacting agents inhibit smooth muscle calcium-stimulated protein kinase and ATPase. Mol Pharmacol 17:66–72
Hill AV (1964) The effect of tension in prolonging the active state in a twitch. Proc R Soc Lond B, Biol Sci 159:589–595
Hinze F, Dieterich C, Radke MH, Granzier H, Gotthardt M (2016) Reducing RBM20 activity improves diastolic dysfunction and cardiac atrophy. J Mol Med 94:1349–1358
Hirt MN, Boeddinghaus J, Mitchell A, Schaaf S, Bornchen C, Muller C, Schulz H, Hubner N, Stenzig J, Stoehr A, Neuber C, Eder A, Luther PK, Hansen A, Eschenhagen T (2014) Functional improvement and maturation of rat and human engineered heart tissue by chronic electrical stimulation. J Mol Cell Cardiol 74:151–161
Ho CY, Lakdawala NK, Cirino AL, Lipshultz SE, Sparks E, Abbasi SA, Kwong RY, Antman EM, Semsarian C, Gonzalez A, Lopez B, Diez J, Orav EJ, Colan SD, Seidman CE (2015) Diltiazem treatment for pre-clinical hypertrophic cardiomyopathy sarcomere mutation carriers: a pilot randomized trial to modify disease expression. JACC Heart Fail 3:180–188
Hobai IA, O'Rourke B (2001) Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation 103:1577–1584
Hoendermis ES, Liu LC, Hummel YM, van der Meer P, de Boer RA, Berger RM, van Veldhuisen DJ, Voors AA (2015) Effects of sildenafil on invasive haemodynamics and exercise capacity in heart failure patients with preserved ejection fraction and pulmonary hypertension: a randomized controlled trial. Eur Heart J 36:2565–2573
Holmes KC, Schroder RR, Sweeney HL, Houdusse A (2004) The structure of the rigor complex and its implications for the power stroke. Phil Trans R Soc Lond B, Biol Sci 359:1819–1828
Hopf AE, Andresen C, Kotter S, Isic M, Ulrich K, Sahin S, Bongardt S, Roll W, Drove F, Scheerer N, Vandekerckhove L, De Keulenaer GW, Hamdani N, Linke WA, Kruger M (2018) Diabetes-induced cardiomyocyte passive stiffening is caused by impaired insulin-dependent titin modification and can be modulated by neuregulin-1. Circ Res 123:342–355
Horowits R, Podolsky RJ (1988) Thick filament movement and isometric tension in activated skeletal muscle. Biophys J 54:165–171
Hougs L, Havndrup O, Bundgaard H, Kober L, Vuust J, Larsen LA, Christiansen M, Andersen PS (2005) One third of Danish hypertrophic cardiomyopathy patients with MYH7 mutations have mutations [corrected] in MYH7 rod region. European journal of human genetics : EJHG 13:161–165
Hulot JS et al (2017) Effect of intracoronary administration of AAV1/SERCA2a on ventricular remodelling in patients with advanced systolic heart failure: results from the AGENT-HF randomized phase 2 trial. Eur J Heart Fail 19:1534–1541
Isaacs WB, Kim IS, Struve A, Fulton AB (1989) Biosynthesis of titin in cultured skeletal muscle cells. J Cell Biol 109:2189–2195
Jessup M, Greenberg B, Mancini D, Cappola T, Pauly DF, Jaski B, Yaroshinsky A, Zsebo KM, Dittrich H, Hajjar RJ (2011) Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 124:304–313
Jewell BR, Wilkie DR (1960) The mechanical properties of relaxing muscle. J Physiol 152:30–47
Jeyaseelan R, Poizat C, Baker RK, Abdishoo S, Isterabadi LB, Lyons GE, Kedes L (1997) A novel cardiac-restricted target for doxorubicin. CARP, a nuclear modulator of gene expression in cardiac progenitor cells and cardiomyocytes. J Biol Chem 272:22800–22808
Jian Z et al (2014) Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Sci Signal 7:ra27
Jiang J, Wakimoto H, Seidman JG, Seidman CE (2013) Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science 342:111–114
Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED, Seidman JG, Seidman CE (2000) Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med 343:1688–1696
Kampourakis T, Zhang X, Sun YB, Irving M (2018) Omecamtiv mercabil and blebbistatin modulate cardiac contractility by perturbing the regulatory state of the myosin filament. J physiol 596:31–46
Karkkainen S, Helio T, Jaaskelainen P, Miettinen R, Tuomainen P, Ylitalo K, Kaartinen M, Reissell E, Toivonen L, Nieminen MS, Kuusisto J, Laakso M, Peuhkurinen K (2004) Two novel mutations in the beta-myosin heavy chain gene associated with dilated cardiomyopathy. Eur J Heart Fail 6:861–868
Kawas RF, Anderson RL, Ingle SRB, Song Y, Sran AS, Rodriguez HM (2017) A small-molecule modulator of cardiac myosin acts on multiple stages of the myosin chemomechanical cycle. J Biol Chem 292:16571–16577
Kellermayer MS, Smith SB, Granzier HL, Bustamante C (1997) Folding-unfolding transitions in single titin molecules characterized with laser tweezers. Science 276:1112–1116
Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, Solaro RJ (2001) Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ Res 88:1059–1065
Koser F, Loescher C, Linke WA (2019) Posttranslational modifications of titin from cardiac muscle: how, where, and what for? FEBS J 286:2240–2260
Kotter S, Gout L, Von Frieling-Salewsky M, Muller AE, Helling S, Marcus K, Dos Remedios C, Linke WA, Kruger M (2013) Differential changes in titin domain phosphorylation increase myofilament stiffness in failing human hearts. Cardiovasc Res 99:648–656
Kruger M, Kotter S, Grutzner A, Lang P, Andresen C, Redfield MM, Butt E, dos Remedios CG, Linke WA (2009) Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res 104:87–94
Labeit S, Gautel M, Lakey A, Trinick J (1992) Towards a molecular understanding of titin. EMBO J 11:1711–1716
Lahmers S, Wu Y, Call DR, Labeit S, Granzier H (2004) Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res 94:505–513
Lange S, Ehler E, Gautel M (2006) From A to Z and back? Multicompartment proteins in the sarcomere. Trends Cell Biol 16:11–18
Lange S, Pinotsis N, Agarkova I, Ehler E (2020) The M-band: the underestimated part of the sarcomere. Biochim Biophys Mol Cell Res 1867:118440
Lee DI, Vahebi S, Tocchetti CG, Barouch LA, Solaro RJ, Takimoto E, Kass DA (2010) PDE5A suppression of acute beta-adrenergic activation requires modulation of myocyte beta-3 signaling coupled to PKG-mediated troponin I phosphorylation. Basic Res Cardiol 105:337–347
Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, Jo SH, Danner T, Zhang M, Rainer PP, Bedja D, Kirk JA, Ranek MJ, Dostmann WR, Kwon C, Margulies KB, Van Eyk JE, Paulus WJ, Takimoto E, Kass DA (2015) Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 519:472–476
Lee LA, Karabina A, Broadwell LJ, Leinwand LA (2019) The ancient sarcomeric myosins found in specialized muscles. Skelet Muscle 9:7
Leite-Moreira AM, Almeida-Coelho J, Neves JS, Pires AL, Ferreira-Martins J, Castro-Ferreira R, Ladeiras-Lopes R, Conceicao G, Miranda-Silva D, Rodrigues P, Hamdani N, Herwig M, Falcao-Pires I, Paulus WJ, Linke WA, Lourenco AP, Leite-Moreira AF (2018) Stretch-induced compliance: a novel adaptive biological mechanism following acute cardiac load. Cardiovasc Res 114:656–667
Lennermann D, Backs J, van den Hoogenhof MMG (2020) New insights in RBM20 cardiomyopathy. Curr Heart Fail Rep 17:234–246
LeWinter MM, Granzier HL (2014) Cardiac titin and heart disease. J Cardiovasc Pharmacol 63:207–212
Li H, Linke WA, Oberhauser AF, Carrion-Vazquez M, Kerkvliet JG, Lu H, Marszalek PE, Fernandez JM (2002) Reverse engineering of the giant muscle protein titin. Nature 418:998–1002
Li D, Morales A, Gonzalez-Quintana J, Norton N, Siegfried JD, Hofmeyer M, Hershberger RE (2010) Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Clin Transl Sci 3:90–97
Li S, Guo W, Dewey CN, Greaser ML (2013) Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res 41:2659–2672
Li EA, Xi W, Han YS, Brozovich FV (2019) Phosphodiesterase expression in the normal and failing heart. Arch Biochem Biophys 662:160–168
Lindner M, Erdmann E, Beuckelmann DJ (1998) Calcium content of the sarcoplasmic reticulum in isolated ventricular myocytes from patients with terminal heart failure. J Mol Cell Cardiol 30:743–749
Linke WA, Popov VI, Pollack GH (1994) Passive and active tension in single cardiac myofibrils. Biophys J 67:782–792
Linke WA, Rudy DE, Centner T, Gautel M, Witt C, Labeit S, Gregorio CC (1999) I-band titin in cardiac muscle is a three-element molecular spring and is critical for maintaining thin filament structure. J Cell Biol 146:631–644
Litvinukova M et al (2020) Cells of the adult human heart. Nature 588:466–472
Louch WE, Bito V, Heinzel FR, Macianskiene R, Vanhaecke J, Flameng W, Mubagwa K, Sipido KR (2004) Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res 62:63–73
Louch WE, Mork HK, Sexton J, Stromme TA, Laake P, Sjaastad I, Sejersted OM (2006) T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol 574:519–533
Lyon AR et al (2020) Investigation of the safety and feasibility of AAV1/SERCA2a gene transfer in patients with chronic heart failure supported with a left ventricular assist device - the SERCA-LVAD TRIAL. Gene Ther 27:579–590
Maatz H, Jens M, Liss M, Schafer S, Heinig M, Kirchner M, Adami E, Rintisch C, Dauksaite V, Radke MH, Selbach M, Barton PJ, Cook SA, Rajewsky N, Gotthardt M, Landthaler M, Hubner N (2014) RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J Clin Investig 124:3419–3430
Maier LS, Bers DM (2007) Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc Res 73:631–640
Makarenko I, Opitz CA, Leake MC, Neagoe C, Kulke M, Gwathmey JK, del Monte F, Hajjar RJ, Linke WA (2004) Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ Res 95:708–716
Marian AJ, Braunwald E (2017) Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circn Res 121:749–770
Maron BJ (2010) Hypertrophic cardiomyopathy. In: Braunwald’s Heart Disease. Elsevier, pp 1582–1594
Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H (2009) Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res 105:219–222
Martin TG, Kirk JA (2020) Under construction: the dynamic assembly, maintenance, and degradation of the cardiac sarcomere. J Mol Cell Cardiol 148:89–102
Maruyama K, Kimura S, Kuroda M, Handa S (1977a) Connectin, an elastic protein of muscle. Its abundance in cardiac myofibrils. J Biochem 82:347–350
Maruyama K, Matsubara S, Natori R, Nonomura Y, Kimura S (1977b) Connectin, an elastic protein of muscle. Characterization and Function. J Biochem 82:317–337
Marx SO, Gaburjakova J, Gaburjakova M, Henrikson C, Ondrias K, Marks AR (2001) Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ Res 88:1151–1158
Masarone D, Kaski JP, Pacileo G, Elliott PM, Bossone E, Day SM, Limongelli G (2018) Epidemiology and clinical aspects of genetic cardiomyopathies. Heart Fail Clin 14:119–128
McDonald KS, Moss RL (1995) Osmotic compression of single cardiac myocytes eliminates the reduction in Ca2+ sensitivity of tension at short sarcomere length. Circulation research 77:199–205
McKillop DF, Geeves MA (1993) Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys J 65:693–701
McNally EM, Golbus JR, Puckelwartz MJ (2013) Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Investig 123:19–26
McNamara JW, Li A, Dos Remedios CG, Cooke R (2015) The role of super-relaxed myosin in skeletal and cardiac muscle. Biophys Rev 7:5–14
McNamara JW, Li A, Lal S, Bos JM, Harris SP, van der Velden J, Ackerman MJ, Cooke R, Dos Remedios CG (2017) MYBPC3 mutations are associated with a reduced super-relaxed state in patients with hypertrophic cardiomyopathy. PloS one 12:e0180064
Mearini G, Stimpel D, Kramer E, Geertz B, Braren I, Gedicke-Hornung C, Precigout G, Muller OJ, Katus HA, Eschenhagen T, Voit T, Garcia L, Lorain S, Carrier L (2013) Repair of Mybpc3 mRNA by 5'-trans-splicing in a mouse model of hypertrophic cardiomyopathy. Mol Ther Nuc Acids 2:e102
Mearini G, Stimpel D, Geertz B, Weinberger F, Kramer E, Schlossarek S, Mourot-Filiatre J, Stoehr A, Dutsch A, Wijnker PJ, Braren I, Katus HA, Muller OJ, Voit T, Eschenhagen T, Carrier L (2014) Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat Commun 5:5515
Merlo M, Sinagra G, Carniel E, Slavov D, Zhu X, Barbati G, Spezzacatene A, Ramani F, Salcedo E, Di Lenarda A, Mestroni L, Taylor MR, Familial Cardiomyopathy R (2013) Poor prognosis of rare sarcomeric gene variants in patients with dilated cardiomyopathy. Clin Transl Sci 6:424–428
Methawasin M, Hutchinson KR, Lee EJ, Smith JE 3rd, Saripalli C, Hidalgo CG, Ottenheijm CA, Granzier H (2014) Experimentally increasing titin compliance in a novel mouse model attenuates the Frank-Starling mechanism but has a beneficial effect on diastole. Circulation 129:1924–1936
Methawasin M, Strom JG, Slater RE, Fernandez V, Saripalli C, Granzier H (2016) Experimentally increasing the compliance of titin through RNA binding Motif-20 (RBM20) inhibition improves diastolic function in a mouse model of heart failure with preserved ejection fractioN. Circulation 134:1085–1099
Nadal-Ginard B, Mahdavi V (1989) Molecular basis of cardiac performance. Plasticity of the myocardium generated through protein isoform switches. J Clin Investig 84:1693–1700
Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, Witt CC, Becker K, Labeit S, Granzier HL (2004) Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 110:155–162
Neagoe C, Kulke M, del Monte F, Gwathmey JK, de Tombe PP, Hajjar RJ, Linke WA (2002) Titin isoform switch in ischemic human heart disease. Circulation 106:1333–1341
Olivotto I et al (2020) Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 396:759–769
Ono K, Yano M, Ohkusa T, Kohno M, Hisaoka T, Tanigawa T, Kobayashi S, Kohno M, Matsuzaki M (2000) Altered interaction of FKBP12.6 with ryanodine receptor as a cause of abnormal Ca(2+) release in heart failure. Cardiovasc Res 48:323–331
Opitz CA, Leake MC, Makarenko I, Benes V, Linke WA (2004) Developmentally regulated switching of titin size alters myofibrillar stiffness in the perinatal heart. Circ Res 94:967–975
O'Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R, Marban E (1999) Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circ Res 84:562–570
Osawa M, Swindells MB, Tanikawa J, Tanaka T, Mase T, Furuya T, Ikura M (1998) Solution structure of calmodulin-W-7 complex: the basis of diversity in molecular recognition. J Mol Biol 276:165–176
Page E, Fozzard HA, Solaro RJ (2002) Handbook of Physiology: The Heart, vol 1. Oxford University Press, New York, p Section 2
Perera S, Holt MR, Mankoo BS, Gautel M (2011) Developmental regulation of MURF ubiquitin ligases and autophagy proteins nbr1, p62/SQSTM1 and LC3 during cardiac myofibril assembly and turnover. Dev Biol 351:46–61
Perrot A, Schmidt-Traub H, Hoffmann B, Prager M, Bit-Avragim N, Rudenko RI, Usupbaeva DA, Kabaeva Z, Imanov B, Mirrakhimov MM, Dietz R, Wycisk A, Tendera M, Gessner R, Osterziel KJ (2005) Prevalence of cardiac beta-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy. J Mol Med 83:468–477
Peter AK, Rossi AC, Buvoli M, Ozeroff CD, Crocini C, Perry AR, Buvoli AE, Lee LA, Leinwand LA (2019) Expression of normally repressed myosin heavy chain 7b in the mammalian heart induces dilated cardiomyopathY. J Am Heart Assoc 8:e013318
Pettinato AM, Ladha FA, Mellert DJ, Legere N, Cohn R, Romano R, Thakar K, Chen YS, Hinson JT (2020) Development of a cardiac sarcomere functional genomics platform to enable scalable interrogation of human TNNT2 variants. Circulation 142:2262–2275
Piot C, LeMaire SA, Albat B, Seguin J, Nargeot J, Richard S (1996) High frequency-induced upregulation of human cardiac calcium currents. Circulation 93:120–128
Pope B, Hoh JF, Weeds A (1980) The ATPase activities of rat cardiac myosin isoenzymes. FEBS lett 118:205–208
Powers JD, Malingen SA, Regnier M, Daniel TL (2021) The sliding filament theory since Andrew Huxley: multiscale and multidisciplinary muscle research. Annu Rev Biophys 50:373–400
Prondzynski M, Kramer E, Laufer SD, Shibamiya A, Pless O, Flenner F, Muller OJ, Munch J, Redwood C, Hansen A, Patten M, Eschenhagen T, Mearini G, Carrier L (2017) Evaluation of MYBPC3 trans-splicing and gene replacement as therapeutic options in human iPSC-derived cardiomyocytes. Mol Ther Nucleic Acids 7:475–486
Pulcastro HC, Awinda PO, Methawasin M, Granzier H, Dong W, Tanner BC (2016) Increased titin compliance reduced length-dependent contraction and slowed cross-bridge kinetics in skinned myocardial strips from Rbm (20DeltaRRM) Mice. Front Physiol 7:322
Redfield MM et al (2013) Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. Jama 309:1268–1277
Reiser PJ, Portman MA, Ning XH, Schomisch Moravec C (2001) Human cardiac myosin heavy chain isoforms in fetal and failing adult atria and ventricles. Am J Physiol Heart Circ Physiol 280:H1814–H1820
Rhee D, Sanger JM, Sanger JW (1994) The premyofibril: evidence for its role in myofibrillogenesis. Cell Motil Cytoskeleton 28:1–24
Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet JP, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M Project EHF(2003) Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 107:2227–2232
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Committee ALQA (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med Off J Am Coll Med Genet 17:405–424
Roberts AM et al (2015) Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med 7:270ra276
Robinson P, Griffiths PJ, Watkins H, Redwood CS (2007) Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res 101:1266–1273
Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song L, Sirabella D, Morikawa K, Teles D, Yazawa M, Vunjak-Novakovic G (2018) Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature 556:239–243
Rosing DR, Kent KM, Borer JS, Seides SF, Maron BJ, Epstein SE (1979) Verapamil therapy: a new approach to the pharmacologic treatment of hypertrophic cardiomyopathy. I. Hemodynamic effects. Circulation 60:1201–1207
Rossi AC, Mammucari C, Argentini C, Reggiani C, Schiaffino S (2010) Two novel/ancient myosins in mammalian skeletal muscles: MYH14/7b and MYH15 are expressed in extraocular muscles and muscle spindles. J Physiol 588:353–364
Rudolph F, Huttemeister J, da Silva LK, Juttner R, Yu L, Bergmann N, Friedrich D, Preibisch S, Wagner E, Lehnart SE, Gregorio CC, Gotthardt M (2019) Resolving titin's lifecycle and the spatial organization of protein turnover in mouse cardiomyocytes. Proc Natl Acad Sci U S A 116:25126–25136
Rudolph F, Fink C, Huttemeister J, Kirchner M, Radke MH, Lopez Carballo J, Wagner E, Kohl T, Lehnart SE, Mertins P, Gotthardt M (2020) Deconstructing sarcomeric structure-function relations in titin-BioID knock-in mice. Nat Commun 11:3133
Scardigli M, Ferrantini C, Crocini C, Pavone FS, Sacconi L (2018) Interplay between sub-cellular alterations of calcium release and T-tubular defects in cardiac diseases. Front Physiol 9:1474
Schafer S et al (2017) Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet 49:46–53
Schmitt JP, Debold EP, Ahmad F, Armstrong A, Frederico A, Conner DA, Mende U, Lohse MJ, Warshaw D, Seidman CE, Seidman JG (2006) Cardiac myosin missense mutations cause dilated cardiomyopathy in mouse models and depress molecular motor function. Proc Natl Acad Sci US A 103:14525–14530
Scholl FA, McLoughlin P, Ehler E, de Giovanni C, Schafer BW (2000) DRAL is a p53-responsive gene whose four and a half LIM domain protein product induces apoptosis. J Cell Biol 151:495–506
Selcen D, Carpen O (2008) The Z-disk diseases. Adv Exp Med Biol 642:116–130
Sequeira V, van der Velden J (2015) Historical perspective on heart function: the Frank-Starling Law. Biophys Rev 7:421–447
Sequeira V, van der Velden J (2017) The Frank-Starling Law: a jigsaw of titin proportions. Biophys Rev 9:259–267
Shiels HA, White E (2008) The Frank-Starling mechanism in vertebrate cardiac myocytes. J Exp Biol 211:2005–2013
Shimkunas R, Hegyi B, Jian Z, Shaw JA, Kazemi-Lari MA, Mitra D, Leach JK, Li X, Jaradeh M, Balardi N, Chen YJ, Escobar AL, Baker AJ, Bossuyt J, Banyasz T, Chiamvimonvat N, Lam KS, Bers DM, Izu LT, Chen-Izu Y (2021) Mechanical load regulates excitation-Ca(2+) signaling-contraction in cardiomyocyte. Circ Res 128:772–774
Silver PJ, Pinto PB, Dachiw J (1986) Modulation of vascular and cardiac contractile protein regulatory mechanisms by calmodulin inhibitors and related compounds. Biochem Pharmacol 35:2545–2551
Slater RE, Strom JG, Methawasin M, Liss M, Gotthardt M, Sweitzer N, Granzier HL (2019) Metformin improves diastolic function in an HFpEF-like mouse model by increasing titin compliance. J Gene Physiol 151:42–52
Soteriou A, Gamage M, Trinick J (1993) A survey of interactions made by the giant protein titin. J Cell Sci 104(Pt 1):119–123
Spotnitz HM, Sonnenblick EH, Spiro D (1966) Relation of ultrastructure to function in the intact heart: sarcomere structure relative to pressure volume curves of intact left ventricles of dog and cat. Circ Res 18:49–66
Starr R, Offer G (1978) The interaction of C-protein with heavy meromyosin and subfragment-2. Biochem J 171:813–816
Sweitzer NK, Moss RL (1993) Determinants of loaded shortening velocity in single cardiac myocytes permeabilized with alpha-hemolysin. Circ Res 73:1150–1162
Teerlink JR et al (2021) Cardiac myosin activation with omecamtiv mecarbil in systolic heart failure. N Eng J Med 384:105–116
Tharp CA, Haywood ME, Sbaizero O, Taylor MRG, Mestroni L (2019) The giant protein titin's role in cardiomyopathy: genetic, transcriptional, and post-translational modifications of TTN and their contribution to cardiac disease. Front Physiol 10:1436
Toepfer CN, Wakimoto H, Garfinkel AC, McDonough B, Liao D, Jiang J, Tai AC, Gorham JM, Lunde IG, Lun M, Lynch TL, McNamara JW, Sadayappan S, Redwood CS, Watkins HC, Seidman JG, Seidman CE (2019) Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Sci Transl Med 11(476):eaat1199
Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC (2016) Cardiac fibrosis: the fibroblast awakens. Circ Res 118:1021–1040
Trombitas K, Freiburg A, Centner T, Labeit S, Granzier H (1999) Molecular dissection of N2B cardiac titin's extensibility. Biophys J 77:3189–3196
Trombitas K, Redkar A, Centner T, Wu Y, Labeit S, Granzier H (2000) Extensibility of isoforms of cardiac titin: variation in contour length of molecular subsegments provides a basis for cellular passive stiffness diversity. Biophys J 79:3226–3234
Udelson JE, Bonow RO, O'Gara PT, Maron BJ, Van Lingen A, Bacharach SL, Epstein SE (1989) Verapamil prevents silent myocardial perfusion abnormalities during exercise in asymptomatic patients with hypertrophic cardiomyopathy. Circulation 79:1052–1060
van den Hoogenhof MMG, Beqqali A, Amin AS, van der Made I, Aufiero S, Khan MAF, Schumacher CA, Jansweijer JA, van Spaendonck-Zwarts KY, Remme CA, Backs J, Verkerk AO, Baartscheer A, Pinto YM, Creemers EE (2018) RBM20 mutations induce an arrhythmogenic dilated cardiomyopathy related to disturbed calcium handling. Circulation 138:1330–1342
van der Velden J, Stienen GJM (2019) Cardiac disorders and pathophysiology of sarcomeric proteins. Physiol Rev 99:381–426
Van Driest SL, Jaeger MA, Ommen SR, Will ML, Gersh BJ, Tajik AJ, Ackerman MJ (2004) Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 44:602–610
van Kampen SJ, van Rooij E (2019) CRISPR craze to transform cardiac biology. Trends Mol Med 25:791–802
Vander Roest AS, Liu C, Morck MM, Kooiker KB, Jung G, Song D, Dawood A, Jhingran A, Pardon G, Ranjbarvaziri S, Fajardo G, Zhao M, Campbell KS, Pruitt BL, Spudich JA, Ruppel KM, Bernstein D (2021) Hypertrophic cardiomyopathy beta-cardiac myosin mutation (P710R) leads to hypercontractility by disrupting super relaxed state. Proc Natl Acad Sci U S A 118(24):e2025030118
Wadmore K, Azad AJ, Gehmlich K (2021) The role of Z-disc proteins in myopathy and cardiomyopathy. Int J Mol Sci 22(6):3058
Waldmuller S, Freund P, Mauch S, Toder R, Vosberg HP (2002) Low-density DNA microarrays are versatile tools to screen for known mutations in hypertrophic cardiomyopathy. Hum Mutat 19:560–569
Walker, C.J., Crocini, C., Ramirez, D. et al. (2021) Nuclear mechanosensing drives chromatin remodelling in persistently activated fibroblasts. Nat Biomed Eng. https://doi.org/10.1038/s41551-021-00709-w
Walker JS, de Tombe PP (2004) Titin and the developing heart. Circ Res 94:860–862
Wang YP, Fuchs F (1995) Osmotic compression of skinned cardiac and skeletal muscle bundles: effects on force generation, Ca2+ sensitivity and Ca2+ binding. J Mol Cell Cardiol 27:1235–1244
Wang K, McCarter R, Wright J, Beverly J, Ramirez-Mitchell R (1991) Regulation of skeletal muscle stiffness and elasticity by titin isoforms: a test of the segmental extension model of resting tension. Proc Natl Acad Sci U S A 88:7101–7105
Wang K, McCarter R, Wright J, Beverly J, Ramirez-Mitchell R (1993) Viscoelasticity of the sarcomere matrix of skeletal muscles. The titin-myosin composite filament is a dual-stage molecular spring. Biophys J 64:1161–1177
Warkman AS, Whitman SA, Miller MK, Garriock RJ, Schwach CM, Gregorio CC, Krieg PA (2012) Developmental expression and cardiac transcriptional regulation of Myh7b, a third myosin heavy chain in the vertebrate heart. Cytoskeleton 69:324–335
Watanabe K, Muhle-Goll C, Kellermayer MS, Labeit S, Granzier H (2002a) Different molecular mechanics displayed by titin's constitutively and differentially expressed tandem Ig segments. J Struct Biol 137:248–258
Watanabe K, Nair P, Labeit D, Kellermayer MS, Greaser M, Labeit S, Granzier H (2002b) Molecular mechanics of cardiac titin's PEVK and N2B spring elements. J Biol Chem 277:11549–11558
Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, Weiss RM, Anderson ME, Cheng H, Song LS (2010) T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res 107:520–531
Wells QS, Becker JR, Su YR, Mosley JD, Weeke P, D'Aoust L, Ausborn NL, Ramirez AH, Pfotenhauer JP, Naftilan AJ, Markham L, Exil V, Roden DM, Hong CC (2013) Whole exome sequencing identifies a causal RBM20 mutation in a large pedigree with familial dilated cardiomyopathy. Circ Cardiovasc Gene 6:317–326
Who CRCWG (2019) World Health Organization cardiovascular disease risk charts: revised models to estimate risk in 21 global regions. Lancet Glob Health 7:e1332–e1345
Willis MS, Schisler JC, Li L, Rodriguez JE, Hilliard EG, Charles PC, Patterson C (2009) Cardiac muscle ring finger-1 increases susceptibility to heart failure in vivo. Circ Res 105:80–88
Willis MS, Wadosky KM, Rodriguez JE, Schisler JC, Lockyer P, Hilliard EG, Glass DJ, Patterson C (2014) Muscle ring finger 1 and muscle ring finger 2 are necessary but functionally redundant during developmental cardiac growth and regulate E2F1-mediated gene expression in vivo. Cell Biochem Funct 32:39–50
Wolny M, Colegrave M, Colman L, White E, Knight PJ, Peckham M (2013) Cardiomyopathy mutations in the tail of beta-cardiac myosin modify the coiled-coil structure and affect integration into thick filaments in muscle sarcomeres in adult cardiomyocytes. J Biol Chem 288:31952–31962
Woodhead JL, Zhao FQ, Craig R, Egelman EH, Alamo L, Padron R (2005) Atomic model of a myosin filament in the relaxed state. Nature 436:1195–1199
Xu C, Craig R, Tobacman L, Horowitz R, Lehman W (1999) Tropomyosin positions in regulated thin filaments revealed by cryoelectron microscopy. Biophys J 77:985–992
Yamamoto T, Yano M, Kohno M, Hisaoka T, Ono K, Tanigawa T, Saiki Y, Hisamatsu Y, Ohkusa T, Matsuzaki M (1999) Abnormal Ca2+ release from cardiac sarcoplasmic reticulum in tachycardia-induced heart failure. Cardiovasc Res 44:146–155
Yano M, Ono K, Ohkusa T, Suetsugu M, Kohno M, Hisaoka T, Kobayashi S, Hisamatsu Y, Yamamoto T, Kohno M, Noguchi N, Takasawa S, Okamoto H, Matsuzaki M (2000) Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca(2+) leak through ryanodine receptor in heart failure. Circulation 102:2131–2136
Yano M, Ikeda Y, Matsuzaki M (2005) Altered intracellular Ca2+ handling in heart failure. J Clin Investig 115:556–564
Yotti R, Seidman CE, Seidman JG (2019) Advances in the genetic basis and pathogenesis of sarcomere cardiomyopathies. Ann Rev Genomics Hum Genet 20:129–153
Zeitz O, Rahman A, Hasenfuss G, Janssen PM (2000) Impact of beta-adrenoceptor antagonists on myofilament calcium sensitivity of rabbit and human myocardium. J Cardiovas Pharmacol 36:126–131
Zhang R, Zhao J, Potter JD (1995) Phosphorylation of both serine residues in cardiac troponin I is required to decrease the Ca2+ affinity of cardiac troponin C. J Biol Chem 270:30773–30780
Zimmer HG (2002) Who discovered the Frank-Starling mechanism? N Physiol Sci 17:181–184
Zou Y, Evans S, Chen J, Kuo HC, Harvey RP, Chien KR (1997) CARP, a cardiac ankyrin repeat protein, is downstream in the Nkx2-5 homeobox gene pathway. Development 124:793–804
Zsebo K, Yaroshinsky A, Rudy JJ, Wagner K, Greenberg B, Jessup M, Hajjar RJ (2014) Long-term effects of AAV1/SERCA2a gene transfer in patients with severe heart failure: analysis of recurrent cardiovascular events and mortality. Circ Res 114:101–108
Funding
Open Access funding enabled and organized by Projekt DEAL. C.C. was supported by the American Heart Association postdoctoral fellowship (grant no. 20POST3521111). M.G. was supported by grants from the DFG, ERC and BMBF.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Crocini, C., Gotthardt, M. Cardiac sarcomere mechanics in health and disease. Biophys Rev 13, 637–652 (2021). https://doi.org/10.1007/s12551-021-00840-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-021-00840-7