
Abstract
In 1995, Yoshida and colleagues proposed the concept of ‘autoimmune pancreatitis’ (AIP). Recently, it is accepted that the existence of two subtypes of AIP—type 1, which involves immunoglobulin G4 (IgG4) as the pancreatic manifestation of IgG4-related disease (IgG4-RD), and type 2, which is characterized by granulocytic epithelial lesions. Type 2 AIP is thought to be rare in Japan. In 2011, the International Consensus Diagnostic Criteria (ICDC) for autoimmune pancreatitis was proposed. In Japan, the clinical diagnostic criteria of AIP 2011 was proposed by the Japan Pancreas Society (JPS) and the Research Committee of Intractable Diseases of the Pancreas. The JPS 2011 is based on ICDC and a simplified checklist of items to diagnose type 1 AIP. Although recent progress in type 1 AIP has resolved clinical features, diagnosis, treatment, and pathogenesis, many clinical and basic issues still remain unclear. Here, we provide an overview of the recent clinical and basic issues associated with type 1 AIP.
Similar content being viewed by others

Introduction
In 1961, Sarles et al. reported a case of chronic idiopathic pancreatitis with hypergammaglobulinemia in which an immune mechanism was supposedly involved [1]. Thirty years later, Kawaguchi et al. described histopathological findings characterized by lymphoplasmacytic infiltration, storiform fibrosis, and obliterative phlebitis as lymphoplasmacytic sclerosing pancreatitis (LPSP) [2]. In 1995, Yoshida et al. proposed the concept of ‘autoimmune pancreatitis’ (AIP) [3] and in 2001 Hamano et al. reported that elevated serum immunoglobulin G4 (IgG4) levels are highly specific and sensitive for the diagnosis of AIP [4]. Thereafter, many investigators reported on the course and features of AIP and it is now accepted as a new clinical entity of pancreatic disorder [5–8]. In 2003, Kamisawa et al. suggested that AIP is a systemic disease, based on their findings that the pancreas and other involved organs showed abundant infiltration of IgG4-positive plasma cells and fibrosis [9].
There have been reports of another unique histological pattern in the resected pancreata of patients with chronic mass-forming nonalcoholic pancreatitis with epithelial destruction by granulocytes in Western countries [10, 11]. This pattern, which includes neutrophil infiltration within the lumen and epithelium of the interlobular ducts, is called idiopathic duct centric pancreatitis (IDCP) [10], i.e., AIP with granulocyte epithelial lesions (AIP with GELs) [11]. In 2011, the International Consensus Diagnostic Criteria (ICDC) for autoimmune pancreatitis proposed the classification of AIP into type 1 AIP (LPSP) and type 2 AIP (IDCP) [12]. As most Japanese AIP cases are type 1 AIP (very few are type 2 AIP [13]), we reviewed the recent advances in the diagnosis and pathophysiology of type 1 AIP.
Diagnosis of type 1 AIP
Currently, we can use two major diagnostic criteria in Japan. The first is ICDC, and the second is the clinical diagnostic criteria of AIP 2011 (JPS 2011), which was proposed by the Japan Pancreas Society (JPS) and the Research Committee of Intractable Diseases of the Pancreas (RCIDP) supported by the Japanese Ministry of Health, Labor, and Welfare [14]. The ICDC criteria correspond to the diagnostic methods of each country (both type 1 and type 2 AIP can be diagnosed). For example, the pancreatogram using endoscopic retrograde cholangiopancreatography (ERCP) has traditionally been an important tool used in the diagnosis of AIP in Japan historically; however, ERCP is not generally used for the diagnosis of AIP in Western countries. Thus, there are many factors that lead to a diagnosis using the ICDC, and a general gastroenterologist may find the ICDC to be complex. As a result, JPS 2011 was proposed based on ICDC and provides a simplified checklist of items in order to diagnose type 1 AIP. The main characteristics of JPS 2011 are (1) ERCP is not essential in diffuse-type 1 AIP; however, to distinguish focal-type AIP, ERCP is still essential, (2) serological findings are narrowed down to only IgG4, (3) clinical and histological features of other organ involvement (sclerosing cholangitis, sclerosing dacryoadenitis/sialoadenitis, retroperitoneal fibrosis) are considered in the diagnosis, (4) resected pancreata can be used for diagnosis, and (5) a steroid trial is added as an optional item.
Sumimoto et al. reported on the accuracy of the existing diagnostic criteria. The authors claim that ICDC has the highest accuracy when compared with other criteria [15], and that the sensitivities of ICDC and JPS 2011 are 95.1 and 86.9 %, respectively. JPS 2011 contains diagnostic criteria for type 1 AIP; however, type 2 AIP can also be identified as a possible diagnosis. JPS 2011 requires ERCP for segmental/focal-type AIP disease, but not for typical diffuse-type AIP. A pancreatogram by endoscopic retrograde pancreatography (ERP) is useful for diagnosis [15, 16], but it has been reported that post-ERCP pancreatitis occurred in 1.1 % of patients in Japan [17]. Endoscopic ultrasound-fine needle aspiration (EUS-FNA) procedures have been increasing in Japan, and may eventually become more common than ERCP. In the future, JPS 2011 will be modified to include EUS-FNA.
At our institute, we initially use CT scans to evaluate the enlarged pancreas followed by evaluation of the main pancreatic duct (MPD) by ERP. In order to distinguish AIP from pancreatic cancer, we essentially perform cytology and/or EUS-FNA. For pancreatic head lesions with obstructive jaundice or biliary enzyme abnormality due to biliary stricture, we first perform diagnostic and therapeutic ERCP. For pancreatic head lesions without obstructive jaundice or pancreatic body and/or tail lesions, we perform EUS-FNA followed by diagnostic and therapeutic ERCP. For pancreatic body or tail lesions, we first perform EUS-FNA [17] (Fig. 1).

Our diagnostic algorithm for pancreatic mass. We initially used CT scans to evaluate enlargement of the pancreas followed by evaluation of the MPD by ERP. In order to distinguish AIP from pancreatic cancer, we performed cytology and/or EUS-FNA. We diagnosed diffuse-type AIP by CT alone, and focal-type AIP using both CT and ERP. For pancreatic head lesions, if there is obstructive jaundice or biliary stricture with biliary enzyme abnormality, we initially perform ERCP; if there is no biliary stricture with obstructive jaundice or biliary enzyme abnormality, we perform EUS-FNA. For the pancreatic body and/or tail lesions, we first perform EUS-FNA. Figure from ref. [15], with kind permission from Elsevier
We speculate that two problems may arise with the spread of EUS-FNA due to the handling of small-tissue samples. One problem is that IgG4-positive cells may be detected in the sample of pancreatic cancer. Second, as opposed to type 1 AIP, type 2 AIP can be diagnosed correctly by viewing neutrophil infiltration. There have been several recent reports of IgG4-positive cells associated with pancreatic ductal adenocarcinoma [18–20]. Fukui et al. reported that of 21 cases of pancreatic ductal adenocarcinoma, the ratio of IgG4/IgG was >40 % in 9 (43 %), 6 (29 %) and 3 (14 %) cases of the main lesion of the cancer, a non-cancerous lesion around the cancer, and an obstructive pancreatitis lesion, respectively, according to the comprehensive diagnostic criteria for IgG4-related disease [21]. Using the comprehensive diagnostic criteria for IgG4-related disease, >40 % of IgG-positive plasma cells are IgG4-positive and each biopsy sample has >10 IgG4-positive cells/high-powered field (hpf). In a report by Fukui et al., 89 % (8 of 9) of type 1 AIP cases had an IgG4/IgG ratio >40 % and >10 IgG4-positive cells/hpf. Five percent (1/21) of the cases with pancreatic cancer in the main pancreatic lesion and obstructive pancreatitis lesion fulfilled two items related to the pathological features of the comprehensive diagnostic criteria of IgG4-related disease.
Another problem with small specimens and EUS-FNA is that neutrophil infiltration is not a characteristic finding of type 2 AIP. In general, it is thought that type 2 AIP is rare in Japan; however, there are some reports of its diagnosis by EUS-FNA [22, 23]. Mitsuyama et al. reported that there is no significant difference in neutrophil infiltration around the intralobular pancreatic ducts and that GELs may even be presented in the intralobular pancreatic ducts of patients with LPSP.
These results show that an AIP diagnosis must be made carefully based on the number of IgG4-positive plasma cells or neutrophils as well as the presence or absence of GELs with a small biopsied sample obtained by EUS-FNA.
Therapy for type 1 AIP
Glucocorticoids are the most common drug for type 1 AIP, and rapid response to glucocorticoids is one of the primary characteristics of type 1 AIP. A poor response to glucocorticoid steroid therapy might indicate misdiagnosis, especially in the case of pancreatic cancer. Although a randomized controlled trial has not yet been conducted, Japanese consensus guidelines have proposed a recommended initial oral prednisolone dose for induction of remission of 0.6 mg/kg/day, which is administered for 2–4 weeks. The dose is then tapered by 5 mg every 1–2 weeks to a maintenance dose (2.5–5 mg/day) that should be continued for 3 years as maintenance therapy [24]. A multicenter study in Japan reported that relapse occurred significantly less often during maintenance steroid therapy (23 %) than after the discontinuation of therapy (34 %) [25]. Tomiyama et al. reported a different useful induction of steroid therapy, i.e., mini-pulse therapy with methylprednisolone (500 mg/day, 3 days/week, 2 cycles) [26], which yielded the same therapeutic effect as oral administration. Moreover, mini-pulse therapy is especially useful in the treatment of biliary lesions. It is worth nothing that maintenance therapy is not commonly used in America and European countries. At the Mayo Clinic, initial therapy with oral prednisolone of 40 mg/day for 4 weeks was tapered by 5 mg/week and discontinued after 11 weeks. Under this regimen, 16 (53 %) of 30 AIP patients associated with sclerosing cholangitis relapsed during a median follow-up period of 29.5 months [27]. Given these discrepancies, it seems that there is a need for international consensus about the role of maintenance therapy.
In most cases of relapsed AIP, re-administration or an increased dose of prednisolone is usually effective in Japan. In Western countries, there have been reports of concomitant use of immunosuppressants such as azathioprine, methotrexate, and mycophenolate mofetil for patients with type 1 AIP and IgG4-related sclerosing cholangitis who relapsed or who were resistant to steroid therapy [28–30]. In 2013, Hart et al. published a relatively large case series (n = 41) and a longer follow-up study [31]. The authors reported that relapse-free survival was similar when relapses were treated with corticosteroids and an immunomodulator compared with corticosteroids alone without maintenance treatment. Additionally, approximately 25 % of patients were unable to tolerate immunomodulator treatment and required drug discontinuation. Rituximab, a monoclonal anti-CD20 antibody, has also been successfully used to treat type 1 AIP patients [31, 32]. A recent clinical trial that evaluated rituximab for the treatment of IgG4-related disease showed effectiveness even without concomitant glucocorticoid therapy [33]. B-cell depletion may be effective for type 1 AIP because of its powerful association with pathogenesis. As rituximab is not yet approved for use in Japan, it is necessary to establish a second-line therapy for the patients who relapse with type 1 AIP.
Prognosis of type 1 AIP
The prognosis of type 1 AIP is thought to be good over the short-term with or without steroid therapy. The long-term prognosis, however, is not clear, because there are many unknown factors including the role of relapse, pancreatic exocrine or endocrine dysfunction, and associated malignancy including pancreatic cancer. Chronic pancreatitis has been reported as one of the risk factors for pancreatic cancer [34]. Some patients with type 1 AIP have been shown to develop pancreatic atrophy or pancreatic stones [31, 35, 36]. Furthermore, Ikeura et al. reported that patients with type 1 AIP have a higher risk of pancreatic cancer, similar to patients with ordinary chronic pancreatitis [37]. Shiokawa et al. reported that the risk of developing various cancers was highest during the first year after AIP diagnosis and speculated that AIP may be a manifestation of paraneoplastic syndrome. It is still unclear whether there is a definitive risk factor for malignancy. However, the risk of patients with type 1 AIP developing cancer is a very important consideration.
Pathophysiology of type 1 AIP
Recent studies have suggested possible multi-pathogenic factors in the development of type 1 AIP; however, the pathogenic mechanism of type 1 AIP still remains unclear. Watanabe et al. reported that activation of toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors in monocytes [38] and basophils [39] of patients with IgG4-RD induced IgG4 production by B cells from healthy controls via production of B-cell activating factor (BAFF). Moreover, Fukui et al. reported that abundant infiltration of TLR-7-positive M2 macrophages was observed in the resected pancreas of patients with type 1 AIP [40]. Thus, an innate immune response may be involved in the development of type 1 AIP.
The role of regulatory T cells (Tregs) and regulatory B cells (Bregs) has received much attention in immune-related diseases. In type 1 AIP, circulatory naive Tregs are significantly decreased in peripheral blood, whereas memory Tregs are significantly increased. Increased peripheral inducible Tregs are positively correlated with serum levels of IgG4 [40]. In addition, increased quantities of inducible costimulator (ICOS)-positive Tregs may influence IgG4 production via interleukin (IL)-10 in type 1 AIP [41]. Bregs have been reported to appear with several surface markers. Sumimoto et al. reported that CD19+CD24+CD38high Bregs increased, whereas CD19+CD24highCD27+ Bregs decreased in type 1 AIP [42]. Recently it was reported that plasmablast may play an important role in IgG4-related disease [43]. When considered in the context of the effectiveness of rituximab, it will be necessary to clarify the role of B cells in type 1 AIP.
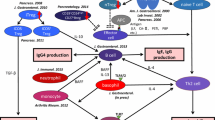
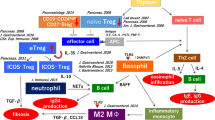
The following is a hypothesis of the pathophysiology of type 1 AIP. Induction of type 1 AIP may be decreased in a number of naive Tregs and CD19+CD24highCD27+ Bregs. On the other hand, inducible Tregs and CD19+CD24+CD38high Bregs can increase reactively. Th2 immune response supports disease progression, and it is thought that both innate and adaptive immunity affect the production of IgG4. ICOS-positive Tregs may regulate by secretion of IL-10. Moreover, basophil and monocytes are also regulated via TLR and NOD-like receptor signaling. Fibrosis may be regulated by TGF-β secreted from ICOS-negative Tregs and M2 macrophage, which perform a suppressive function against inflammation. M2 macrophages are also well known to accelerate Th2 immune response. Finally, M2 macrophages may also contribute to a Th2 immune response in type 1 AIP (Fig. 2).

Proposed pathophysiology of type 1 AIP. A decreased number of naive regulatory T cells (Tregs) and CD19+CD24highCD27+ regulatory B cells (Bregs) may be involved in the induction of type 1 AIP. Inducible regulatory T cells (iTregs) and CD19+CD24+CD38high Bregs increased reactively. Disease progression was supported by an increased Th2 immune response. The production of IgG4 may be regulated by IL-10 secreted from ICOS-positive Tregs; basophil and monocytes also regulate the production of IgG4 via TLR and NOD-like receptor signaling. Fibrosis may be regulated by TGF-β secreted from ICOS-negative Tregs and M2 macrophages. M2 macrophages may also contribute to the Th2 immune response in type 1 AIP. AIP autoimmune pancreatitis, IgG4 immunoglobulin G4, TLR toll-like receptor, NOD nucleotide-binding oligomerization domain, and ICOS inducible costimulator. Figure from ref. [44], with kind permission from Wolters Kluwer Health, Inc.
Conclusion
Type 1 AIP is now recognized as a pancreatic lesion of IgG4-related disease. However, many clinical and basic issues still remain unclear in cases of type 1 AIP including clinical features, diagnosis, treatment, prognosis, and pathogenesis. We believe we have provided a foundation to clarify some of these issues.
Abbreviations
- AIP:
-
Autoimmune pancreatitis
- IgG4:
-
Immunoglobulin G4
- LPSP:
-
Lymphoplasmacytic sclerosing pancreatitis
- IDCP:
-
Idiopathic duct centric pancreatitis
- GEL:
-
Granulocytic epithelial lesion
- ERCP:
-
Endoscopic retrograde cholangiopancreatography
- EUS-FNA:
-
Endoscopic ultrasound-guided fine needle aspiration
- TLR:
-
Toll-like receptor
- NOD:
-
Nucleotide-binding oligomerization domain
- ICOS:
-
Inducible costimulator
- Tregs:
-
Regulatory T cells
- Bregs:
-
Regulatory B cells
References
Sarles H, Sarles JC, Muratore R, et al. Chronic inflammatory sclerosis of the pancreas—an autonomous pancreatic disease? Am J Dig Dis. 1961;6:688–98.
Kawaguchi K, Koike M, Tsuruta K, et al. Lymphoplasmacytic sclerosing pancreatitis with cholangitis: a variant of primary sclerosing cholangitis extensively involving pancreas. Hum Pathol. 1991;22:387–95.
Yoshida K, Toki F, Takeuchi T, et al. Chronic pancreatitis caused by an autoimmune abnormality. Proposal of the concept of autoimmune pancreatitis. Dig Dis Sci. 1995;40:1561–8.
Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732–8.
Ito T, Nakano I, Koyanagi S, et al. Autoimmune pancreatitis as a new clinical entity. Three cases of autoimmune pancreatitis with effective steroid therapy. Dig Dis Sci. 1997;42:1458–68.
Horiuchi A, Kawa S, Akamatsu T, et al. Characteristic pancreatic duct appearance in autoimmune chronic pancreatitis: a case report and review of the Japanese literature. Am J Gastroenterol. 1998;93:260–3.
Uchida K, Okazaki K, Konishi Y, et al. Clinical analysis of autoimmune-related pancreatitis. Am J Gastroenterol. 2000;95:2788–94.
Okazaki K, Uchida K, Chiba T. Recent concept of autoimmune-related pancreatitis. J Gastroenterol. 2001;36:293–302.
Kamisawa T, Funata N, Hayashi Y, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003;38:982–4.
Notohara K, Burgart LJ, Yadav D, et al. Idiopathic chronic pancreatitis with periductal lymphoplasmacytic infiltration: clinicopathologic features of 35 cases. Am J Surg Pathol. 2003;27:1119–27.
Zamboni G, Luttges J, Capelli P, et al. Histopathological features of diagnostic and clinical relevance in autoimmune pancreatitis: a study on 53 resection specimens and 9 biopsy specimens. Virchows Arch. 2004;445:552–63.
Shimosegawa T, Chari ST, Frulloni L, et al. International consensus diagnostic criteria for autoimmune pancreatitis: guidelines of the International Association of Pancreatology. Pancreas. 2011;40:352–8.
Hart PA, Kamisawa T, Brugge WR, et al. Long-term outcomes of autoimmune pancreatitis: a multicentre, international analysis. Gut. 2013;62:1771–6.
The Japan Pancreas Society, the Ministry of Health and Welfare Investigation Research Team for Intractable Pancreatic Disease. Clinical Diagnostic Criteria for Autoimmune Pancreatitis 2011 (Proposal). Suizou (Japanese). 2012;27:17–25.
Sumimoto K, Uchida K, Mitsuyama T, et al. A proposal of a diagnostic algorithm with validation of International Consensus Diagnostic Criteria for autoimmune pancreatitis in a Japanese cohort. Pancreatology. 2013;13:230–7.
Sugumar A, Levy MJ, Kamisawa T, et al. Endoscopic retrograde pancreatography criteria to diagnose autoimmune pancreatitis: an international multicentre study. Gut. 2011;60:666–70.
Arata S, Takada T, Hirata K, et al. Post-ERCP pancreatitis. J Hepatobiliary Pancreat Sci. 2010;17:70–8.
Zhang L, Notohara K, Levy MJ, et al. IgG4-positive plasma cell infiltration in the diagnosis of autoimmune pancreatitis. Mod Pathol. 2007;20:23–8.
Dhall D, Suriawinata AA, Tang LH, et al. Use of immunohistochemistry for IgG4 in the distinction of autoimmune pancreatitis from peritumoral pancreatitis. Hum Pathol. 2010;41:643–52.
Fukui Y, Uchida K, Sakaguchi Y, et al. Possible involvement of Toll-like receptor 7 in the development of type 1 autoimmune pancreatitis. J Gastroenterol. 2015;50:435–44.
Umehara H, Okazaki K, Masaki Y, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012;22:21–30.
Kanno A, Ishida K, Hamada S, et al. Diagnosis of autoimmune pancreatitis by EUS-FNA by using a 22-gauge needle based on the International Consensus Diagnostic Criteria. Gastrointest Endosc. 2012;76:594–602.
Ishikawa T, Itoh A, Kawashima H, et al. Endoscopic ultrasound-guided fine needle aspiration in the differentiation of type 1 and type 2 autoimmune pancreatitis. World J Gastroenterol. 2012;18:3883–8.
Kamisawa T, Okazaki K, Kawa S, et al. Amendment of the Japanese Consensus Guidelines for Autoimmune Pancreatitis, 2013 III. Treatment and prognosis of autoimmune pancreatitis. J Gastroenterol. 2014;49(6):961–70.
Kamisawa T, Shimosegawa T, Okazaki K, et al. Standard steroid treatment for autoimmune pancreatitis. Gut. 2009;58:1504–7.
Tomiyama T, Uchida K, Matsushita M, et al. Comparison of steroid pulse therapy and conventional oral steroid therapy as initial treatment for autoimmune pancreatitis. J Gastroenterol. 2011;46:696–704.
Ghazale A, Chari ST, Zhang L, et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology. 2008;134:706–15.
Ghazale A, Chari ST, Zhang L, et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology. 2008;134:706–15.
Sandanayake NS, Church NI, Chapman MH, et al. Presentation and management of post-treatment relapse in autoimmune pancreatitis/immunoglobulin G4-associated cholangitis. Clin Gastroenterol Hepatol. 2009;7:1089–96.
Raina A, Yadav D, Krasinskas AM, et al. Evaluation and management of autoimmune pancreatitis: experience at a large US center. Am J Gastroenterol. 2009;104:2295–306.
Hart PA, Topazian MD, Witzig TE, et al. Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: the Mayo Clinic experience. Gut. 2013;62:1607–15.
Topazian M, Witzig TE, Smyrk TC, et al. Rituximab therapy for refractory biliary strictures in immunoglobulin G4-associated cholangitis. Clin Gastroenterol Hepatol. 2008;6:364–6.
Carruthers MN, Topazian MD, Khosroshahi A, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015;74:1171–7.
Lowenfels AB, Maisonneuve P, Cavallini G, et al. Pancreatitis and the risk of pancreatic cancer. N Engl J Med. 1993;328:1422–7.
Maruyama M, Watanabe T, Kanai K, et al. Autoimmune pancreatitis can develop into chronic pancreatitis. Orphanet J Rare Dis. 2014;9:77.
Uchida K, Yazumi S, Nishio A, et al. Long-term outcome of autoimmune pancreatitis. J Gastroenterol. 2009;44:726–32.
Ikeura T, Miyoshi H, Uchida K, et al. Relationship between autoimmune pancreatitis and pancreatic cancer: a single-center experience. Pancreatology. 2014;14:373–9.
Watanabe T, Yamashita K, Fujikawa S, et al. Involvement of activation of toll-like receptors and nucleotide-binding oligomerization domain-like receptors in enhanced IgG4 responses in autoimmune pancreatitis. Arthritis Rheum. 2012;64:914–24.
Watanabe T, Yamashita K, Sakurai T, et al. Toll-like receptor activation in basophils contributes to the development of IgG4-related disease. J Gastroenterol. 2013;48:247–53.
Miyoshi H, Uchida K, Taniguchi T, et al. Circulating naïve and CD4 + CD25high regulatory T cells in patients with autoimmune pancreatitis. Pancreas. 2008;36:133–40.
Kusuda T, Uchida K, Miyoshi H, et al. Involvement of inducible costimulator- and interleukin 10-positive regulatory T cells in the development of IgG4-related autoimmune pancreatitis. Pancreas. 2011;40:1120–30.
Sumimoto K, Uchida K, Kusuda T, et al. The role of CD19 + CD24high CD38high and CD19 + CD24high CD27 + regulatory B cells in patients with type 1 autoimmune pancreatitis. Pancreatology. 2014;14:193–200.
Mattoo H, Mahajan VS, Della-Torre E, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol. 2014;134:679–87.
Okazaki K, Uchida K. Autoimmune pancreatitis: the past, present, and future. Pancreas. 2015;44:1006–16.
Acknowledgments
This study was partially supported by (1) JSPS KAKENHI Grant Numbers 26461038, 15K09052, and (2) a Grant-in-Aid for ‘Research for Intractable Diseases’ Program from the Ministry of Labor and Welfare of Japan.
Reprinted from ref. [15] with permission from Elsevier, and from ref. [44] with permission from Wolters Kluwer Health, Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Kazushige Uchida, Hideaki Miyoshi, Tsukasa Ikeura, Masaaki Shimatani, Makoto Takaoka and Kazuichi Okazaki declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Uchida, K., Miyoshi, H., Ikeura, T. et al. Clinical and pathophysiological issues associated with type 1 autoimmune pancreatitis. Clin J Gastroenterol 9, 7–12 (2016). https://doi.org/10.1007/s12328-016-0628-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12328-016-0628-9