
Abstract
Introduction
Etrolizumab is a novel, dual-action anti-β7 integrin antibody studied in phase 3 trials in patients with inflammatory bowel disease. An autoinjector (AI) is being developed in parallel to complement the prefilled syringe with needle safety device (PFS-NSD) for subcutaneous (SC) administration in these trials. Here we demonstrate the comparable pharmacokinetics, tolerability, and safety of both devices.
Methods
This randomized, open-label, two-part study in healthy participants evaluated the comparability of etrolizumab exposure between the AI and the PFS-NSD. Part 1 (pilot) involved a small number of participants, and initial results were used to finalize the design of the larger part 2 (pivotal) study. In both parts, participants were randomly assigned to receive a single SC dose of etrolizumab 105 mg by AI or PFS-NSD. Randomization was stratified by body weight. Primary pharmacokinetic outcomes were Cmax, AUClast, and AUC0–inf.
Results
One hundred and eighty healthy participants (part 1, n = 30; part 2, n = 150) received a single SC dose of etrolizumab by AI or PFS-NSD. Primary pharmacokinetic results from part 1 supported modification of the part 2 study design. Results from part 2 demonstrated that etrolizumab exposure was equivalent between devices, with geometric mean ratios (GMRs) between AI and PFS-NSD of 102% (90% confidence interval [CI] 94.2–111) for Cmax, 98.0% (90% CI 89.3–107) for AUClast, and 97.6% (90% CI 88.6–107) for AUC0–inf. Median tmax and mean terminal t1/2 were also similar between devices. GMRs and 90% CIs of all primary pharmacokinetic parameters were fully contained within the predefined equivalence limits (80–125%).
Conclusion
This pharmacokinetic study demonstrated that single SC injections of etrolizumab 105 mg using an AI or a PFS-NSD resulted in equivalent etrolizumab exposure and similar safety and tolerability in healthy participants. Taken together, these results support the use of an AI for etrolizumab administration.
Trial Registration
NCT02996019.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Etrolizumab is a novel, subcutaneously administered, dual-action anti-β7 integrin antibody in development for patients with inflammatory bowel disease. |
An autoinjector (AI) is being developed to complement the prefilled syringe with needle safety device (PFS-NSD). |
This open-label study assessed the pharmacokinetics, safety, and tolerability of etrolizumab delivered by PFS-NSD or AI in 180 healthy volunteers. |
What was learned from the study? |
Single injections of etrolizumab 105 mg using an AI or a PFS-NSD resulted in equivalent etrolizumab exposure and similar safety and tolerability in healthy volunteers. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article, go to https://doi.org/10.6084/m9.figshare.13799711.
Introduction
Inflammatory bowel disease (IBD) is a chronic gastrointestinal disease that can dramatically affect quality of life and often necessitates invasive surgery [1,2,3]. The two major forms of IBD are ulcerative colitis (UC) and Crohn’s disease, two distinct conditions with partially overlapping symptoms and etiologies [4]. Current pharmacologic therapies for IBD are not curative, may lose efficacy over time, and can result in systemic side effects [4,5,6].
Etrolizumab is a novel, humanized, anti-β7 integrin antibody in development for patients with UC and Crohn’s disease. Etrolizumab selectively inhibits both α4β7:MAdCAM-1-mediated lymphocyte trafficking to the gut mucosa and αEβ7:E-cadherin-mediated lymphocyte retention in the intraepithelial space, thereby reducing consequent inflammatory effects on the gut lining [7,8,9]. The safety and efficacy of etrolizumab in patients with UC were demonstrated in the phase 2 study EUCALYPTUS [10]. Etrolizumab is under development for Crohn’s disease [11]; phase 3 studies in patients with moderately to severely active UC have demonstrated that etrolizumab is well tolerated. In most of these clinical trials, etrolizumab is administered as a 105-mg dose given once per month by subcutaneous (SC) injection using a prefilled syringe with needle safety device (PFS-NSD).
Several studies have confirmed that many patients and caregivers prefer prefilled autoinjectors (AIs) to other types of self-administration devices. Patients report increased convenience and ease of use, reduced risk for dosage error, reduction in injection-associated pain, and increased compliance with AIs [12,13,14,15]. This may be especially true in patients with needle or injection anxiety who may prefer a device that consistently keeps the needle out of sight. Recently, a randomized crossover study demonstrated that more than 70% of the studied patients with UC receiving golimumab (Simponi; Janssen Biotech) preferred treatment administration using an AI over a PFS, reporting that the AI device was easier to use [15].
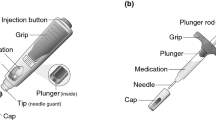
In parallel with the etrolizumab phase 3 studies, an AI is being developed with the goal of improving the overall self-injection experience for patients receiving etrolizumab once regulatory approval is received (Fig. 1). The etrolizumab AI automatically inserts the needle on contact with skin, keeping the needle out of view at all times. Audio and visual features are included to signal the completion of the injection and to assist users with self-injection.

Prefilled etrolizumab autoinjector
The tolerability, usage, and performance of the AI were first evaluated in a first-in-human phase 1 study (NCT02629744). Results of this study are published elsewhere (Tyrrell et al., Adv Ther 2021 (accepted)); in brief, the first-in-human study demonstrated that single SC injections of etrolizumab 105 mg using the AI were well tolerated, associated with only mild pain, and not associated with significant usage errors (i.e., errors related to tasks required to perform the injection and where the error could have a reasonable likelihood of potentially negative clinical consequences) in healthy participants.
The pharmacokinetic (PK) comparability study (NCT02996019) presented here aimed to demonstrate the comparability of etrolizumab exposure following SC administration using the AI and the PFS-NSD and to evaluate the safety and tolerability of etrolizumab following SC injection using both devices. Part 1 of the study was an exploratory pilot cohort used to evaluate the geometric mean ratio (GMR) and variability of PK parameters for etrolizumab administration with the AI versus the PFS-NSD. Those results informed the study design, including sample size and study duration for part 2 (the pivotal cohort). In part 2, the study aimed to demonstrate exposure comparability between a single dose of etrolizumab administered SC by AI or PFS-NSD.
Methods
Study Design and Procedures
This was a randomized, multicenter, open-label, parallel-group study conducted in healthy participants at three clinical sites within the USA (Fig. 2). The design of this study was based on US Food and Drug Administration (FDA) guidance for bioavailability studies [16], and consisted of a pilot cohort (part 1) and a pivotal cohort (part 2) with a sample size sufficient for 80% power to detect the exposure difference (if any) between the two device groups. In both parts, healthy participants were randomly assigned 1:1 to receive a single dose of etrolizumab 105 mg SC by either AI (test device) or PFS-NSD (reference device). Etrolizumab was administered by a health care professional into the participant’s abdomen. Randomization in both cohorts was stratified by body weight (≤ 79.9 vs ≥ 80 kg).

Participant disposition. AI autoinjector, AUC area under the curve, PFS-NSD prefilled syringe with needle safety device, PK pharmacokinetic, SC subcutaneous. aExcluded because of eligibility criteria (weight restriction). Participants were excluded from specific PK analyses because of insufficient PK data for calculations
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all participants included in the study. Study protocol, informed consent forms, information given to participants, participant recruitment materials, and all relevant supporting information were approved by the institutional review board (Midlands Independent Review Board, Overland Park, KS, USA) before study initiation.
Participants
Eligible healthy participants were men and women between 18 and 55 years of age with a body mass index (BMI) between 18.0 and 30.0 kg/m2. Volunteers with prior exposure to etrolizumab or other anti-integrin agents (e.g., natalizumab, vedolizumab, efalizumab), anti-mucosal vascular addressin cell adhesion molecule 1 (MAdCAM-1) agents, immunosuppressive agents (e.g., methotrexate, azathioprine, mercaptopurine), or rituximab were not eligible for inclusion. Volunteers were also ineligible for inclusion if they had a history of moderate or severe allergic or anaphylactic/anaphylactoid reactions to chimeric, human, or humanized antibodies; fusion or murine proteins; or hypersensitivity to etrolizumab (active drug substance) or any of the excipients (l-histidine, l-arginine, succinic acid, polysorbate 20).
On the basis of results from the pilot cohort, participants in the pivotal cohort also had to have a body weight between 60 and 100 kg (inclusive) at the time of study entry. Participants had to be in good health (no clinically significant findings from medical history, physical examination, 12-lead electrocardiography, or vital signs).
Assessments and Outcome Measures
Blood samples for determination of etrolizumab serum concentrations in parts 1 and 2 were collected predose and 6 h postdose on day 1, then on days 2, 4, 6, 8, 11, 15, 29, 43, 57, and 71 (study completion). The GMR and variability of the maximum etrolizumab concentration (Cmax), area under the concentration–time curve from the time of drug administration to the time of last measurable concentration (AUClast), AUC extrapolated to infinity (AUC0–inf), and ratio of AUClast to AUC0–inf (AUCR) of etrolizumab were measured in part 1. For part 2, Cmax, AUClast, and AUC0–inf were measured as primary endpoints. Secondary PK parameters included the time to maximum concentration of etrolizumab (tmax), the terminal elimination half-life (t1/2), and AUCR.
Blood samples for determination of antidrug antibodies (ADAs) in parts 1 and 2 were collected before dosing on day 1 and again on days 29, 57, and 71. Data from all ADA-positive participants were included in the final PK statistical analysis unless the participant met the predefined exclusion criteria for PK analysis.
Safety and tolerability assessments included the incidence, nature, and severity of adverse events (AEs), graded according to National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03. The incidence of injection site reactions, changes in vital signs, physical examination findings, clinical laboratory results, and ADAs were also assessed.
Bioanalytical Methods
Etrolizumab concentrations were measured using a validated fluorescence immunoassay (Gyrolab; Gyros Protein Technologies) that used a minimum required dilution (MRD) of 1/100, with a minimum quantifiable concentration of 80 ng/mL etrolizumab in sera from patients with UC or Crohn’s disease and from healthy volunteers.
Anti-etrolizumab antibodies in serum were detected using a validated assay. This colorimetric enzyme-linked immunosorbent assay used a 1/20 MRD and a monoclonal anti-etrolizumab control antibody. Relative sensitivity of the method was determined to be 12.0 ng/mL in the sera of healthy volunteers. Drug tolerance of the assay was established: 28 ng/mL of the positive control ADA could be detected in the presence of 50 μg/mL etrolizumab.
Sample analyses for etrolizumab concentrations and ADA levels were performed at ICON (Whitesboro, NY).
Sample Size Determination
Enrollment of up to 30 healthy participants in part 1 was planned to ensure at least 12 participants in each arm (24 total) had evaluable PK profiles to enable estimation of the GMR and coefficient of variation (CV%) for PK parameters (Cmax, AUClast, and AUC0–inf). Part 2 planned for enrollment of 146 participants. Assuming a dropout rate or unevaluable PK profiles from approximately 10% of participants through day 71, approximately 131 healthy participants with evaluable full PK profiles were expected to provide at least 80% power to demonstrate exposure comparability for Cmax, AUClast, and AUC0–inf based on the GMR and PK variability outcomes from the part 1 pilot cohort.
Pharmacokinetic Analysis
PK parameters were determined from the serum etrolizumab concentrations using noncompartmental methods and were performed using Phoenix WinNonlin (version 6.4; Centara USA, Inc.).
Statistical Analysis
The analysis population consisted of all participants who received an SC injection of etrolizumab and who had an evaluable PK profile, defined as sufficient samples available to accurately determine key PK parameters. In particular, participants who discontinued the study early, on or before day 15, were considered not to have an evaluable PK profile. Participants without an available sample to determine the concentration of day 71 were excluded from statistical analysis of AUClast; participants with fewer than three available samples among days 28, 43, 57, and 71 were excluded from statistical analysis of AUC0–inf.
Descriptive, exploratory analysis of PK parameters was carried out with data from part 1 (pilot), with a focus on evaluating GMR, CV%, and distribution of AUCR to inform the part 2 (pivotal) sample size and final study design. Only data from the pivotal cohort (part 2) were included in the comparability statistical analysis. In part 2, an analysis of variance, including treatment as the fixed effect, was performed to assess comparability of Cmax, AUClast, and AUC0–inf between the AI and PFS-NSD groups. Data for Cmax, AUClast, and AUC0–inf were natural log (ln)-transformed before analysis, and the 90% CIs of the GMRs for the AI group compared with those from the PFS-NSD group were calculated by taking the antilog of the corresponding 90% CIs for the differences between the means (log scale). Exposure between the AI and PFS-NSD groups met PK comparability criteria if the 90% CIs of the GMRs for Cmax, AUClast, and AUC0–inf were all within the prespecified limits of 80–125%. These limits and the overall approach of evaluating PK comparability via 90% CIs of GMRs for selected PK parameters were adopted in accordance to guidance from the FDA for bioequivalence studies [16, 17].
Results
Pharmacokinetics: Pilot Study (Part 1)
All 30 participants enrolled and randomly assigned in part 1 received a single 105-mg SC dose of etrolizumab by AI (n = 15) or PFS-NSD (n = 15). Twenty-seven participants completed the study; two participants (one in each arm) discontinued earlier because of loss of follow-up and one (PFS-NSD arm) discontinued because of a serious AE (seizure) 32 days after receiving etrolizumab; this AE was not reported by the investigator as related to etrolizumab treatment. Participant characteristics are shown in Table 1. In the overall cohort, most participants were male (63.3%) and white (70.0%). The body weight and age of participants differed between treatment arms; the mean body weight was 79.7 kg in the AI arm and 74.3 kg in the PFS-NSD arm, and the mean age was 42 years in the AI arm and 34 years in the PFS-NSD arm.
Given that body weight appears to impact etrolizumab exposure, especially AUC0–inf (Fig. 3), the imbalance in body weight between treatment arms could potentially bias assessment of GMR values of primary PK parameters and PK variability. Therefore, participants in the pilot cohort with a body weight lower than 60 kg (n = 4) or higher than 100 kg (n = 1) were excluded from the exploratory analysis, with the aim to evaluate GMR and CV% of PK parameters used to guide the determination of final sample size for pivotal study (part 2). The PK-evaluable population meeting the body weight restriction of 60–100 kg in part 1 included 14 participants in the AI arm and 11 participants in the PFS-NSD arm. As expected, when body weight range between the two arms was balanced, GMR (CV) values of the AI versus PFS-NSD groups were 0.96 (29.9), 1.02 (33.4), and 1.02 (34.2) for Cmax, AUClast, and AUC0–inf, respectively (Table 2). Hence, the sample size for the pivotal cohort was calculated on the basis of these GMR and CV% values, and a body weight restriction was added for the part 2 study as an inclusion criterion. Furthermore, all participants had AUCR values greater than 80% (data not shown), which met the requirement for a bioequivalence study as stated in the FDA guideline [17], and hence resulted in modification of the last day of the pivotal study from day 85 (the originally planned date) to day 71.

Impact of body weight on etrolizumab a Cmax and b AUC0–inf. AI autoinjector, AUC area under the curve, AUC0–infAUC extrapolated to infinity, Cmax maximum concentration, PFS-NSD prefilled syringe with needle safety device, SC subcutaneous
Pharmacokinetics: Pivotal Study (Part 2)
In the pivotal cohort, 150 (100%) of the enrolled and randomly assigned participants received a single SC 105-mg dose of etrolizumab by AI (n = 74) or PFS-NSD (n = 76). Eight participants discontinued the study, five because of loss of follow-up (three in the AI arm, two in the PFS-NSD arm), one because of the protocol violation of not meeting body weight criteria (PFS-NSD arm), and two (one in each arm) because they withdrew. In part 2 (including both arms), 53.3% of participants were male, 60.7% were white, and 36.0% were African American. Treatment arms were well balanced for body weight and age; mean body weight was 76.2 kg in the AI arm and 76.3 kg in the PFS-NSD arm, and mean age was 35 years in the AI arm and 37 years in the PFS-NSD arm (Table 1).
Etrolizumab serum concentrations over time for the AI and PFS-NSD groups are shown in Fig. 4; PK parameters are summarized in Table 3. GMRs between the AI and PFS-NSD groups were 102% (90% CI 94.2–111) for Cmax, 98.0% (90% CI 89.3–107) for AUClast, and 97.6% (90% CI 88.6–107) for AUC0–inf (Table 4). The 90% CIs of the GMRs for each of these primary PK parameters were within the predefined equivalence limits of 80–125%, which meets the predefined comparability criteria and supports equivalent exposure of etrolizumab between two device groups. The results also demonstrated comparable median time to maximum observed concentration (tmax, 5.04 vs 6.97 days) and mean terminal elimination half-life (t1/2, 11.8 vs 12.2 days) between AI and PFS-NSD groups. PK profiles of ADA-positive participants appear to be similar to those of ADA-negative participants (Fig. 5).

Etrolizumab serum concentrations (arithmetic mean ± standard deviation) over time with AI and PFS-NSD on a linear scale (a) and a semilogarithmic scale (b) in the pivotal study. AI autoinjector, PFS-NSD prefilled syringe with needle safety device

Etrolizumab serum concentrations over time by ADA status with (a) AI and (b) PFS-NSD. ADA antidrug antibody, AI autoinjector, PFS-NSD prefilled syringe with needle safety device
Safety
Twenty-nine participants from the pilot cohort (15 in AI group, 14 in PFS-NSD group) and 148 participants in the pivotal cohort (73 in AI group, 75 in PFS-NSD group) were evaluable for the postbaseline ADA assessment. At baseline, no patients in the pilot cohort and 5.3% of patients in the pivotal cohort (8/150; five in AI group, three in PFS-NSD group) had a positive ADA test result. The overall incidence of treatment-emergent ADAs among evaluable participants after baseline was 20.7% (6/29 participants) in the pilot cohort and 29.7% (44/148 participants) in the pivotal cohort and was similar between both groups in the pilot cohort (3/15 [20%] vs 3/14 [21.4%] participants) and the pivotal cohort (20/73 [27.4%] vs 24/75 [32.0%] participants).
Treatment-emergent adverse events (TEAEs) were experienced by 53.3% of participants in the pilot cohort and 32.7% in the pivotal cohort, and most were mild (Table 5). One participant in the PFS-NSD arm of the pilot cohort had a serious AE (grade 3 seizure) approximately 32 days after injection that led to study discontinuation. This patient reported a previous history of seizure, and this AE was reported by the investigator as not related to etrolizumab. Ten participants (5.6%) had TEAEs related to injection site reactions (ISRs) during the study. Nine participants in the pilot cohort (five [33.3%] with AI and four [26.7%] with PFS-NSD) and one participant in the pivotal cohort (1.4% in the AI arm) had ISRs that were reported as TEAEs. Most TEAEs related to ISRs occurred on day 1 and were assessed by the investigators as mild. More ISRs were reported in the pilot cohort, but this likely represented a reporting bias because the injection sites were closely monitored at predefined time points during this part of the study. Six participants (40.0%) in the AI arm and three (20.0%) in the PFS-NSD arm of the pilot cohort and seven participants (9.5%) in the AI arm and ten (13.2%) in the PFS-NSD arm of the pivotal cohort experienced TEAES thought by the investigator to be related to etrolizumab.
Discussion
The pivotal cohort in this study confirmed comparable etrolizumab exposure between the AI and PFS-NSD groups after a single SC dose of etrolizumab in healthy participants. The GMRs observed with each of the primary PK parameters were between 98% and 102%, with 90% CIs within the prespecified equivalence limits. These GMRs for all primary exposure parameters are impressive, given the GMRs obtained when comparing other AIs and PFS-NSDs in healthy participants. For example, two recent studies used healthy volunteers to compare AI and PFS devices delivering a biosimilar of adalimumab, an antitumor necrosis factor inhibitor indicated for the treatment of UC and Crohn’s disease. The first study compared AI and PFS devices for the SC administration of the adalimumab biosimilar SB5 and demonstrated GMRs of 102%, 107%, and 110% for Cmax, AUClast, and AUC0–inf, respectively, with 90% CIs within equivalence limits of 80–125% [18]. In a similar study of the adalimumab biosimilar BI 695501, GMRs between AI and PFS devices were 100% (90% CI 82.1–122.3) for AUC0–inf and 110% (90% CI 96.8–125.4) for Cmax, the latter of which was above the upper 90% CI equivalence limit of 125% (Voltaire®-AI study) [19].
The pilot cohort was valuable for informing the final design of the pivotal cohort. The pilot cohort primarily evaluated GMRs and the variability of PK parameters, which are the key assumptions used in the sample size estimation. Preliminary results from the first-in-human etrolizumab AI study raised uncertainty regarding etrolizumab exposure with AI administration (Tyrrell et al., Adv Ther 2021 (accepted)). To minimize the risk for underpowering the pivotal PK comparability study, a small pilot cohort was added to the original study protocol with the intent of gaining certainty around GMR values of the primary PK parameters and PK variability after the administration of etrolizumab by AI or PFS-NSD. GMR and PK variability values obtained from the pilot cohort (see Table 2) provided added confidence in estimating the sample size for the pivotal cohort.
Although body weight was stratified at randomization, the final body weight distribution range was still imbalanced in the pilot cohort, which might have biased the final GMR outcome of the pilot cohort. To minimize such bias, only data from participants with a body weight from 60 to 100 kg (a common body weight range for both arms within the pilot cohort) were used for the estimation of GMR and PK variabilities. This body weight restriction was implemented in the pivotal cohort as an inclusion criterion. The pilot cohort also evaluated a study duration of 10 weeks (70 days), 2 weeks shorter than the planned duration of the original study, as suggested by the FDA. As expected, all evaluable participants in the pilot cohort had AUCR values greater than 80%, the value required by the FDA guideline for PK comparability studies [17]. This result suggested that the 10-week study duration was long enough to capture more than 80% of AUC0–inf and, hence, the duration of the pivotal study could be shortened from 12 to 10 weeks without the risk of not meeting the requirement of less than 20% extrapolated AUC for the calculated AUC0–inf.
The overall incidence of treatment-emergent ADAs among postbaseline evaluable participants was similar when comparing the AI and PFS-NSD groups in both the pilot cohorts as well as the pivotal cohorts. This suggests that the devices themselves are comparable in immunogenicity risk. However, the rate of ADAs was greater than 20% in both the pilot and the pivotal cohorts of this study, compared with 5% (2/38) of patients with UC who received single or multiple etrolizumab doses in a phase 1 trial and 5% (4/81) of patients with UC who received multiple etrolizumab doses in a randomized phase 2 trial [10, 20]. Potential factors leading to the relatively higher ADA rate in this PK comparability study may include (1) healthy participants may have better overall immune competence than patients with IBD; (2) the lack of immunosuppressant use in this study population may result in increased ADA production; and (3) the single SC dose regimen studied here differs from the intended clinical use (i.e., repeated dosing once every 4 weeks). Given these unique clinical settings, the ADA response from the single SC dose regimen evaluated here is likely not clinically relevant with regard to the intended use of this molecule in the treatment of IBD.
Regardless of the relatively high incidence of ADAs after a single SC injection of etrolizumab (greater than 20%) in this study, the impact of ADA positivity on PK appears to be minimal given that the PK profiles of ADA-positive participants largely overlapped with those observed in ADA-negative participants (Fig. 5a, b). Moreover, the variability (CV%) of AUC0–inf or AUClast values ranged from 33% to 36% in the overall pivotal part 2 study population, which was in line with those observed for other monoclonal antibodies [19, 21]. Such a small exposure variability further confirms that the total exposure (AUC0–inf) between ADA-positive and ADA-negative participants was very similar. Data from all evaluable participants, regardless of their ADA status, were included in the comparability statistical assessment, and the final outcome showed a high degree of exposure similarity between groups.
The exposure comparability testing of the AI and PFS-NSD groups and the sample size determination in the pivotal study design following a single SC dose of etrolizumab were based on data from a subset of participants within a body weight range of 60–100 kg. Although our finding of comparable etrolizumab exposure between the AI and PFS-NSD was based on data from participants within a defined body weight range, it can be applied to anyone outside this body weight range because the current study identified no exposure difference attributed solely to the drug delivery device. Furthermore, a similar study for another therapeutic antibody (biosimilar to adalimumab [BI 695501]) that did not restrict by body weight suggests that this may be a fair assumption [19]. In that study, a similar relationship between drug exposure and body weight was observed, but when comparing AI and PFS-NSD devices for adalimumab SC administration, exposure comparability was still achieved regardless of body weight. Clearance of etrolizumab is known to be significantly impacted by body weight [22], a finding confirmed in the pilot cohort of this study.
In these healthy participants, a single SC dose of etrolizumab was generally safe and well tolerated when administered using an AI or a PFS-NSD. TEAE rates were comparable between the AI and the PFS-NSD in the pivotal cohort (9.5% vs 13.2%). Most TEAEs were mild or moderate and usually resolved by the end of the study. One serious AE (seizure) led to study discontinuation in the PFS-NSD arm of the pilot cohort; however, this was not considered related to treatment with etrolizumab.
This study is not without limitations. Healthy volunteers were used in this study to align with the FDA guidance documents for determining bioavailability [16]. It is likely that certain characteristics of healthy volunteers within the pivotal cohort (enrolled with defined body weight restrictions) will differ from those of the intended users (patients with IBD). However, this should not impact the AI use in patients owing to the comparable PK, safety, and tolerability between two devices.
Conclusion
Results from this PK comparability study demonstrated that etrolizumab exposure was similar when administered SC by either AI or PFS-NSD, with 90% CI for exposure parameters (AUClast, Cmax, AUC0–inf) contained within equivalence limits (80–125%) between devices. In addition, single SC doses of etrolizumab administered by AI were generally well tolerated in healthy participants and were not associated with more AEs than doses administered by PFS-NSD injection.
This study also highlights the value of a pilot cohort in facilitating the design of a pivotal PK comparability study because data from the pilot cohort increased confidence in the study design and minimized assumption bias.
References
Casellas F, López-Vivancos J, Vergara M, Malagelada JR. Impact of inflammatory bowel disease on health-related quality of life. Dig Dis. 1999;17:208–18.
Borren NZ, van der Woude CJ, Ananthakrishnan AN. Fatigue in IBD: epidemiology, pathophysiology and management. Nat Rev Gastroenterol Hepatol. 2019;16:247–59.
Carter MJ, Lobo AJ, Travis SP, IBD Section of the British Society of Gastroenterology. Guidelines for the management of inflammatory bowel disease in adults. Gut. 2004;53(suppl 5):V1–16.
Zhang YZ, Li YY. Inflammatory bowel disease: pathogenesis. World J Gastroenterol. 2014;20:91–9.
Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–78.
Rogler G. Gastrointestinal and liver adverse effects of drugs used for treating IBD. Best Pract Res Clin Gastroenterol. 2010;24:157–65.
Cepek KL, Parker CM, Madara JL, Brenner MB. Integrin alpha E beta 7 mediates adhesion of T lymphocytes to epithelial cells. J Immunol. 1993;150(part 1):3459–70.
Andrew DP, Berlin C, Honda S, et al. Distinct but overlapping epitopes are involved in α4β7-mediated adhesion to vascular cell adhesion molecule-1, mucosal addressin-1, fibronectin, and lymphocyte aggregation. J Immunol. 1994;153:3847–61.
Zundler S, Becker E, Schulze LL, Neurath MF. Immune cell trafficking and retention in inflammatory bowel disease: mechanistic insights and therapeutic advances. Gut. 2019;68:1688–700.
Vermeire S, O’Byrne S, Keir M, et al. Etrolizumab as induction therapy for ulcerative colitis: a randomised, controlled, phase 2 trial. Lancet. 2014;384:309–18.
Sandborn WJ, Vermeire S, Tyrrell H, et al. Etrolizumab for the treatment of ulcerative colitis and Crohn’s disease: an overview of the phase 3 clinical program. Adv Ther. 2020;37:3417–31.
Kivitz A, Cohen S, Dowd JE, et al. Clinical assessment of pain, tolerability, and preference of an autoinjection pen versus a prefilled syringe for patient self-administration of the fully human, monoclonal antibody adalimumab: the TOUCH trial. Clin Ther. 2006;28:1619–29.
Kivitz A, Segurado OG. HUMIRA pen: a novel autoinjection device for subcutaneous injection of the fully human monoclonal antibody adalimumab. Expert Rev Med Devices. 2007;4:109–16.
Borras-Blasco J, Gracia-Perez A, Rosique-Robles JD, Castera MD, Abad FJ. Acceptability of switching adalimumab from a prefilled syringe to an autoinjection pen. Expert Opin Biol Ther. 2010;10:301–7.
Vermeire S, D’Heygere F, Nakad A, et al. Preference for a prefilled syringe or an auto-injection device for delivering golimumab in patients with moderate-to-severe ulcerative colitis: a randomized crossover study. Patient Prefer Adherence. 2018;12:1193–202.
US Food and Drug Administration. Guidance for industry: bioavailability studies submitted in NDAs or INDs—general considerations. Published February 21, 2019. https://www.fda.gov/media/121311/download. Accessed 1 Feb 2021.
US Food and Drug Administration. Guidance for industry: statistical approaches to establishing bioequivalence. Published February 2001. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/statistical-approaches-establishing-bioequivalence. Accessed 18 Dec 2020.
Shin D, Lee Y, Jeong D, Ellis-Pegler R. Comparative pharmacokinetics of an adalimumab biosimilar SB5 administered via autoinjector or prefilled syringe in healthy subjects. Drug Des Devel Ther. 2018;12:3799–805.
Ramael S, Van Hoorick B, Tiessen R, et al. Similar pharmacokinetics of the adalimumab (Humira®) biosimilar BI 695501 whether administered via subcutaneous autoinjector or prefilled syringe (VOLTAIRE®-AI and VOLTAIRE®-TAI): phase 1, randomized, open-label, parallel-group trials. Rheumatol Ther. 2018;5:403–21.
Rutgeerts PJ, Fedorak RN, Hommes DW, et al. A randomised phase I study of etrolizumab (rhuMAb beta7) in moderate to severe ulcerative colitis. Gut. 2013;62:1122–30.
Anumolu SS, Lindgren S, Vemula J, et al. Bioequivalence of canakinumab injected subcutaneously via an autoinjector device or a prefilled safety syringe device in healthy subjects. Clin Pharmacol Drug Dev. 2018;7:829–36.
Tang MT, Keir ME, Erickson R, et al. Review article: nonclinical and clinical pharmacology, pharmacokinetics and pharmacodynamics of etrolizumab, an antiβ7 integrin therapy for inflammatory bowel disease. Aliment Pharmacol Ther. 2018;47:1440–52.
Acknowledgements
The authors thank the study investigators, study site personnel, and study participants for their valuable contributions.
Funding
Sponsorship for this study, including the journal’s Rapid Service and Open Access fee, was funded by F. Hoffmann-La Roche, Ltd., which funded this study, participated in data analysis and interpretation, and reviewed and approved this manuscript for publication. All authors had full access to all the data in this study and take complete responsibility for the integrity of the data and the accuracy of the data analysis.
Editorial Assistance
Editorial assistance in the preparation of this article was provided by Stacie Dilks, PhD, of ApotheCom (San Diego, CA). Support for this assistance was funded by F. Hoffmann-La Roche, Ltd.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship of this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
W. Zhang, H. T. Ding, M. Abouhossein, and R. Ravanello are employees of Genentech, Inc., and receive salary and stock options. R. Erickson is a former employee of Genentech, Inc., and received salary and stock options during employment. H. Tyrrell and J. Pulley are employees of Roche Products, Ltd., and receive salary and stock options. A. Boruvka is an employee of Hoffmann-La Roche, Ltd., and receives salary and stock options. M. T. Tang was an employee of Genentech, Inc., during the time of the study.
Compliance with Ethics Guidelines
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all participants included in the study. Study protocol, informed consent forms, information given to participants, participant recruitment materials, and all relevant supporting information were approved by the institutional review board (Midlands Independent Review Board, Overland Park, KS, USA) before study initiation.
Data Availability
Data generated during this study are available from the corresponding author upon reasonable request.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Zhang, W., Tyrrell, H., Ding, H.T. et al. Comparable Pharmacokinetics, Safety, and Tolerability of Etrolizumab Administered by Prefilled Syringe or Autoinjector in a Randomized Trial in Healthy Volunteers. Adv Ther 38, 2418–2434 (2021). https://doi.org/10.1007/s12325-021-01661-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-021-01661-6