
Abstract
Purpose
To investigate the effectiveness and safety of bevacizumab–paclitaxel combination therapy as first- or second-line chemotherapy for HER2-negative locally advanced or metastatic breast cancer in daily clinical practice.
Methods
In this prospective multicenter observational study, bevacizumab–paclitaxel was administered at the discretion of attending physicians. Cohorts A and B had hormone receptor-positive and triple-negative breast cancer (TNBC), respectively. Primary endpoint was overall survival (OS). Multivariate analyses were conducted to identify prognostic factors.
Results
Between November 2012 and October 2014, 767 patients were enrolled from 155 institutions across Japan. Effectiveness was analyzed in 754 eligible patients (cohort A, 539; cohort B, 215) and safety in 750 treated patients (median observation period, 19.7 months). Median OS (95% CI) was 21.7 (19.8–23.6) months in eligible patients; 25.2 (22.4–27.4) months and 13.2 (11.3–16.6) months in cohorts A and B, respectively; and 24.4 (21.9–27.2) months and 17.6 (15.2–20.0) months in patients receiving first- and second-line therapy, respectively. Factors affecting OS (hazard ratio 95% CI) were TNBC (1.75, 1.44–2.14), second-line therapy (1.35, 1.13–1.63), ECOG performance status ≥ 1 (1.28, 1.04–1.57), taxane-based chemotherapy (0.65, 0.49–0.86), cancer-related symptoms (0.56, 0.46–0.68), and visceral metastasis (0.52, 0.40–0.66). Incidences of grade ≥ 3 AEs hypertension, neutropenia, peripheral neuropathy, proteinuria, and bleeding were 35.7%, 27.2%, 7.2%, 3.7%, and 0.3%, respectively.
Conclusions
In Japanese clinical practice, combined bevacizumab–paclitaxel was as effective as in previous studies. Factors that independently predicted poor prognosis in the present study are consistent with those identified previously.
Trial registration
Trial no. UMIN000009086.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bevacizumab is a humanized monoclonal antibody for vascular endothelial growth factor (VEGF), which is the most important regulator for angiogenesis in both healthy and pathological states [1]. Its enhanced expression is observed in many types of tumors and promotes tumor growth and metastasis [2]. Bevacizumab binds to VEGF, thereby inhibiting VEGF binding to VEGF receptors 1 and 2 on endothelial cells. The consequent inhibition of tumor angiogenesis at the tumor site is understood to suppress the growth of cancer cells [3]. Additionally, normalization of abnormal vessels in the tumor tissue reduces its interstitial pressure, thereby facilitating penetration by anticancer agents in combination with bevacizumab [4].
A meta-analysis on addition of bevacizumab to chemotherapy for patients with locally advanced or metastatic breast cancer (LA/mBC) showed that addition of bevacizumab to first- or second-line chemotherapy significantly prolongs progression-free survival (PFS) and overall response rate (ORR) but not overall survival (OS) [5]. However, another meta-analysis of factors indicating poor prognosis in patients with LA/mBC showed that addition of bevacizumab to first-line chemotherapy improves 1-year OS and OS in patients with poor prognostic factors, as compared with chemotherapy alone [6]. Regarding adverse events (AEs), addition of bevacizumab increases the incidence of hypertension, proteinuria, and bleeding; however, the incidence of thromboembolism or gastrointestinal perforation is unchanged and that of treatment-related deaths is low [5].
The JO19901 study, carried out in Japan, was a phase II study of bevacizumab plus paclitaxel in chemotherapy-naive patients with HER2-negative LA/mBC [7]. The primary efficacy endpoint, median PFS, was 12.9 months. Regarding secondary endpoints, ORR was 74% and median OS was 35.8 months. Regarding safety, no new serious AEs were detected. Thus, the study confirmed the reproducibility in Japanese patients of the efficacy and safety results achieved for bevacizumab plus paclitaxel combination therapy in studies conducted outside Japan.
Although several cohort studies have been carried out in other countries [8,9,10,11], clinical experience of bevacizumab plus paclitaxel combination therapy in Japan has been limited to the small number of patients in the JO19901 study, which enrolled 120 patients [7]. Therefore, we conducted a prospective multicenter observational study to investigate the effectiveness and safety of this combination as first- or second-line therapy for LA/mBC in daily clinical practice. Two cohorts, one comprising patients with hormone receptor-positive breast cancer and the other comprising those with triple-negative breast cancer, were established to enable comparison of prognostic factors in patients with each of these cancer subtypes and in patients receiving first- or second-line therapy.
Patients and methods
Study design
In this multicenter prospective observational cohort study, patients who met the following inclusion criteria were enrolled: histologically confirmed HER2-negative LA/mBC with confirmed HR status; Eastern Cooperative Oncology Group (ECOG) performance status (PS), 0–3; no history of second-line chemotherapy for LA/mBC; and sufficient bone marrow and major organ functions determined by the attending physician. Exclusion criteria included history of hypersensitivity to the ingredients of bevacizumab or paclitaxel, history of hemoptysis, uncontrolled hypertension, thromboembolism, positive urinary protein test result (≥ 2 +), gastrointestinal perforation, and severe fistula.
Patients were enrolled through central registration and classified by HR status: cohort A comprised patients with HR-positive breast cancer, and cohort B, those with triple-negative breast cancer (TNBC). First-line therapy was defined as treatment for patients who had not previously received chemotherapy for LA/mBC. Second-line therapy was defined as treatment for disease progression after or during receipt of first-line chemotherapy for LA/mBC. In cases of relapse during adjuvant chemotherapy, the first treatment after the relapse was considered the second-line therapy.
Written informed consent was obtained from all patients. The study protocol, procedures, and consent forms were approved by the institutional review board of each participating institution. The study has been registered with the University Hospital Medical Information Network Clinical Trials Registry (https://www.umin.ac.jp/ctr/index-j.htm; trial no. UMIN000009086).
Study treatment
Because the study was an observational study conducted in a clinical setting, dosage, treatment schedule, and criteria for dose reduction, interruption, and discontinuation were not specified. However, the study protocol recommended the following standard treatment regimen, which was used in the JO19901 study [7]: bevacizumab 10 mg/kg given every 2 weeks, and paclitaxel 90 mg/m2 given every week for 3 weeks, followed by a 1-week rest. Each combination of bevacizumab and paclitaxel administered as above for 4 weeks was deemed one cycle.
In cases of discontinuation of either drug due to AEs, the other drug could be continued as monotherapy. The protocol did not specify any treatment after discontinuation.
Study assessment
At screening on registration, medical history, symptoms of cancer, physical findings, pathological findings relating to the primary and metastatic lesions, presence or absence of measurable lesions, and previous treatments were recorded. During the treatment period, treatment schedule, treatment discontinuations, dose reductions, treatment interruption, concomitant drugs, and the last dosing date were recorded by electronic data capture.
Regarding safety, the incidence of five selected AEs of bevacizumab plus paclitaxel (i.e. neutropenia, hypertension, proteinuria, bleeding, and peripheral neuropathy), of any grade, was recorded. For other AEs, only those of grade ≥ 3 were recorded. AEs were evaluated based on CTCAE version 4.0 (Japanese Clinical Oncology Group edition) [12]. Effectiveness was evaluated and recorded in accordance with the Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1 (Japanese Clinical Oncology Group edition) [13]. At the end of the observational period, patients’ disease progression, death, and post-treatment status were recorded.
Analysis populations and endpoints
The eligible patient population (used for the effectiveness analysis) was defined as patients who were registered according to the registration procedure, excluding those with ineligible cases or registration error. The treated patient population (used for the safety analysis and the sensitive analysis) was defined as patients who received bevacizumab plus paclitaxel combination therapy at least once. All evaluations were done by attending physicians.
The primary endpoint was OS, defined as the period between date of registration and death from any cause. Secondary endpoints were PFS, ORR, and safety. PFS was defined as the period between the registration date and the day when disease progression was determined (if that occurred first) or death (all causes).
Statistical analyses
This was an observational study conducted in the setting of daily clinical practice; therefore, the sample size was determined based on feasibility, considering the number of participating institutions, length of the registration period, and epidemiology of patients with HER2-negative LA/mBC. Consequently, the target numbers of patients were determined as 500 for cohort A and 250 for cohort B.
Expected median OS in each cohort according to treatment line (i.e. first- or second-line therapy) was estimated based on data from the prospective studies [7, 8, 14, 15, 17, 18]. Consequently, the expected median OS was 29.0 months and 18.0 months in patients receiving the study treatment as first-line and second-line therapy, respectively, in cohort A, and 17.0 months and 13.0 months in those receiving it as first-line and second-line therapy, respectively, in cohort B. Because the present study included patients who received the study treatment as both first- and second-line therapy, the ratio of first-line therapy patients to second-line therapy patients was assumed to be 5:5–7:3. Therefore, median OS was estimated to be 23.8 months in cohort A patients and 15.2 months in cohort B patients.
For the eligible patient population, cumulative survival curves for OS, median OS, and survival rate in each year were estimated using the Kaplan–Meier method, and Greenwood’s formula was used to construct 95% confidential intervals (CIs). Subgroup analysis was performed by Cox regression analysis to identify important prognostic factors. Sensitivity analysis was also performed, using data from the treated patient population. The same analyses were performed for PFS as those for OS. ORR was calculated as the proportion of patients achieving complete or partial response as the best overall response in patients with measurable lesions. CIs were calculated using the Clopper–Pearson method.
Safety was assessed using data from the treated patient population. The numbers of AEs, their grades, and their causal relation with the study drug were tabulated.
Results
Study population and baseline patient characteristics
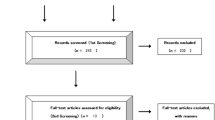
A total of 767 patients were enrolled from 155 institutions across Japan between November 2012 and October 2014. Patient disposition is shown in Supplementary Fig. 1. Of these, the eligible patient population comprised 754 patients after exclusion of ineligible cases. Within this group, 539 (71.5%) were in cohort A and 215 (28.5%) in cohort B. The numbers of patients receiving the study treatment as first- and second-line therapy were 478 (63.4%) and 276 (36.6%), respectively. The treated patient population, that is, those who received the study treatment at least once, comprised 750 patients.
Baseline characteristics of the eligible patient population are shown in Table 1 and Supplementary Table 1a. Median age was 58 years. Most patients had distant metastasis (86.1%). Of these patients, most had visceral metastasis (91.7%), with ≥ 3 organs affected in a minority of cases (14.0%). Symptoms related to cancer (e.g. pain, dyspnea, pleural effusion, ascites, skin ulcer, and tumor fever) were experienced by 57.6% of eligible patients. Baseline characteristics of the treated patient population are shown in Supplementary Table 1c and are similar to those of the eligible patient population.
The proportions of cohort B patients with distant metastasis and metastasis to ≥ 3 organs were generally higher in those receiving first-line therapy than in those receiving second-line therapy; however, there were no differences for the other prognostic factors (Supplementary Table 1a).
Treatment exposure
Most patients received treatment in accordance with the treatment regimen used in the JO19901 study [7]. Details of treatment exposure in eligible patients are shown in Table 2 and Supplementary Table 2a. Median duration of bevacizumab and paclitaxel exposure was 5.1 and 4.9 months, respectively. Contrary to our expectation, duration of bevacizumab monotherapy after discontinuation of bevacizumab in combination with paclitaxel was extremely short and about 90% of cases discontinued bevacizumab at almost the same time as paclitaxel was discontinued (Table2, Supplementary Table 2a–c).
Of the 754 eligible patients, 748 (99.2%) discontinued the study treatment; of these, 28.6% and 32.9% discontinued bevacizumab and paclitaxel, respectively, due to AEs. Regarding discontinuations due to other reasons, those recorded for ≥ 1% of patients included patient request (4.9%), maximum response (2.8%), breast surgery (2.7%), completion of scheduled treatment (2.3%), and treatment for other disease (1.5%).
The dose of bevacizumab or paclitaxel was reduced due to AEs in 1.5% and 33.1%, respectively, and it was suspended due to AEs in 19.7% and 29.7%, respectively.
When the treatment schedule of bevacizumab plus paclitaxel was the same as that in the JO19901 study [7], relative dose intensity of bevacizumab and paclitaxel was 99.2% and 90.9%, respectively.
Details of treatment exposure for patents in the treated patient population are shown in Supplementary Table 2b, c. Treatment exposure in this population was similar to that in the eligible patient population.
Effectiveness
Overall survival
Median observation period was 19.7 months. Events occurred in 496 of the 754 eligible patients (65.8%) during observation. Median OS was 21.7 months (95% CI 19.8–23.6 months), 25.2 months (95% CI 22.4–27.4 months), 13.2 months (95% CI 11.3–16.6 months), 24.4 months (95% CI 21.9–27.2 months), and 17.6 months (95% CI 15.2–20.0 months) in the full eligible patient population, in cohort A, in cohort B, in patients receiving the study treatment as first-line chemotherapy, and in those receiving it as second-line chemotherapy, respectively (Fig. 1a–c). Additionally, 1-year OS was 71.0%, 77.6%, 54.3%, 74.1%, 65.7%, in the full eligible patient population, in cohort A, in cohort B, in patients receiving the study treatment as first-line chemotherapy, and in those receiving it as second-line chemotherapy, respectively. Details of OS for eligible populations by cohort and treatment-line are shown in Supplementary Table 3 and Fig. 1d, e. Interestingly, OS was significantly longer in patients receiving the study treatment as first-line therapy than in those receiving it as second-line therapy in cohort A (log-rank test p < 0.0001, Fig. 2d), but not in cohort B (p = 0.3583, Fig. 1e).

Overall survival in the eligible patient population: a all eligible patients; b cohort A (patients with hormone receptor-positive breast cancer) versus cohort B (patients with triple-negative breast cancer); c, all eligible patients receiving first-line versus second-line therapy; d first-line versus second-line therapy in cohort A; e first-line versus second-line therapy in cohort B
The results of multivariate analysis for OS in the eligible patient population are summarized in Table 3a. In decreasing order of hazard ratio (HR), the baseline characteristics independently associated with OS were TNBC, second-line therapy, ECOG PS ≥ 1, neoadjuvant or adjuvant taxane-based chemotherapy, cancer-related symptoms, and visceral metastasis.
The results of multivariate analysis in the 521 patients with recurrent breast cancer are summarized in Table 3b. The following baseline characteristics were identified as independent predictors of OS: ECOG PS ≥ 1, neoadjuvant or adjuvant taxane-based chemotherapy, visceral metastasis, cancer-related symptoms, and disease-free interval (DFI) ≤ 24 months.
The results of univariate and multivariate analyses of baseline characteristics associated with OS by cohort are shown in Supplementary Tables 4 and 5.
Progression-free survival and objective response rates
Median PFS was 8.5 months (95% CI 7.8–9.2 months), 9.4 months (95% CI 8.7–10.7 months), 6.0 months (95% CI 5.5–7.4 months), 9.3 months (95% CI 8.5–10.7 months), and 7.2 months (95% CI 6.0–8.4 months) in the full eligible patient population, in cohort A, in cohort B, in patients receiving the study treatment as first-line chemotherapy, and in those receiving it as second-line chemotherapy, respectively (Supplementary Fig. 2a, 2b, 2c). Details of PFS for eligible populations by cohort and treatment line are shown in Supplementary Table 6 and Supplementary Fig. 2d, 2e.
Multivariate analysis identified the following baseline characteristics independently associated with PFS (Table 4a): TNBC, ECOG PS ≥ 1, history of endocrine therapy, cancer-related symptoms, history of neoadjuvant or adjuvant chemotherapy, history of neoadjuvant or adjuvant taxane-based chemotherapy, and visceral metastasis.
Multivariate analysis also identified several baseline characteristics as independent predictors of prognosis in the 521 patients with recurrent breast cancer (Table 4b): ECOG PS ≥ 1, cancer-related symptoms, and DFI ≤ 24 months.
In the sensitivity analysis, the results for OS and PFS in treated patients were similar to those for the eligible patient population (Supplementary Tables 3 and 6).
ORR in patients with measurable lesions was 56.1%, 59.3%, 48.8%, 62.2%, and 45.1% in the full eligible patient population, in cohort A, in cohort B, in patients receiving the study treatment as first-line chemotherapy, and in those receiving it as second-line chemotherapy, respectively (Table 5a). ORRs by cohort and treatment line are summarized in Table 5b.
Safety
The great majority of eligible patients (96.3%) experienced at least one AE, and 63.1% experienced one or more grade ≥ 3 AEs. Table 6 lists the AEs in treated patients. Incidences of grade ≥ 3 AEs hypertension, neutropenia, peripheral neuropathy, proteinuria, and bleeding were 35.7%, 27.2%, 7.2%, 3.7%, and 0.3%, respectively.
Serious AEs were recorded in 66 patients (8.8%) including 15 patients with infection, five patients with congestive heart failure and 4 patients with drug-induced pneumonitis, fracture, gastrointestinal perforation, or liver dysfunction (Supplementary Table 7). Treatment-related deaths occurred in 6 patients (0.8%); the causes were liver failure (3 patients), acute gastroenteritis and heart failure (1 patient), gastrointestinal bleeding (1 patient), and gastrointestinal perforation (1 patient). Of the 3 deaths due to liver failure, one had liver failure associated with disease progression, and the other two had so-called pseudocirrhosis, which is associated with liver atrophy due to acute tumor response by chemotherapy on massive liver metastases and disorder of subsequent liver regeneration process.
Discussion
The B-SHARE study was a prospective observational study to investigate the effectiveness and safety of bevacizumab combined with paclitaxel as first- or second-line chemotherapy for HER2-negative LA/mBC under real-world clinical conditions in Japan. During the median observation period of 19.7 months, median OS for eligible patients was 21.7 months, and median OS for eligible patients receiving first-line therapy was 24.4 months. These results are within the range (21.6–30.2 months) achieved in previous phase III studies [14,15,16] and observational studies [8,9,10,11, 17]. Although there have been no previous observational studies on bevacizumab plus paclitaxel as second-line therapy, median OS for eligible patients receiving second-line therapy in the present study (17.6 months) was similar to the 18.0 months achieved in the RIBBON-2 study conducted as second-line chemotherapy [18], in which the efficacy and safety of bevacizumab combined with standard chemotherapy was compared with standard chemotherapy alone.
The 74.1% 1-year median OS for first-line therapy was a good result and similar to that determined by a meta-analysis of data from randomized controlled studies of bevacizumab combined with chemotherapy as first-line therapy (i.e. 71%) [6], showing that bevacizumab combined with chemotherapy may improve 1-year OS when compared with chemotherapy alone in high-risk patients.
The multivariate analysis results for OS in eligible patients identified TNBC, second-line therapy, poor PS, perioperative history of taxane therapy, cancer-related symptoms, DFI ≤ 2 years (i.e. recurrent breast cancer), and visceral metastasis as independent factors for poor prognosis. This is similar to the findings of previous studies on chemotherapy with [19] or without bevacizumab [20,21,22,23].
OS was significantly longer in patients receiving first-line therapy than in those receiving second-line therapy in cohort A but not in cohort B. Regarding baseline characteristics in cohort B, the proportions of patients with distant metastasis and metastasis to ≥ 3 organs were higher in those receiving first-line therapy than in those receiving second-line therapy, but no differences were found for the other factors. After completion of the study treatment, a greater proportion of patients receiving first-line therapy in cohort B were transferred to best supportive care compared with those in cohort A (33.1% and 21.2%, respectively). These findings suggest that patients with TNBC are less likely than those with hormone receptor-positive cancer to continue therapy because of many poor prognostic factors, but when patients were able to undergo second-line and subsequent therapy, they are likely to have a better prognosis.
As for first-line therapy, median PFS in eligible patients was 9.3 months and ORR in those with measurable lesions was 62.2%. As with OS, the results were consistent with those of previous randomized controlled studies [14, 15, 24, 25] and observational studies [8,9,10,11, 17]. For second-line therapy, median PFS was 7.2 months and ORR was 45.1%, similar to the results of the RIBBON-2 study [18].
The multivariate analysis results for PFS in eligible patients, including those with advanced disease, identified TNBC, poor PS, history of endocrine therapy, cancer-related symptoms, history of perioperative chemotherapy, history of perioperative taxane, and visceral metastasis as factors indicating poor prognosis. However, in patients with recurrent breast cancer, poor PS, cancer-related symptoms, and DFI ≤ 2 years were independent factors for poor prognosis. Therefore, poor prognostic factors for PFS differed with patient background. Although poor PS and cancer-related symptoms may be considered mutually associated, they were independent poor prognostic factors for both OS and PFS, regardless of whether the cancer was advanced or recurrent. The possibility that cancer-related symptoms are a poor prognostic factor in LA/mBC is supported by several other studies [23, 26, 27].
Despite the similarity in effectiveness (i.e. OS, PFS, and ORR) shown in the present study to that obtained in randomized controlled studies [14,15,16, 24] and observational studies [8,9,10,11, 17], the dosing period for bevacizumab in first-line therapy (5.3 months) was shorter than in randomized controlled studies [24, 25]. In fact, the bevacizumab dosing period tends to be shorter in observational studies [8,9,10,11, 17] than in randomized controlled studies [24, 25]. However, the bevacizumab dosing period in a retrospective cohort study [11] using information from the French Epidemiological Strategy and Medical Economics database was similar to that of the present study. The shorter dosing period in the present study compared with in randomized controlled studies may have been due to differences in patient selection (with poorer PS) and adherence to treatment. In the present study, 15.3% of patients were aged ≥ 70 years, and 8.0% had PS of ≥ 2. About 30% of patients discontinued treatment because of AEs, which is similar to that in the randomized controlled studies, whereas about 20% discontinued treatment without having disease progress (e.g. undergoing surgery after tumor shrinkage or switching to endocrine therapy).
No new AEs related to bevacizumab plus paclitaxel were detected in the present study. Incidence of all grades of AEs (96.3%) and those of grade ≥ 3 (63.1%) were higher than in previous randomized controlled studies [14, 18, 24, 25] and observational studies [8,9,10, 17]. However, there was no increase in the incidence of serious AEs or treatment-related deaths. We experienced 2 cases of treatment-related death due to so-called pseudocirrhosis during treatment of bevacizumab plus paclitaxel. Pseudocirrhosis is characterized by morphological changes in the liver that resembling cirrhosis on the radiological findings without typical histopathology of cirrhosis [28]. Pseudocirrhosis as adverse events by chemotherapy is not rare and an important complication of chemotherapy in patients with liver metastases. Recently, Oliai et al. [29] reported that pseudocirrhosis developed in 37 (55%) of 67 metastatic breast cancer patients with liver metastasis and was associated with poor prognosis in patients with live metastasis. They also described that chemotherapy agents associated with the development of pseudocirrhosis were albumin-bound paclitaxel, capecitabine, cisplatin, everolimus and vinorelbine. This adverse event is not bevacizumab-specific. However, the possibility that bevacizumab may inhibit the process of liver regeneration after treatment-induced hepatic injury cannot be ruled out.
The present study had several limitations. First, it was a single-arm observational study of bevacizumab plus paclitaxel combination therapy, so there was no direct comparison in terms of the effectiveness and safety between bevacizumab plus paclitaxel and paclitaxel alone. Second, treatment effectiveness (PFS and ORR) was assessed by attending physicians, and HR and HER2 status were also assessed at each facility. Central assessment or review was not done for the evaluation of effectiveness and those receptors status. Third, most patients received treatment in accordance with the treatment regimen used in the JO19901 study. Therefore, we could not examine the relationship between the dosage or the schedule of bevacizumab plus paclitaxel and its effectiveness to find the optimal use of this combination. Fourth, because the present study was done under daily clinical conditions, discontinuation due to the wishes of the patient or the decision of the attending physician was possible, regardless of whether the effects of treatment were sustained. During the course of treatment, various strategies were adopted after tumor reduction due to study treatment, such as discontinuation of treatment, switching to hormonal therapy for maintenance, or surgical intervention, which are uncommon in randomized controlled studies. The limitations of the present study make it difficult to obtain a true result for PFS and ORR. However, OS is a robust endpoint and we consider the OS reported here to be close to its true value, because it was achieved in patients treated with bevacizumab plus paclitaxel under real clinical conditions.
In conclusion, bevacizumab plus paclitaxel as first- or second-line chemotherapy in Japanese patients with HER2-negative LA/mBC was as effective as in previous randomized controlled studies and prospective observational studies. Furthermore, the good tolerability of this regimen was confirmed.
References
Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400.
Toi M, Matsumoto T, Bando H. Vascular endothelial growth factor: its prognostic, predictive, and therapeutic implications. Lancet Oncol. 2001;2:667–73. https://doi.org/10.1016/S1470-2045(01)00556-3.
Presta LG, Chen H, O’Connor SJ, Chisholm V, Meng YG, Krummen L, et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593–9.
Willett CG, Boucher Y, di Tomaso E, Duda DG, Munn LL, Tong RT, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10:145–7. https://doi.org/10.1038/nm988.
Wagner AD, Thomssen C, Haerting J, Unverzagt S. Vascular-endothelial-growth-factor (VEGF) targeting therapies for endocrine refractory or resistant metastatic breast cancer. Cochrane Database Syst Rev. 2012;11:CD008941. https://doi.org/10.1002/14651858.CD008941.pub2.
Miles DW, Diéras V, Cortés J, Duenne AA, Yi J, O’Shaughnessy J. First-line bevacizumab in combination with chemotherapy for HER2-negative metastatic breast cancer: pooled and subgroup analyses of data from 2447 patients. Ann Oncol. 2013;24:2773–800. https://doi.org/10.1093/annonc/mdt276.
Aogi K, Masuda N, Ohno S, Oda T, Iwata H, Kashiwaba M, et al. First-line bevacizumab in combination with weekly paclitaxel for metastatic breast cancer: efficacy and safety results from a large, open-label, single-arm Japanese study. Breast Cancer Res Treat. 2011;129:829–38. https://doi.org/10.1007/s10549-011-1685-x.
Smith IE, Pierga JY, Biganzoli L, Cortés-Funes H, Thomssen C, Pivot X, Fabi A, et al. First-line bevacizumab plus taxane-based chemotherapy for locally recurrent or metastatic breast cancer: safety and efficacy in an open-label study in 2,251 patients. Ann Oncol. 2011;22:595–602. https://doi.org/10.1093/annonc/mdq430.
Dank M, Budi L, Piko B, Mangel L, Erfan J, Cseh J, et al. First-line bevacizumab-paclitaxel in 220 patients with metastatic breast cancer: results from the AVAREG study. Anticancer Res. 2014;34:1275–80.
Schneeweiss A, Förster F, Tesch H, Aktas B, Gluz O, Geberth M, et al. First-line bevacizumab-containing therapy for HER2-negative metastatic breast cancer: final results from a prospective German study. Anticancer Res. 2016;36:967–74.
Delaloge S, Pérol D, Courtinard C, Brain E, Asselain B, Bachelot T, et al. Paclitaxel plus bevacizumab or paclitaxel as first-line treatment for HER2-negative metastatic breast cancer in a multicenter national observational study. Ann Oncol. 2016;27:1725–32. https://doi.org/10.1093/annonc/mdw260.
National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE), version 4.0 (Japanese Clinical Oncology Group edition). 2020. https://www.jcog.jp/doctor/tool/CTCAEv4J_20170912_version.pdf
Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1 (Japanese Clinical Oncology Group edition). 2020. https://www.jcog.jp/doctor/tool/RECISTv11J_20100810.pdf
Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666–76. https://doi.org/10.1056/NEJMoa072113.
Gray R, Bhattacharya S, Bowden C, Miller K, Comis RL. Independent review of E2100: a phase III trial of bevacizumab plus paclitaxel versus paclitaxel in women with metastatic breast cancer. J Clin Oncol. 2009;27:4966–72. https://doi.org/10.1200/JCO.2008.21.6630.
Miles D, Cameron D, Hilton M, Garcia J, O’Shaughnessy J. Overall survival in MERiDiAN, a double-blind placebo-controlled randomised phase III trial evaluating first-line bevacizumab plus paclitaxel for HER2-negative metastatic breast cancer. Eur J Cancer. 2018;90:153–5. https://doi.org/10.1016/j.ejca.2017.10.018.
Smith I, Pierga JY, Biganzoli L, Cortes-Funes H, Thomssen C, Saracchini S, et al. Final overall survival results and effect of prolonged (≥1 year) first-line bevacizumab-containing therapy for metastatic breast cancer in the ATHENA trial. Breast Cancer Res Treat. 2011;130:133–143. https://doi.org/10.1007/s10549-011-1695-8.
Brufsky AM, Hurvitz S, Perez E, Swamy R, Valero V, O’Neill V, et al. RIBBON-2: a randomized, double-blind, placebo-controlled, phase III trial evaluating the efficacy and safety of bevacizumab in combination with chemotherapy for second-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol. 2011;29:4286–93. https://doi.org/10.1200/JCO.2010.34.1255.
Llombart-Cussac A, Pivot X, Biganzoli L, Cortes-Funes H, Pritchard KI, Pierga JY, et al. A prognostic factor index for overall survival in patients receiving first-line chemotherapy for HER2-negative advanced breast cancer: an analysis of the ATHENA trial. Breast. 2014;23:656–62. https://doi.org/10.1016/j.breast.2014.06.017.
Yamamoto N, Watanabe T, Katsumata N, Omuro Y, Ando M, Fukuda H, et al. Construction and validation of a practical prognostic index for patients with metastatic breast cancer. J Clin Oncol. 1998;16:2401–8. https://doi.org/10.1200/JCO.1998.16.7.2401.
Kramer JA, Curran D, Piccart M, de Haes JC, Bruning P, Klijn J, et al. Identification and interpretation of clinical and quality of life prognostic factors for survival and response to treatment in first-line chemotherapy in advanced breast cancer. Eur J Cancer. 2000;36:1498–506.
Sledge GW, Neuberg D, Bernardo P, Ingle JN, Martino S, Rowinsky EK, et al. Phase III trial of doxorubicin, paclitaxel, and the combination of doxorubicin and paclitaxel as front-line chemotherapy for metastatic breast cancer: an intergroup trial (E1193). J Clin Oncol. 2003;21:588–92. https://doi.org/10.1200/JCO.2003.08.013.
Smyth EN, Shen W, Bowman L, Peterson P, John W, Melemed A, et al. Patient-reported pain and other quality of life domains as prognostic factors for survival in a phase III clinical trial of patients with advanced breast cancer. Health Qual Life Outcomes. 2016;14:52. https://doi.org/10.1186/s12955-016-0449-z.
Robert NJ, Diéras V, Glaspy J, Brufsky AM, Bondarenko I, Lipatov ON, et al. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J Clin Oncol. 2011;29:1252–60. https://doi.org/10.1200/JCO.2010.28.0982.
Miles D, Cameron D, Bondarenko I, Manzyuk L, Alcedo JC, Lopez RI, et al. Bevacizumab plus paclitaxel versus placebo plus paclitaxel as first-line therapy for HER2-negative metastatic breast cancer (MERiDiAN): a double-blind placebo-controlled randomised phase III trial with prospective biomarker evaluation. Eur J Cancer. 2017;70:146–55. https://doi.org/10.1016/j.ejca.2016.09.024.
Niikura N, Liu J, Hayashi N, Palla SL, Tokuda Y, Hortobagyi GN, et al. Retrospective analysis of antitumor effects of zoledronic acid in breast cancer patients with bone-only metastases. Cancer. 2012;118:2039–47. https://doi.org/10.1002/cncr.26512.
Niikura N, Hayashi N, Masuda N, Takashima S, Nakamura R, Watanabe K, et al. Treatment outcomes and prognostic factors for patients with brain metastases from breast cancer of each subtype: a multicenter retrospective analysis. Breast Cancer Res Treat. 2014;147:103–12. https://doi.org/10.1007/s10549-014-3090-8.
Adike A, Karline N, Menias C, Carey EJ. Pseudocirrhosis: a case series and literature review. Case Rep Gastroenterol. 2016;10:381–91.
Oliai C, Douek ML, Rhoane C, Bhutada A, Ge PS, Runyon BA, et al. Clinical features of pseudocirrhosis in metastatic breast cancer. Breast Cancer Res Treat. 2019;177:409–17.
Acknowledgements
The authors thank patients who participated in this study and their families, as well as the ranchers and medical staff involved in the study.
Funding
The study was funded by Chugai Pharmaceutical Co., Ltd., Tokyo, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Yamamoto Y reports grants and personal fees from Daiichi-Sankyo, grants and personal fees from Eisai, grants and personal fees from Eli Lilly, grants and personal fees from Takeda, personal fees from Sysmex, personal fees from GE Health Care Japan, personal fees from AstraZeneca, grants and personal fees from Pfizer, grants and personal fees from Novartis, grants and personal fees from Nihon Kayaku, grants and personal fees from Kyowa-Kirin, grants and personal fees from Taiho, grants and personal fees from Chugai, outside the submitted work; and A board member of the Japanese Breast Cancer Society, A board member of the Japan Breast Cancer Research Group. Yamashiro H reports personal fees from Chugai, personal fees from Daiich Sankyo, personal fees from Pfizer, personal fees from Kyowa Kirin, personal fees from Eisai, personal fees from Eli Lilly, personal fees from Takeda, personal fees from Taiho outside the submitted work; Toh U reports remuneration from Chugai, Kyowa Kirin, Daiichi Sankyo, Taiho, Nihon Kayaku and Eisai, outside the submitted work; Kondo N reports personal fees from Chugai, personal fees from Eli Lilly, personal fees from Pfizer, personal fees from AstraZeneca, outside the submitted work; Nakamura R reports personal fees from Chugai, outside the submitted work; Kashiwaba M reports Speaker's bureaus from Chugai, Novartis, Kyowa Kirin, Pfizer, AstraZeneca, Taiho, Eisai, Daiichi Sankyo and Shionogi, outside the submitted work; Takahashi M reports personal fees from Chugai, grants and personal fees from Nippon Kayaku, outside the submitted work; Tsugawa K reports grants and personal fees from AstraZeneca, grants and personal fees from Chugai, grants and personal fees from Eisai, grants and personal fees from Taiho, grants and personal fees from Takeda, grants and personal fees from Nippon Kayaku, grants from MSD, personal fees from Eli Lilly, personal fees from Daiichi Sankyo, personal fees from Pfizer, during the conduct of the study; Ishikawa T reports grants and other from Eisai, grants and other from Nihon Kayaku, grants and other from Chugai, grants and other from Taiho, grants from Sanofi, grants and other from Eli Lilly, other from Pfizer, outside the submitted work; Nakayama T reports personal fees from Chugai, personal fees from Novartis, personal fees from Eli Lilly, personal fees from AstraZeneca, personal fees from Taiho, personal fees from Eisai, personal fees from Takeda, outside the submitted work; Ohtani S reports other from Chugai, other from Eisai, other from AstraZeneca, other from Pfizer, other from Eli Lilly, outside the submitted work; Takano T reports grants and personal fees from Daiichi Sankyo, grants and personal fees from Kyowa Kirin, grants and personal fees from Eisai, personal fees from Pfizer, personal fees from Eli Lilly, grants from Ono, grants from MSD, grants from Merck Serono, grants from Taiho, grants from Novartis, grants from Chugai, outside the submitted work; Fujisawa T reports personal fees from Chugai, personal fees from Eli Lilly, during the conduct of the study; Toyama T reports grants and personal fees from Chugai, grants and personal fees from Novartis, grants and personal fees from Eisai, grants and personal fees from AstraZeneca, personal fees from Eli Lilly, personal fees from Kyowa Kirin, personal fees from Taiho, personal fees from Daiichi Sankyo, personal fees from Nippon Kayaku, personal fees from Pfizer, personal fees from Takeda, during the conduct of the study; Kawaguchi H reports personal fees from Pfizer, personal fees from Chugai, personal fees from AstraZeneca, personal fees from Eli Lilly, personal fees from Eisai, personal fees from Kyowa Kirin, personal fees from Novartis, personal fees from Taiho, personal fees from Takeda, personal fees from Nippon Chemiphar, personal fees from Daiichi Sankyo, during the conduct of the study; Mashino K reports personal fees from Chugai, outside the submitted work; Tanino Y reports grants from Sysmex Corporation, other from Ono, other from Chugai, other from Novartis, other from Pfizer, other from Daiichi-Sankyo, other from Eli Lilly, other from Taiho, other from Eisai, outside the submitted work; Dr. Morita reports personal fees from AstraZeneca, personal fees from Bristol-Myers Squibb Company, personal fees from Chugai, personal fees from Eisai, personal fees from Eli Lilly, personal fees from MSD, personal fees from Pfizer, personal fees from Taiho, outside the submitted work; Toi M reports grants and personal fees from Chugai, grants and personal fees from Takeda, grants and personal fees from Pfizer, grants and personal fees from Kyowa Kirin, grants and personal fees from C & C Res Lab, grants and personal fees from Taiho, grants from JBCRG association, grants and personal fees from Eisai, grants and personal fees from Daiichi Sankyo, grants and personal fees from AstraZeneca, personal fees from Eli Lilly, personal fees from MSD, personal fees from Genomic Health, personal fees from Novartis, personal fees from Konica Minolta, grants from Astellas, outside the submitted work; and Board of directors; JBCRG association, Organisation for Oncology and Translational Research, Kyoto Breast cancer Research Network. Ohno S reports personal fees from Chugai, grants and personal fees from Eisai, grants and personal fees from Taiho, personal fees from AstraZeneca, personal fees from Pfizer, personal fees from Eli Lilly, personal fees from Kyowa Kirin, personal fees from Nippon Kayaku, outside the submitted work;
Ethical statement
The study was carried out in accordance with the Declaration of Helsinki and the Ethical Guidelines for Clinical Research of the Ministry of Health, Labour and Welfare of Japan. The study protocol, procedures, and consent forms were approved by the institutional review board of each participating institution.
Informed consent
Written informed consent was obtained from all patients.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Yamamoto, Y., Yamashiro, H., Toh, U. et al. Prospective observational study of bevacizumab combined with paclitaxel as first- or second-line chemotherapy for locally advanced or metastatic breast cancer: the JBCRG-C05 (B-SHARE) study. Breast Cancer 28, 145–160 (2021). https://doi.org/10.1007/s12282-020-01138-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12282-020-01138-4