
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive lethal malignancy, characterized by late diagnosis, aggressive growth, and therapy resistance, leading to a poor overall prognosis. Emerging evidence shows that the peripheral nerve is an important non-tumor component in the tumor microenvironment that regulates tumor growth and immune escape. The crosstalk between the neuronal system and PDAC has become a hot research topic that may provide novel mechanisms underlying tumor progression and further uncover promising therapeutic targets. In this review, we highlight the mechanisms of perineural invasion and the role of various types of tumor innervation in the progression of PDAC, summarize the potential signaling pathways modulating the neuronal-cancer interaction, and discuss the current and future therapeutic possibilities for this condition.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The tumor microenvironment (TME) is a complex acidic environment consisting of tumor and non-tumor cell types [1, 2], which plays a crucial role in the development and progression of tumors [3]. As an important part of the non-tumor element in the TME, the role and mechanism of tumor innervation have been increasingly investigated in various tumors including lung cancer [4, 5], melanoma [6,7,8,9,10,11,12], thyroid cancer [13,14,15], prostate cancer [16,17,18,19,20,21], breast cancer [22,23,24,25], ovarian carcinoma [26,27,28], head and neck cancer [29,30,31,32,33], gastric cancer [34,35,36,37,38,39,40,41,42,43], and pancreatic cancer [44,45,46,47], among others (Table 1). The crosstalk between the nervous system and cancer has favored the establishment of an interdisciplinary field—cancer neuroscience—and may provide additional potential therapeutic strategies.
Pancreatic ductal adenocarcinoma (PDAC) accounts for 90% of pancreatic cancers and is a highly malignant solid tumor characterized by an insidious onset, strong invasiveness, and a high recurrence or metastasis rate [48]. The standard treatment for PDAC involves a combination of surgery, chemotherapy, and radiation therapy, with the choice of therapy dependent on the stage and location of the cancer, as well as the overall health of the patient. Immune checkpoint inhibitors, such as nivolumab and pembrolizumab, have been shown to increase overall survival in some patients with advanced-stage disease [49]. However, due to a lack of effective screening methods, 80% of patients with PDAC are at an advanced stage when diagnosed, losing the chance of receiving resectable surgery. Resistance to chemotherapy and radiation therapy remains a great challenge to the treatment of PDAC. These factors contribute to the poor prognosis of PDAC patients, with an overall 5-year survival rate of only around 10% in 2021 [48]. Therefore, novel targets and therapies are required to enhance the outcome of PDAC.
Perineural invasion (PNI) is a typical characteristic of PDAC, defined as a tumor near the nerve where the tumor cells are located in at least 33% of the nerve circumference or any of the three layers of the nerve sheath [50]. PNI has been reported in about 80%–100% of patients with PDAC and is associated with postoperative recurrence and metastasis [51,52,53,54]. Meanwhile, tumor cells can produce and release neurotrophic factors like nerve growth factor (NGF) or brain-derived neurotrophic factor to promote tumor innervation [1, 55,56,57]. In the LSL-KrasG12D/+, LSL-Trp53R172H/+, Pdx-1-Cre (KPC) mouse model of PDAC, the number of sympathetic nerve fibers is tripled, and the number and density of calcitonin gene-related peptide (CGRP)-positive sensory nerves is increased by five times [58]. Analysis of clinical samples from patients with PDAC also illustrates a negative correlation between the density of nerve fibers in the tumor and survival [59]. Deep exploration of the interaction between nerve and tumor cells could lead to the identification of novel strategies for the treatment of PDAC.
In this review, we exhibit the mechanisms of PNI in PDAC and the role of different types of nerves innervating PDAC in tumor progression, summarize the potential mechanisms underlying the neuronal-cancer interaction, and discuss the current and potential therapeutic possibilities for PDAC.
Innervation of Normal Pancreas and Pancreatitis
The pancreas receives both autonomic and sensory innervation. The autonomic innervation consists of sympathetic and parasympathetic nerves. The sympathetic nerves permeate the pancreatic ganglion, vascular system, endocrine islets, ducts, and lymph nodes [60]. On the other hand, activation of the parasympathetic nervous system promotes the release of digestive enzymes and reduces glucose-triggered insulin secretion. In terms of sensory nerves, substance P (SP) and CGRP-positive nerve fibers are distributed throughout the exocrine tissues and most islets [61]. Myelinated sensory fibers along with thinly-myelinated and unmyelinated peptidergic sensory fibers are present in the parenchyma of the head, body, and tail of the pancreas. The relative density of these sensory fibers is highest in the head and decreases towards the tail. In contrast, the post-ganglionic sympathetic fibers are relatively evenly distributed throughout the parenchyma of the pancreas [62]. The presence of autonomic and sensory nerves in the pancreas is crucial for maintaining its normal functions. The sympathetic and parasympathetic nerves work in tandem to regulate digestive processes and insulin secretion. The sensory nerves play a role in detecting changes in the environment and transmitting information about the state of the pancreas to the central nervous system.
The phenomenon of increased number and diameter of pancreatic nerve fibers was first discovered in individuals with chronic pancreatitis (CP) [63]. CP is considered to be a high-risk factor for PDAC and has been shown to play a crucial role in the progression of this disease [64] (Fig. 1). In studies conducted on adult mice with PDAC, researchers found that expression of the K-Ras (G12V) mutation did not result in a tumor unless the mice also had CP [65]. This highlights the importance of considering CP as a risk factor for PDAC. In addition, research has shown that the expression of NGF and its receptor tropomyosin receptor kinase A (TrkA) is significantly higher in individuals with CP than in those with a normal pancreas. The increased expression of NGF is higher in metaplastic ductal cells and acinar cells that have dedifferentiated into tubular structures [66]. Further reports demonstrated that actively growing nerves in CP are associated with an activated NGF/TrkA pathway and a pain syndrome [66, 67]. As the normal pancreas with CP and intraepithelial neoplasia progresses into PDAC, pancreatic innervation is constantly remodeled and plays a crucial role in the worsening of the malignancy. The size (nerve hypertrophy) and number (nerve density) of pancreatic nerves are increased, the proportion of autonomic nerve fibers and sensory nerve fibers is altered (nerve remodeling), and there is infiltration of inflammatory cells around the nerve (pancreatic neuritis) or by PDAC cells (PNI) [51, 63, 68,69,70,71,72,73].

Timeline for major findings leading to the identification of crosstalk between peripheral innervation and PDAC. Abbreviations: PDAC, pancreatic ductal adenocarcinoma; PNI, perineural invasion
Perineural Invasion (PNI) in PDAC
General Background
PNI is a process in which cancer cells invade and spread along peripheral nerves. This histological characteristic has been found in a variety of tumors, including cancers of the head and neck, prostate, tongue, and pancreas. PNI involves a complex interplay between nerves and various cell types present in the TME, including Schwann cells, macrophages, and cancer-associated fibroblasts [74, 75]. Tumor cells interact closely with nerve components by releasing neurotrophic factors or exosomes and produce perineural niches, which provide a favorable environment for their survival and invasion and in turn, trigger the growth of nerves and stimulate the development of neural progenitor cells [76]. The underlying molecular mechanisms of PNI are governed by various factors such as NGF, glial cell line-derived neurotrophic factor (GDNF), and their corresponding receptors [77]. The cancer cells can cause damage to the neuronal sheath, activating nociceptive nerve fibers as a result of cancer-secreted mediators or stimuli from the extracellular matrix. This leads to the release of pro-inflammatory neuropeptides from peripheral nerve endings, further enhancing the spread of the tumor and causing pain. PNI has been demonstrated to be an independent predictor of poor prognosis among patients with oral squamous cell carcinomas, and nerve-tumor distance is a sensitive criterion to reclassify PNI [78]. A meta-analysis has shown that the presence of PNI is associated with a higher risk of biochemical recurrence of prostate cancer after radical prostatectomy or radiotherapy [79]. PNI is also an independent risk factor affecting the poor prognosis of patients with gastric cancer and colorectal cancer [80, 81].
Mechanisms of PNI in PDAC
Pancreatic cancer cells can reach the peripheral nerve at a short distance, which is the anatomical basis for why pancreatic cancer is prone to PNI. PDAC has a distinctive chronic inflammatory microenvironment that triggers the abnormal growth and malignant transformation of pancreatic cells. Chemokines, significant components of this environment, are known to contribute to both local invasion and distant metastasis of tumor cells [82]. Among them, CX3CL1 is a transmembrane chemokine highly expressed by numerous neurons, and it mediates the adhesion of endothelial cells to peripheral nerves. The overexpression of its receptor CX3CR1 in PDAC is associated with PNI and early postoperative recurrence [83]. PDAC cells can migrate to nerves that express CX3CL1 ligands by activating Gi protein and adhesion molecules [84]. The CXCL12/CXCR4 axis, another widespread chemokine signaling pathway, also plays a critical role in the tumor-matrix interaction and the neural infiltration of PDAC [85]. Aside from chemokines, Semaphorin 3D (SEMA3D) from tumor cells activates Plexin D1 (PLXND1) on dorsal root ganglion (DRG) neurons to increase the migration and invasion activity of pancreatic cancer cells. Increased expression levels of SEMA3D and PLXND1 have been confirmed in human PDAC specimens associated with PNI [86]. Nerve-derived glutamate also upregulates hexokinase 2 expression through mRNA m6A modification via N-methyl-d-aspartate receptor subunit 2B and the downstream Ca2+ pathway and ultimately promotes PNI [87] (Fig. 2A).

PNI in PDAC. A Direct interaction between PDAC cells and neurons in PNI. The neurogenic chemokines CXCL12 and CX3CL1 promote tumor cell invasion of nerves through their respective receptors CXCR4 and CX3CR1. Glutamate from neurons up-regulates HK2 expression through NMDAR2B and mRNA m6A modification of downstream Ca2+-dependent CaMKII/ERK-MAPK pathways, enhances glycolysis in nerve cells, and ultimately promotes PNI. Tumor cell-derived SEMA3D activates PLXND1 on DRG neurons to increase the migration and invasive activity of PDAC. The upregulation of CD74 on PDAC enhances its invasive capability and GDNF secretion via the AKT/EGR-1 pathway, thereby enhancing PNI. B Communication between PDAC cells and Schwann cells in PNI. The chemokine CCL7 produced by Schwann cells enhances the migration, invasion, and TIMP1 expression of PDAC cells through the CCR1/STAT2 pathway, and TIMP1 further stimulates the proliferation and migration of Schwann cells via the CD63/PI3K/AKT signal. CCL2 from Schwann cells drives CCR2-expressing IMs to differentiate into macrophages and enhance neural invasion through CTSB-mediated processes. In addition, high expression of MMP1 in PDAC promotes Schwann cell differentiation by stimulating the NT-3/TrkC signaling pathway. C Role of PSCs and acinar cells in PNI. PSCs induce PDAC cells to produce miR-21-5P exosomes through the NGF-TrkA axis, which further augments the Warburg effect of neurons and promotes PNI. HGF from PSCs activates the mTOR-NGF pathway through the c-Met receptor on PDAC cells, which boosts PNI. The pancreatitis-associated protein (PAP/REG3A) produced by inflammatory acinar cells in the microenvironment around the tumor promotes PNI by activating the JAK/STAT signaling pathway in PDAC. Abbreviations: PDAC, pancreatic ductal adenocarcinoma; NGF, nerve growth factor; Trk, tropomyosin receptor kinase; HK2, hexokinase 2; NMDAR2B, N-methyl-d-aspartate receptor subunit 2B; HGF, hepatocyte growth factor; IM, inflammatory monocytes; CTSB, cathepsin B; MMP, matrix metalloproteinase; PLXND1, Plexin D1; SEMA3D, Semaphorin 3D; GDNF, glial cell line-derived neurotrophic factor; PNI, perineural invasion; TIMP1, tissue inhibitor of metalloproteinases 1; PSC, pancreatic stellate cells; PAP, pancreatitis-associated protein; REG3A, regenerating islet-derived protein 3 alpha
Schwann cells interacting with PDAC cells engage in the occurrence and development of PNI. The paracrine NGF of tumor cells activates Schwann cell autophagy, enhances the chemical attraction to tumor cells, and accelerates the removal and phagocytosis of myelin debris to promote early axonal and myelin regeneration [88, 89]. CCL7 secreted by Schwann cells enhances the migration, invasion, and tissue inhibitor of metalloproteinases 1 (TIMP1) expression of PDAC cells through the CCR1/STAT2 pathway, and TIMP1 further promotes Schwann cell proliferation and migration through CD63/PI3K/AKT signaling [90]. In addition, high expression of matrix metalloproteinase (MMP)1 in PDAC promotes the epithelial-mesenchymal transition and Schwann cell differentiation by stimulating the NT-3/TrkC signaling pathway [91]. CCL2 released by Schwann cells drives CCR2-expressing inflammatory monocytes (IM), preferentially recruiting them to the PNI site, where they differentiate into macrophages and enhance neural invasion through cathepsin B (CTSB)-mediated processes [92] (Fig. 2B).
Other cell types overexpressing NGF and GDNF like stromal pancreatic stellate cells (PSCs) or acinar cells in the TME also contribute to the PNI. The tumor-derived exosome miR-21-5p stimulated by NGF from PSCs activates the Warburg effect in neurons, upregulates the expression of nociceptor genes, and promotes the PNI [93]. Hepatocyte growth factor (HGF) produced by PSCs binds to the receptor c-Met on PDAC cells and endothelial cells [94] and activates the mTOR/NGF axis to boost PNI [95]. The up-regulation of CD74 enhances the migration and invasion of PDAC cells and promotes the production of GDNF through the AKT/EGR-1/GDNF axis to promote neural plasticity [96]. Moreover, the inflammatory acinar cells within the pancreas contribute to PNI through the production of pancreatitis-associated protein (pancreatitis-associated protein/regenerating islet-derived protein 3 alpha) (PAP/REG3A), which activates the JAK/STAT signaling pathway in cancer cells [86] (Fig. 2C).
Clinical Significance
The occurrence of PNI in PDAC ranges from 70% to 95%, making it one of the most common features of these patients [51]. In patients with resectable PDAC, PNI represents a major determinant of tumor recurrence and post-operative survival, particularly in the early stages, where the invasion of nerves by cancer cells plays a driving role in disease progression [53]. In the setting of pre-operative gemcitabine-based chemoradiation therapy, PNI in resected PDAC specimens is significantly associated with disease-free survival and predicts the pattern of recurrence [97]. A meta-analysis of fourteen studies concluded that pre-operative PNI is also a promising marker for the prognosis of PDAC patients who undergo curative resection without neoadjuvant treatment [98]. Taken together, PNI in PDAC is an important prognostic factor, and early detection and management of PNI may help to improve clinical outcomes and survival in these patients.
Sympathetic Innervation in PDAC
General Background
The role of sympathetic innervation in various cancer types has been extensively investigated. Sympathetic activation increases the growth of primary tumors and elicits relevant symptoms, and tumor cells spread to normal adjacent tissues through adrenergic signaling pathways [46, 99, 100]. All β1, β2, and β3 adrenergic receptors are expressed in peripheral blood monocytes, activated T cells, monocytes, and monocyte-induced dendritic cells, and combinatorial sympathetic and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) blockade can inhibit the growth of murine melanoma [101]. Analysis of tumor samples from mice and patients shows an increase in the density of infiltrating autonomic nerve fibers [102], and that the autonomic innervation in the prostate regulates the development and spread of prostate cancer [17]. Specifically, sympathetic nerve fiber density is significantly higher in prostate tumors than in normal para-tumor tissue [102], and activation of sympathetic adrenalin signals is necessary for the early stages of prostate cancer and the initiation of the angiogenic switch. Angiogenesis is inhibited when the loss of β-adrenergic receptor signaling increases the oxidative phosphorylation of endothelial cells by increasing the expression of mitochondrial cytochrome c oxidase assembly factor 6 [20]. In addition, the adrenergic signal is closely associated with the malignant invasion of the tumor [102]. The adrenergic signal up-regulates the expression of CCL2 in lung stromal cells before metastasis, increases the infiltration of monocytes and macrophages into lung tissue, and promotes the colonization of tumor cells through lung metastasis [23].
Mechanisms in PDAC
The pancreas is innervated by sympathetic nerve fibers that release both adrenergic and neurotrophic factors which drive the cancer-nerve feedforward loop [100]. Tumor-derived neurotrophic factors bind to corresponding receptors like TrkA on sympathetic nerves, induce neurogenesis and axonogenesis [103, 104], and thus increase cancerous innervation [51, 57, 100]. The secretion of neurotrophic factors or induction of tumor cell-derived exosomes by PSCs also potentiates nerve proliferation and increases tumor innervation [104,105,106] (Fig. 3A). Catecholamines like norepinephrine (NE) from sympathetic nerves act on ADRB2 from PDAC cells and promotes their PNI, invasion, and metastasis via the activation of the ADRB2/PKA/STAT3 signaling pathway, which increases the production of NGF and MMP2/9 [107]. Meanwhile, the ADRB2-Akt pathway in PDAC activated by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) mediates smoking-induced tumor stemness and gemcitabine resistance by increasing octamer-binding transcription factor-4 (OCT-4), SRY-Box transcription factor 2 (SOX-2), and Nanog in pancreatic cancer cells [108] (Fig.3B). The orthotopic mouse model of breast cancer shows negligible effects of circulating epinephrine on β2-adrenergic signaling [109]. In addition to acting directly on cancer cells, sympathetic nerves have also been shown to regulate the immune function of tumor-infiltrating lymphocytes through ADRB2 on CD8+T cells in melanoma [101]. The density of PD-L1+ tumor-associated nerves is inversely correlated with that of CD8+ tumor-associated lymphocytes and predicts higher biochemical recurrence [110].

Sympathetic innervation and signaling pathways between NE and PDAC cells. A PDAC cells and pancreatic stellate cells release NGF to act on the TrkA receptors on the sympathetic nerve and promote the sympathetic innervation of the tumor. On the contrary, the neurotransmitter NE released by the sympathetic nerve promotes the proliferation of tumor cells through the ADRB2 receptors on tumor cells and endothelial cells and inhibits immune function by binding to receptors on immune cells to promote tumor progression. B The combination of NE and ADRB2 activates the downstream PKA-STAT3 signaling pathway, increases the phosphorylation level of STAT3, and promotes the release of NGF, MMP2, and MMP9 from tumor cells. Activation of ADRB2 by NNK triggers the downstream Akt pathway which in turn increases the expression of ADRB2. The levels of OCT-4, SOX-2, and Nanog are also up-regulated and thus promote the tumor stemness and chemoresistance of PDAC. Abbreviations: PDAC, pancreatic ductal adenocarcinoma; NE, norepinephrine; NGF, nerve growth factor; TrkA, tropomyosin receptor kinase A; ADRB, β-adrenergic receptor; MMP, matrix metalloproteinase; NNK, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone; OCT-4, octamer-binding transcription factor-4; SOX-2, SRY-Box transcription factor 2
Therapeutic Implications
Since sympathetic activation in TME leads to the progression of PDAC, blocking the adrenergic signaling pathway may be a potential therapeutic strategy. Pharmacological ablation of sympathetic nerves by 6-hydroxydopamine [5, 23] results in an increased proportion of neutrophils in the spleen of infected and uninfected mice, suggesting that sympathetic nerves may also be involved in the inhibition of neutrophil infiltration during infection [111]. In addition, the application of adrenaline-signaling pathway blockers [112, 113] such as propranolol [23, 114] can reverse the effect of chronic stress on the progression of PDAC [46]. When combined with gemcitabine, it reduces NGF expression and nerve density and improves the survival rate of KPC mice [100]. Combined sympathetic and CTLA-4 blockade inhibits murine melanoma growth by targeting infiltrating T cells [101]. Other drugs that target adrenergic signals including antipsychotics and tricyclic antidepressants have been shown to reduce the risk of colorectal cancer and glioma and are associated with increased survival [115,116,117,118,119,120].
Parasympathetic Innervation in PDAC
General Background
In many solid tumors, parasympathetic input is provided by the vagus nerve, which has been shown to modulate tumor growth in an organ-specific way. The stomach is innervated predominantly by the parasympathetic nervous system, where choline can stimulate the gastric epithelium to overexpress NGF, which leads to further enlargement of the enteric nerve and promotes canceration [39]. Acetylcholine can also promote the self-renewal and immune escape of CD133+ thyroid cancer cells through activation of the CD133/PI3K/Akt pathway [14]. In human prostate cancer cell lines and mouse models of prostate cancer, cholinergic signals are transduced in the tumor stroma through the muscarinic cholinergic receptor 1 (CHRM1) to promote tumor invasion [121]. The ability of muscarinic agonists to stimulate growth and muscarinic receptor antagonists to inhibit tumor growth has also been demonstrated for breast, melanoma, lung, colon, ovarian, and brain cancer [122].
Mechanisms in PDAC
In PDAC, over-expressed parasympathetic and cholinergic receptors have been detected in tumor tissue from patient and mouse models [123, 124]. Patients with PDAC and high parasympathetic density showed higher tumor budding and earlier recurrence rates than patients with low parasympathetic density [123]. The cholinergic signal enhances tumor growth by inhibiting the T cell response in the orthotopic PDAC model. When the parasympathetic nerve is stimulated, acetylcholine is released from the postganglionic fibers. Acetylcholine inhibits the recruitment of CD8+ T cell infiltration to PDAC through histone deacetylase 1-mediated CCL1, and directly inhibits CD8+ T-cell production of IFNγ in a concentration-dependent manner, reducing the Th1/Th2 ratio in the TME. In contrast, in tumor-bearing mice, vagotomy blockade not only reduces PNI but also increases CD8+ T cell infiltration and mouse survival [125] (Fig. 4A). Nicotine also promotes the metastasis of pancreatic cancer via the activation of the nicotinic acetylcholine receptor/JAK2 /STAT3 downstream signaling cascade and the upregulation of MUC4 expression [126] (Fig. 4B). However, Renz and colleagues showed that subdiaphragmatic vagotomy accelerates tumorigenesis and a muscarinic agonist suppresses tumorigenesis via MAPK and PI3K/AKT signaling [127] (Fig. 4C), suggesting that parasympathetic innervation may play distinct roles during the initiative and progressive stages of PDAC.

Parasympathetic innervation and crosstalk with PDAC cells. A PDAC cells release the neurotrophic factor NGF, which combines with TrkA on the parasympathetic nerve and promotes the proliferation of the parasympathetic nerve and the innervation of PDAC, resulting in an increase in the level of acetylcholine (ACh) and promotes the growth of PDAC cells. In addition, parasympathetic nerves can also promote the transformation of Th1 to Th2 immune cells by releasing chemokines and inhibiting the release of IFNγ from CD8 + T cells, resulting in immunosuppression. B The activation of the α7 subunit of nAChRs by nicotine increases the expression of MUC4 through JAK2/STAT3 downstream signaling and in cooperation with the MEK/ERK1/2 pathway. MUC4 upregulation further promotes the metastasis of PDAC via the activation of downstream effectors, such as HER2, c-Src, and FAK. C Activation of ACh receptors by muscarinic agonists inhibits downstream EGFR/MAPK and PI3K/AKT signaling pathways and inhibits the proliferation of PDAC cells. Abbreviations: PDAC, pancreatic ductal adenocarcinoma; NGF, nerve growth factor; TrkA, tropomyosin receptor kinase A; nAChR, nicotinic acetylcholine receptor; ACh, acetylcholine
Therapeutic Implications
Blocking parasympathetic innervation with bilateral subdiaphragmatic vagotomy improves the survival of PDAC mice [47]. Similarly, abrogation of cholinergic input by vagotomy or chemical denervation inhibits the growth of gastric cancer by blocking the M3 receptor-mediated Wnt pathway [39]. It also enhances the therapeutic effect of systemic chemotherapy and prolongs survival. The inhibitory effect induced by denervation is related to the inhibition of Wnt signaling and stem cell expansion [128]. Carbachol is a selective CHRM3 agonist, which enhances prostate cancer growth via the CaM/CaMKK-mediated phosphorylation of Akt. Blocking CHRM3 by darifenacin treatment inhibits prostate cancer growth and castration resistance in vitro and in vivo [129]. In this line, other studies have also reported that CHRM1 is involved in regulating the migration and invasion of prostate cancer through the Hedgehog signaling pathway. The selective CHRM1 antagonist pirenzepine inhibits the migration and invasion of cancer cells [121]. Furthermore, the application of the CHRM inhibitors Pirenzepine [17] and Darifenacin [129] reduces migration and invasion, thereby suppressing cancer cell proliferation.
Sensory Innervation in PDAC
General Background
The role and mechanism of sensory innervation in tumor progression have been increasingly investigated recently. In head and neck cancer, loss of tumor protein 53 leads to adrenergic transdifferentiation of tumor-associated sensory nerves through loss of the microRNA miR-34a, and tumor growth is suppressed by sensory denervation [130]. Melanoma cells interact with nociceptive sensory neurons, leading to increases in their neurite outgrowth and release of CGRP, which may further increase the exhaustion of cytotoxic CD8+ T cells and promote tumor immune escape [131]. In oral mucosa carcinomas, the low-glucose environment drives the production of NGF, which may further promote the release of CGRP from nociceptive nerves. CGRP subsequently induces cytoprotective autophagy in cancer cells that thrive in nutrient-poor environments [132]. CGRP is also an important neurotransmitter in the neural-immune axis, negatively regulating the infection-related immune response [133,134,135]. In CGRP-knockout mice with oral squamous cell carcinoma, the tumor burden is significantly reduced with increased tumor-infiltrating lymphocytes [29].
Mechanisms in PDAC
Neurotrophic factors derived from PDAC cells can induce the proliferation of nerve fibers including sensory nerves. In turn, sensory nerves promote the migration and invasion of cancer cells in vitro and in vivo by releasing neurotrophic factors or chemokines [58, 86, 136]. In the nutrient-poor microenvironment of PDAC, the sprouting sensory nerve could also secret exogenous serine to maintain the survival of cancer cells [103] (Fig. 5A). In PDAC patient samples, high expression of neurotrophic factors has been confirmed to be associated with PNI [86]. Transient receptor potential vanilla 1 (TRPV1) is an ion channel expressed on nociceptive sensory neurons and mediates thermal pain. TRPV1 can be activated by the acidic environment of the TME [137], resulting in increased release of SP and CGRP from nociceptive neurons. In the early stage of primary PDAC formation, MMP1 induces protease-activated receptor-1 (PAR1) expression in DRGs to release SP by activating the AKT pathway, thereby activating PDAC cells expressing neurokinin 1 receptor (NK-1R) and enhancing cell migration, invasion, and PNI through the SP/NK1R/ERK signal. In addition, SP can also induce the expression of MMP2 in tumor cells [138, 139]. Organoid culture experiments have also confirmed that sensory neurons promote the proliferation of pancreatic intraepithelial neoplasms (PanIN)-like organs through SP-NK1-R signaling and STAT3 activation. In the genetically engineered mouse model of PDAC, sensory denervation leads to a loss of STAT3 activation and slows down the progression of PanIN to tumors [140] (Fig. 5B).

Molecular mechanisms by which sensory neurons promote PDAC progression. A PDAC cells release NGF, promote the sprouting of sensory nerves via TrKA, resulting in increased levels of CGRP and SP, and promote the growth of PDAC cells by binding to the SP receptors NK-1Rs on tumor cells. Sensory nerves also secret exogenous serine to maintain the survival of PDAC. B TRPV1 is activated by the acidic environment of TME, resulting in the increasing release of SP and CGRP from nociceptive neurons. MMP1 binding to its receptor PAR1 in DRG neurons mediates PNI of PDAC cells by activating the Akt pathway and induces the release of SP. SP promotes the migration, invasion, and PNI of PDAC cells through NK-1Rs by the activation of downstream ERK signaling. It also fuels the progress of PanIN by activating the STAT3 signaling pathway. Abbreviations: PDAC, pancreatic ductal adenocarcinoma; NGF, nerve growth factor; TrkA, tropomyosin receptor kinase A; NK-1R, neurokinin 1 receptor; CGRP, calcitonin gene-related peptide; SP, substance P; TME, tumor microenvironment; TRPV1, transient receptor potential vanilla 1; PAR1, protease-activated receptor-1; MMP, matrix metalloproteinase; PanIN, pancreatic intraepithelial neoplasms. PNI, perineural invasion
Therapeutic Implications
Drugs targeting nociceptor nerves, neuropeptides, and their receptor pathways are mainly used for pain treatment. But they now appear to have great potential in treating cancer. In acute myeloid leukemia and Ewing sarcoma, the efficacy of some drugs targeting CGRP and its receptors calcitonin receptor-like receptor (CALCRL) and receptor activity-modifying protein 1 (RAMP1) has been verified. The CGRP antagonist olcegepant increases differentiation and reduces the burden of leukemia and key stem cell characteristics in mouse models of acute myeloid leukemia, while small molecule inhibitors targeting CGRP receptors reduce the growth of Ewing sarcoma [141,142,143]. Also, TRPV1 is desensitized by capsaicin, and capsaicin or resiniferatoxin has been used as an alternative pharmacological method to block pain by depleting CGRP and SP without stimulation or toxicity. In addition, intravesical injection of resiniferatoxin improves bladder function in patients with an overactive bladder. In the bone cancer model, intrathecal injection of resiniferatoxin effectively relieves pain and improves function without significant long-term side-effects. These suggest the multiple therapeutic effects of targeting sensory nerves [15, 44, 144,145,146,147,148,149,150].
Pain Relief Targeting the Nerves Innervating PDAC
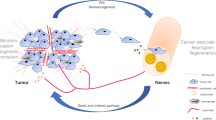
Cancer cells communicate with their surrounding environment [151]. Non-tumor cells in the TME may directly or indirectly interact with cancer cells, affecting the proliferation, migration, invasion, or drug resistance of PDAC. Evidence shows that sympathetic, parasympathetic, and sensory nerves undergo different forms of neuronal remodeling during the development of normal pancreatic tissue into PDAC. This has been confirmed in animal experiments and clinical pathological samples. Interstitial components such as nerve fibers in the TME play a direct or indirect role in promoting neurogenesis and tumor growth through various neurotransmitters, neurotrophic factors, and chemokines. The neural supply of amino-acids (such as serine) to the nutritionally deficient TME is also an important factor in the progression of PDAC [103]. Therefore, targeting nerves may be a promising strategy to treat cancer and immune evasion in the TME [152].
Pain is one of the common clinical symptoms of advanced PDAC. The abdominal pain symptoms can arise from various causes including tissue damage, inflammation, ductal obstruction and infiltration, and/or a direct mass effect on nerves in the celiac plexus [70]. At present, clinical treatments for pancreatic cancer pain mainly depend on opioids and surgery. Commonly-used analgesics are bucinnazine hydrochloride and morphine, but long-term use usually causes drug tolerance and adverse drug reactions. Surgical treatment can be categorized into celiac plexus neurolysis (CPN) and celiac ganglion neurolysis (CGN) [153,154,155,156], which are variations of an interventional technique for the diagnosis and treatment of concealed abdominal pain. Also, botulinum toxin is used as a preventive strategy for precancerous lesions and local treatment of low-risk tumors in prostate cancer, or as an adjunct to tumor treatment to reduce recurrence rates [157]. Neurolytic agents such as ethanol and phenol are used to permanently destroy the celiac plexus. Local anesthetics, most commonly bupivacaine or lidocaine, are used in combination with steroids and ethanol for the sake of reducing pain and the usage of painkillers [158, 159]. However, short-term back pain may occur at the injection site within 72 hours after celiac nerve block [156]. Other common side-effects include postural hypotension and diarrhea, which may be related to blocking or damaging sympathetic signals. Severe postoperative complications include lower limb paralysis and multiple organ failure, pain, and loss of temperature sensation. Other cases have been reported in which celiac trunk thrombosis after celiac artery spasm causes liver and spleen infarction, as well as stomach and proximal small intestine infarction [160]. In a prospective study of patients with unresectable PDAC and abdominal pain, compared with CPN, CGN shortened the median survival time and did not improve pain, quality of life, or frequency of adverse events [161]. Therefore, celiac nerve block should be carefully considered.
To this end, safer and more effective treatments for PDAC-related pain are urgently needed. Deep exploration of cancer-nerve crosstalk may provide potential targets [162, 163], such as neurotransmitters, neurotrophic factors, and chemokines. The effectiveness and safety of these strategies have been verified in preclinical animal models. Drugs currently known to regulate sympathetic or parasympathetic signals, such as the selective or non-selective β-blocker propranolol or metoprolol, or parasympathetic-like drugs, tend to have an antinociceptive effect with promising suppression of PDAC progression [164]. In turn, lidocaine or bupivacaine treatment has proved effective in inhibiting tumor growth and nerve fiber formation as well as cancer pain relief [165, 166]. Similarly, targeted neurotrophic factor therapy has also demonstrated tumor-suppressive effects in triple-negative breast cancer [167]. However, differences in cholinergic responses between cancers such as gastric and pancreatic cancers need to be carefully identified. In addition, capsaicin or resiniferatoxin targeting nociceptor sensory nerves could reduce the production of CGRP and SP, thus inhibiting PDAC growth and attenuating cancer pain. In addition to existing methods, recently developed neural engineering techniques allow the selective manipulation of the specific type of nerve fibers in the TME, in order to control the cancer progression and pain [152, 168].
Conclusions and Perspectives
Here we highlight the crucial role of tumor-innervating nerves as key TME components regulating the initiation and progression of PDAC as well as other cancer types. In addition, sympathetic, parasympathetic, or sensory innervation modulates distinct signaling pathways of tumor survival or immune escape. Selective peripheral nerve blockade or abrogation, and drugs targeting neuropeptides and their receptor pathways may be promising treatments for PDAC and cancer pain. However, it remains unclear how sensory nerves regulate the infiltration and function of immunological components in the TME of PDAC. Moreover, the direct or indirect modulation of cancer cells, stromal cells, and immune cells by tumor innervation interacting as a network in the TME warrants specific identification and detailed illustration. Recently, innervated wild-type or KPC murine pancreatic organoids have been well established, providing an ex vivo model to further study pancreatic neuropathy [169]. Future research is also needed to determine optimal strategies for tumor innervation based on current findings and to explore potential synergistic benefits when combined with chemotherapy or immunotherapy.
References
Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res 2019, 79: 4557–4566.
Demir IE, Friess H, Ceyhan GO. Nerve-cancer interactions in the stromal biology of pancreatic cancer. Front Physiol 2012, 3: 97.
Sejda A, Sigorski D, Gulczyński J, Wesołowski W, Kitlińska J, Iżycka-Świeszewska E. Complexity of neural component of tumor microenvironment in prostate cancer. Pathobiology 2020, 87: 87–99.
Xu R, Shang C, Zhao J, Han Y, Liu J, Chen K. Activation of M3 muscarinic receptor by acetylcholine promotes non-small cell lung cancer cell proliferation and invasion via EGFR/PI3K/AKT pathway. Tumor Biol 2015, 36: 4091–4100.
Xia Y, Wei Y, Li ZY, Cai XY, Zhang LL, Dong XR, et al. Catecholamines contribute to the neovascularization of lung cancer via tumor-associated macrophages. Brain Behav Immun 2019, 81: 111–121.
Vats K, Kruglov O, Sahoo B, Soman V, Zhang J, Shurin GV, et al. Sensory nerves impede the formation of tertiary lymphoid structures and development of protective antimelanoma immune responses. Cancer Immunol Res 2022, 10: 1141–1154.
Horvathova L, Padova A, Tillinger A, Osacka J, Bizik J, Mravec B. Sympathectomy reduces tumor weight and affects expression of tumor-related genes in melanoma tissue in the mouse. Stress 2016, 19: 528–534.
Valles SL, Benlloch M, Rodriguez ML, Mena S, Pellicer JA, Asensi M, et al. Stress hormones promote growth of B16–F10 melanoma metastases: An interleukin 6- and glutathione-dependent mechanism. J Transl Med 2013, 11: 72.
Oppitz M, Busch C, Garbe C, Drews U. Distribution of muscarinic receptor subtype M3 in melanomas and their metastases. J Cutan Pathol 2008, 35: 809–815.
Sereni F, Monte MD, Filippi L, Bagnoli P. Role of host β1- and β2-adrenergic receptors in a murine model of B16 melanoma: Functional involvement of β3-adrenergic receptors. Naunyn-Schmiedeberg’s Arch Pharmacol 2015, 388: 1317–1331.
Lammerding-Köppel M, Noda S, Blum A, Schaumburg-Lever G, Rassner G, Drews U. Immunohistochemical localization of muscarinic acetylcholine receptors in primary and metastatic malignant melanomas. J Cutan Pathol 1997, 24: 137–144.
Keskinov AA, Tapias V, Watkins SC, Ma Y, Shurin MR, Shurin GV. Impact of the sensory neurons on melanoma growth in vivo. PLoS ONE 2016, 11: e0156095.
Rowe CW, Dill T, Griffin N, Jobling P, Faulkner S, Paul JW, et al. Innervation of papillary thyroid cancer and its association with extra-thyroidal invasion. Sci Rep 2020, 10: 1539.
Wang Z, Liu W, Wang C, Li Y, Ai Z. Acetylcholine promotes the self-renewal and immune escape of CD133+ thyroid cancer cells through activation of CD133-Akt pathway. Cancer Lett 2020, 471: 116–124.
Xu S, Zhang L, Cheng X, Yu H, Bao J, Lu R. Capsaicin inhibits the metastasis of human papillary thyroid carcinoma BCPAP cells through the modulation of the TRPV1 channel. Food Funct 2018, 9: 344–354.
Dwivedi S, Bautista M, Shrestha S, Elhasasna H, Chaphekar T, Vizeacoumar FS, et al. Sympathetic signaling facilitates progression of neuroendocrine prostate cancer. Cell Death Discov 2021, 7: 364.
Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, et al. Autonomic nerve development contributes to prostate cancer progression. Science 2013, 341: 1236361. https://doi.org/10.1126/science.1236361.
Huang Z, Li G, Zhang Z, Gu R, Wang W, Lai X, et al. β2AR-HIF-1α-CXCL12 signaling of osteoblasts activated by isoproterenol promotes migration and invasion of prostate cancer cells. BMC Cancer 2019, 19: 1142.
Ayala GE, Wheeler TM, Shine HD, Schmelz M, Frolov A, Chakraborty S, et al. In vitro dorsal root Ganglia and human prostate cell line interaction: Redefining perineural invasion in prostate cancer. Prostate 2001, 49: 213–223.
Zahalka AH, Arnal-Estapé A, Maryanovich M, Nakahara F, Cruz CD, Finley LWS, et al. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science 2017, 358: 321–326.
Wang Q, Chen J, Zhang M, Wang H, Zeng Y, Huang Y, et al. Autophagy induced by muscarinic acetylcholine receptor 1 mediates migration and invasion targeting Atg5 via AMPK/mTOR pathway in prostate cancer. J Oncol 2022, 2022: 6523195.
Kamiya A, Hayama Y, Kato S, Shimomura A, Shimomura T, Irie K, et al. Genetic manipulation of autonomic nerve fiber innervation and activity and its effect on breast cancer progression. Nat Neurosci 2019, 22: 1289–1305.
Chen H, Liu D, Guo L, Cheng X, Guo N, Shi M. Chronic psychological stress promotes lung metastatic colonization of circulating breast cancer cells by decorating a pre-metastatic niche through activating β-adrenergic signaling. J Pathol 2018, 244: 49–60.
Bruzzone A, Sarappa MG, Castillo LF, Lüthy IA. Involvement of α2- and β2-adrenoceptors on breast cancer cell proliferation and tumour growth regulation. Br J Pharmacol 2012, 166: 721–736.
Erin N, Barkan GA, Clawson GA. Vagus nerve regulates breast cancer metastasis to the adrenal gland. Anticancer Res 2013, 33: 3675–3682.
Lee JW, Shahzad MMK, Lin YG, Armaiz-Pena G, Mangala LS, Han HD, et al. Surgical stress promotes tumor growth in ovarian carcinoma. Clin Cancer Res 2009, 15: 2695–2702.
Zakut H, Ehrlich G, Ayalon A, Prody CA, Malinger G, Seidman S, et al. Acetylcholinesterase and butyrylcholinesterase genes coamplify in primary ovarian carcinomas. J Clin Invest 1990, 86: 900–908.
Han GH, Chay DB, Nam S, Cho H, Chung JY, Kim JH. Prognostic significance of transient receptor potential vanilloid type 1 (TRPV1) and phosphatase and tension homolog (PTEN) in epithelial ovarian cancer. Cancer Genomics Proteomics 2020, 17: 309–319.
McIlvried LA, Atherton MA, Horan NL, Goch TN, Scheff NN. Sensory neurotransmitter calcitonin gene-related peptide modulates tumor growth and lymphocyte infiltration in oral squamous cell carcinoma. Adv Biol (Weinh) 2022, 6: e2200019.
Kwon SY, Chun KJ, Kil HK, Jung N, Shin HA, Jang JY, et al. β2-adrenergic receptor expression and the effects of norepinephrine and propranolol on various head and neck cancer subtypes. Oncol Lett 2021, 22: 804.
Hsu CC, Tsai KY, Su YF, Chien CY, Chen YC, Wu YC, et al. α7-Nicotine acetylcholine receptor mediated nicotine induced cell survival and cisplatin resistance in oral cancer. Arch Oral Biol 2020, 111: 104653.
Shang ZJ, Liu K, Liang DF. Expression of beta2-adrenergic receptor in oral squamous cell carcinoma. J Oral Pathol Med 2009, 38: 371–376.
Zhang Y, Chen M, Liu Z, Wang X, Ji T. The neuropeptide calcitonin gene-related peptide links perineural invasion with lymph node metastasis in oral squamous cell carcinoma. BMC Cancer 2021, 21: 1254.
Liu Y, Hao Y, Zhao H, Zhang Y, Cheng D, Zhao L, et al. PlexinA1 activation induced by β2-AR promotes epithelial-mesenchymal transition through JAK-STAT3 signaling in human gastric cancer cells. J Cancer 2022, 13: 2258–2270.
Lu Y, Zhang Y, Zhao H, Li Q, Liu Y, Zuo Y, et al. Chronic stress model simulated by salbutamol promotes tumorigenesis of gastric cancer cells through β2-AR/ERK/EMT pathway. J Cancer 2022, 13: 401–412.
Lu YJ, Geng ZJ, Sun XY, Li YH, Fu XB, Zhao XY, et al. Isoprenaline induces epithelial-mesenchymal transition in gastric cancer cells. Mol Cell Biochem 2015, 408: 1–13.
Shan T, Cui X, Li W, Lin W, Li Y, Chen X, et al. Novel regulatory program for norepinephrine-induced epithelial-mesenchymal transition in gastric adenocarcinoma cell lines. Cancer Sci 2014, 105: 847–856.
Shi M, Liu D, Duan H, Han C, Wei B, Qian L, et al. Catecholamine up-regulates MMP-7 expression by activating AP-1 and STAT3 in gastric cancer. Mol Cancer 2010, 9: 269.
Hayakawa Y, Sakitani K, Konishi M, Asfaha S, Niikura R, Tomita H, et al. Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell 2017, 31: 21–34.
Yu H, Xia H, Tang Q, Xu H, Wei G, Chen Y, et al. Acetylcholine acts through M3 muscarinic receptor to activate the EGFR signaling and promotes gastric cancer cell proliferation. Sci Rep 2017, 7: 40802.
Yang T, He W, Cui F, Xia J, Zhou R, Wu Z, et al. MACC1 mediates acetylcholine-induced invasion and migration by human gastric cancer cells. Oncotarget 2016, 7: 18085–18094.
Xie R, Xu J, Xiao Y, Wu J, Wan H, Tang B, et al. Calcium promotes human gastric cancer via a novel coupling of calcium-sensing receptor and TRPV4 channel. Cancer Res 2017, 77: 6499–6512.
Tang B, Wu J, Zhu MX, Sun X, Liu J, Xie R, et al. VPAC1 couples with TRPV4 channel to promote calcium-dependent gastric cancer progression via a novel autocrine mechanism. Oncogene 2019, 38: 3946–3961.
Saloman JL, Albers KM, Li D, Hartman DJ, Crawford HC, Muha EA, et al. Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. Proc Natl Acad Sci U S A 2016, 113: 3078–3083.
Partecke LI, Speerforck S, Käding A, Seubert F, Kühn S, Lorenz E, et al. Chronic stress increases experimental pancreatic cancer growth, reduces survival and can be antagonised by beta-adrenergic receptor blockade. Pancreatology 2016, 16: 423–433.
Kim-Fuchs C, Le CP, Pimentel MA, Shackleford D, Ferrari D, Angst E, et al. Chronic stress accelerates pancreatic cancer growth and invasion: A critical role for beta-adrenergic signaling in the pancreatic microenvironment. Brain Behav Immun 2014, 40: 40–47.
Partecke LI, Käding A, Trung DN, Diedrich S, Sendler M, Weiss F, et al. Subdiaphragmatic vagotomy promotes tumor growth and reduces survival via TNFα in a murine pancreatic cancer model. Oncotarget 2017, 8: 22501–22512.
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA A Cancer J Clin 2021, 71: 7–33.
Hosein AN, Dougan SK, Aguirre AJ, Maitra A. Translational advances in pancreatic ductal adenocarcinoma therapy. Nat Cancer 2022, 3: 272–286.
Liebig C, Ayala G, Wilks JA, Berger DH, Albo D. Perineural invasion in cancer: A review of the literature. Cancer 2009, 115: 3379–3391.
Ferdoushi A, Griffin N, Marsland M, Xu X, Faulkner S, Gao F, et al. Tumor innervation and clinical outcome in pancreatic cancer. Sci Rep 2021, 11: 7390.
Kondo N, Murakami Y, Uemura K, Hashimoto Y, Nakagawa N, Sasaki H, et al. An increased number of perineural invasions is independently associated with poor survival of patients with resectable pancreatic ductal adenocarcinoma. Pancreas 2015, 44: 1345–1351.
Crippa S, Pergolini I, Javed AA, Honselmann KC, Weiss MJ, Di Salvo F, et al. Implications of perineural invasion on disease recurrence and survival after pancreatectomy for pancreatic head ductal adenocarcinoma. Ann Surg 2020, 276: 378–385.
Nagakawa T, Mori K, Nakano T, Kadoya M, Kobayashi H, Akiyama T, et al. Perineural invasion of carcinoma of the pancreas and biliary tract. Br J Surg 2005, 80: 619–621.
Wang H, Zheng Q, Lu Z, Wang L, Ding L, Xia L, et al. Role of the nervous system in cancers: A review. Cell Death Discov 2021, 7: 76.
Bizzozero L, Pergolizzi M, Pascal D, Maldi E, Villari G, Erriquez J, et al. Tumoral neuroligin 1 promotes cancer-nerve interactions and synergizes with the glial cell line-derived neurotrophic factor. Cells 2022, 11: 280.
Li JH, Ma QY, Shen SG, Hu HT. Stimulation of dorsal root ganglion neurons activity by pancreatic cancer cell lines. Cell Biol Int 2008, 32: 1530–1535.
Hirth M, Gandla J, Höper C, Gaida MM, Agarwal N, Simonetti M, et al. CXCL10 and CCL21 promote migration of pancreatic cancer cells toward sensory neurons and neural remodeling in tumors in mice, associated with pain in patients. Gastroenterology 2020, 159: 665-681.e13.
Iwasaki T, Hiraoka N, Ino Y, Nakajima K, Kishi Y, Nara S, et al. Reduction of intrapancreatic neural density in cancer tissue predicts poorer outcome in pancreatic ductal carcinoma. Cancer Sci 2019, 110: 1491–1502.
Radzimirska M, Kuchinka J, Nowak E, Trybus W, Szczurkowski A. Cholinergic and adrenergic innervation of the pancreas in Chinchilla (Chinchilla laniger Molina). Folia Histochem Cytobiol 2020, 58: 54–60.
Makhmutova M, Caicedo A. Optical imaging of pancreatic innervation. Front Endocrinol (Lausanne) 2021, 12: 663022.
Lindsay TH, Halvorson KG, Peters CM, Ghilardi JR, Kuskowski MA, Wong GY, et al. A quantitative analysis of the sensory and sympathetic innervation of the mouse pancreas. Neuroscience 2006, 137: 1417–1426.
Bockman DE, Buchler M, Malfertheiner P, Beger HG. Analysis of nerves in chronic pancreatitis. Gastroenterology 1988, 94: 1459–1469.
Dhiraj Yadav. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144: 1252–1261.
Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11: 291–302.
Friess H, Zhu ZW, di Mola FF, Kulli C, Graber HU, Andren-Sandberg Å, et al. Nerve growth factor and its high-affinity receptor in chronic pancreatitis. Ann Surg 1999, 230: 615.
Cournoyer A, Fournier D, Benoit-Biancamano MO. Neural hypertrophy and hyperplasia in a case of chronic ovine pancreatitis. J Comp Pathol 2021, 185: 1–7.
Lindsay TH, Jonas BM, Sevcik MA, Kubota K, Halvorson KG, Ghilardi JR, et al. Pancreatic cancer pain and its correlation with changes in tumor vasculature, macrophage infiltration, neuronal innervation, body weight and disease progression. Pain 2005, 119: 233–246.
Ceyhan GO, Demir IE, Rauch U, Bergmann F, Müller MW, Büchler MW, et al. Pancreatic neuropathy results in neural remodeling and altered pancreatic innervation in chronic pancreatitis and pancreatic cancer. Am J Gastroenterol 2009, 104: 2555–2565.
Ceyhan GO, Bergmann F, Kadihasanoglu M, Altintas B, Demir IE, Hinz U, et al. Pancreatic neuropathy and neuropathic pain—a comprehensive pathomorphological study of 546 cases. Gastroenterology 2009, 136: 177-186.e1.
Stopczynski RE, Normolle DP, Hartman DJ, Ying H, DeBerry JJ, Bielefeldt K, et al. Neuroplastic changes occur early in the development of pancreatic ductal adenocarcinoma. Cancer Res 2014, 74: 1718–1727.
Demir IE, Friess H, Ceyhan GO. Neural plasticity in pancreatitis and pancreatic cancer. Nat Rev Gastroenterol Hepatol 2015, 12: 649–659.
Lu M, Xiu DR, Guo LM, Yuan CH, Zhang LF, Tao LY. Extrapancreatic neuropathy correlates with early liver metastasis in pancreatic head adenocarcinoma. Onco Targets Ther 2019, 12: 11083–11095.
Faulkner S, Jobling P, March B, Jiang CC, Hondermarck H. Tumor neurobiology and the war of nerves in cancer. Cancer Discov 2019, 9: 702–710.
Reavis HD, Chen HI, Drapkin R. Tumor innervation: Cancer has some nerve. Trends Cancer 2020, 6: 1059–1067.
Mauffrey P, Tchitchek N, Barroca V, Bemelmans AP, Firlej V, Allory Y, et al. Progenitors from the central nervous system drive neurogenesis in cancer. Nature 2019, 569: 672–678.
Almagro J, Messal HA, Zaw Thin M, van Rheenen J, Behrens A. Tissue clearing to examine tumour complexity in three dimensions. Nat Rev Cancer 2021, 21: 718–730.
Schmitd LB, Perez-Pacheco C, Bellile EL, Wu W, Casper K, Mierzwa M, et al. Spatial and transcriptomic analysis of perineural invasion in oral cancer. Clin Cancer Res 2022, 28: 3557–3572.
Zhang LJ, Wu B, Zha ZL, Qu W, Zhao H, Yuan J, et al. Perineural invasion as an independent predictor of biochemical recurrence in prostate cancer following radical prostatectomy or radiotherapy: A systematic review and meta-analysis. BMC Urol 2018, 18: 5.
Deng J, You Q, Gao Y, Yu Q, Zhao P, Zheng Y, et al. Prognostic value of perineural invasion in gastric cancer: A systematic review and meta-analysis. PLoS One 2014, 9: e88907.
Al-Sukhni E, Attwood K, Gabriel EM, LeVea CM, Kanehira K, Nurkin SJ. Lymphovascular and perineural invasion are associated with poor prognostic features and outcomes in colorectal cancer: A retrospective cohort study. Int J Surg 2017, 37: 42–49.
Marchesi F, Piemonti L, Mantovani A, Allavena P. Molecular mechanisms of perineural invasion, a forgotten pathway of dissemination and metastasis. Cytokine Growth Factor Rev 2010, 21: 77–82.
Marchesi F, Piemonti L, Fedele G, Destro A, Roncalli M, Albarello L, et al. The chemokine receptor CX3CR1 is involved in the neural tropism and malignant behavior of pancreatic ductal adenocarcinoma. Cancer Res 2008, 68: 9060–9069.
Xu Q, Wang Z, Chen X, Duan W, Lei J, Zong L, et al. Stromal-derived factor-1α/CXCL12-CXCR4 chemotactic pathway promotes perineural invasion in pancreatic cancer. Oncotarget 2015, 6: 4717–4732.
Zaitseva L, Murray MY, Shafat MS, Lawes MJ, MacEwan DJ, Bowles KM, et al. Ibrutinib inhibits SDF1/CXCR4 mediated migration in AML. Oncotarget 2014, 5: 9930–9938.
Jurcak NR, Rucki AA, Muth S, Thompson E, Sharma R, Ding D, et al. Axon guidance molecules promote perineural invasion and metastasis of orthotopic pancreatic tumors in mice. Gastroenterology 2019, 157: 838-850.e6.
Li F, He C, Yao H, Zhao Y, Ye X, Zhou S, et al. Glutamate from nerve cells promotes perineural invasion in pancreatic cancer by regulating tumor glycolysis through HK2 mRNA-m6A modification. Pharmacol Res 2023, 187: 106555.
Zhang W, He R, Yang W, Zhang Y, Yuan Q, Wang J, et al. Autophagic Schwann cells promote perineural invasion mediated by the NGF/ATG7 paracrine pathway in pancreatic cancer. J Exp Clin Cancer Res 2022, 41: 48.
Li R, Li D, Wu C, Ye L, Wu Y, Yuan Y, et al. Nerve growth factor activates autophagy in Schwann cells to enhance myelin debris clearance and to expedite nerve regeneration. Theranostics 2020, 10: 1649–1677.
Tian Z, Ou G, Su M, Li R, Pan L, Lin X, et al. TIMP1 derived from pancreatic cancer cells stimulates Schwann cells and promotes the occurrence of perineural invasion. Cancer Lett 2022, 546: 215863.
Xu X, Lu X, Chen L, Peng K, Ji F. Downregulation of MMP1 functions in preventing perineural invasion of pancreatic cancer through blocking the NT-3/TrkC signaling pathway. J Clin Lab Anal 2022, 36: e24719.
Bakst RL, Xiong H, Chen CH, Deborde S, Lyubchik A, Zhou Y, et al. Inflammatory monocytes promote perineural invasion via CCL2-mediated recruitment and cathepsin B expression. Cancer Res 2017, 77: 6400–6414.
Peng T, Guo Y, Gan Z, Ling Y, Xiong J, Liang X, et al. Nerve growth factor (NGF) encourages the neuroinvasive potential of pancreatic cancer cells by activating the Warburg effect and promoting tumor derived exosomal miRNA-21 expression. Oxid Med Cell Longev 2022, 2022: 8445093.
Pothula SP, Xu Z, Goldstein D, Pirola RC, Wilson JS, Apte MV. Targeting HGF/c-MET axis in pancreatic cancer. Int J Mol Sci 2020, 21: 9170.
Qin T, Xiao Y, Qian W, Wang X, Gong M, Wang Q, et al. HGF/c-Met pathway facilitates the perineural invasion of pancreatic cancer by activating the mTOR/NGF axis. Cell Death Dis 2022, 13: 387.
Zhang JF, Tao LY, Yang MW, Xu DP, Jiang SH, Fu XL, et al. CD74 promotes perineural invasion of cancer cells and mediates neuroplasticity via the AKT/EGR-1/GDNF axis in pancreatic ductal adenocarcinoma. Cancer Lett 2021, 508: 47–58.
Takahashi H, Ohigashi H, Ishikawa O, Gotoh K, Yamada T, Nagata S, et al. Perineural invasion and lymph node involvement as indicators of surgical outcome and pattern of recurrence in the setting of preoperative gemcitabine-based chemoradiation therapy for resectable pancreatic cancer. Ann Surg 2012, 255: 95–102.
Zhao P, Wu Z, Wang Z, Wu C, Huang X, Tian B. Prognostic role of the prognostic nutritional index in patients with pancreatic cancer who underwent curative resection without preoperative neoadjuvant treatment: A systematic review and meta-analysis. Front Surg 2022, 9: 992641.
Tong F, He Q, Du WJ, Yang H, Du D, Pu S, et al. Sympathetic nerve mediated spinal Glia activation underlies itch in a cutaneous T-cell lymphoma model. Neurosci Bull 2022, 38: 435–439.
Renz BW, Takahashi R, Tanaka T, Macchini M, Hayakawa Y, Dantes Z, et al. β2 adrenergic-neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell 2018, 34: 863–867.
Wang B, Xu Z, Sunthamala N, Yaguchi T, Huang J, Kawakami Y, et al. Combinatorial sympathetic and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) blockades inhibit the murine melanoma growth by targeting infiltrating T cells. Transl Cancer Res 2021, 10: 899–913.
Ayala GE, Dai H, Powell M, Li R, Ding Y, Wheeler TM, et al. Cancer-related axonogenesis and neurogenesis in prostate cancer. Clin Cancer Res 2008, 14: 7593–7603.
Banh RS, Biancur DE, Yamamoto K, Sohn ASW, Walters B, Kuljanin M, et al. Neurons release serine to support mRNA translation in pancreatic cancer. Cell 2020, 183: 1202-1218.e25.
Jiang J, Bai J, Qin T, Wang Z, Han L. NGF from pancreatic stellate cells induces pancreatic cancer proliferation and invasion by PI3K/AKT/GSK signal pathway. J Cell Mol Med 2020, 24: 5901–5910.
Mazaki AI, Yamauchi K, Orita S, Inage K, Suzuki M, Fujimoto K, et al. Nerve growth factor in breast cancer cells promotes axonal growth and expression of calcitonin gene-related peptide in a rat model of spinal metastasis. Anticancer Res 2022, 42: 581–587.
Madeo M, Colbert PL, Vermeer DW, Lucido CT, Cain JT, Vichaya EG, et al. Cancer exosomes induce tumor innervation. Nat Commun 2018, 9: 4284.
Guo K, Ma Q, Li J, Wang Z, Shan T, Li W, et al. Interaction of the sympathetic nerve with pancreatic cancer cells promotes perineural invasion through the activation of STAT3 signaling. Mol Cancer Ther 2013, 12: 264–273.
Chen X, Zhang W, Liu R, Zhu Z, Gong M, Wang Q, et al. NNK from tobacco smoking enhances pancreatic cancer cell stemness and chemoresistance by creating a β2AR-Akt feedback loop that activates autophagy. Mol Oncol 2022, 16: 2881–2895.
Walker AK, Martelli D, Ziegler AI, Lambert GW, Phillips SE, Hill SJ, et al. Circulating epinephrine is not required for chronic stress to enhance metastasis. Psychoneuroendocrinology 2019, 99: 191–195.
Mo RJ, Han ZD, Liang YK, Ye JH, Wu SL, Lin SX, et al. Expression of PD-L1 in tumor-associated nerves correlates with reduced CD8+ tumor-associated lymphocytes and poor prognosis in prostate cancer. Int J Cancer 2019, 144: 3099–3110.
Rice PA, Boehm GW, Moynihan JA, Bellinger DL, Stevens SY. Chemical sympathectomy alters numbers of splenic and peritoneal leukocytes. Brain Behav Immun 2002, 16: 62–73.
Melhem-Bertrandt A, Chavez-Macgregor M, Lei X, Brown EN, Lee RT, Meric-Bernstam F, et al. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J Clin Oncol 2011, 29: 2645–2652.
Watkins JL, Thaker PH, Nick AM, Ramondetta LM, Kumar S, Urbauer DL, et al. Clinical impact of selective and nonselective beta-blockers on survival in patients with ovarian cancer. Cancer 2015, 121: 3444–3451.
Hwa YL, Shi Q, Kumar SK, Lacy MQ, Gertz MA, Kapoor P, et al. Beta-blockers improve survival outcomes in patients with multiple myeloma: A retrospective evaluation. Am J Hematol 2017, 92: 50–55.
Grytli HH, Fagerland MW, Fosså SD, Taskén KA. Association between use of β-blockers and prostate cancer-specific survival: A cohort study of 3561 prostate cancer patients with high-risk or metastatic disease. Eur Urol 2014, 65: 635–641.
Saloman JL, Albers KM, Rhim AD, Davis BM. Can stopping nerves, stop cancer? Trends Neurosci 2016, 39: 880–889.
Barak Y, Achiron A, Mandel M, Mirecki I, Aizenberg D. Reduced cancer incidence among patients with schizophrenia. Cancer 2005, 104: 2817–2821.
Chou FH, Tsai KY, Su CY, Lee CC. The incidence and relative risk factors for developing cancer among patients with schizophrenia: A nine-year follow-up study. Schizophr Res 2011, 129: 97–103.
Chubak J, Boudreau DM, Rulyak SJ, Mandelson MT. Colorectal cancer risk in relation to antidepressant medication use. Int J Cancer 2011, 128: 227–232.
Pottegård A, García Rodríguez LA, Rasmussen L, Damkier P, Friis S, Gaist D. Use of tricyclic antidepressants and risk of glioma: A nationwide case-control study. Br J Cancer 2016, 114: 1265–1268.
Yin QQ, Xu LH, Zhang M, Xu C. Muscarinic acetylcholine receptor M1 mediates prostate cancer cell migration and invasion through hedgehog signaling. Asian J Androl 2018, 20: 608–614.
Calaf GM, Crispin LA, Muñoz JP, Aguayo F, Bleak TC. Muscarinic receptors associated with cancer. Cancers 2022, 14: 2322.
Zhang L, Guo L, Tao M, Fu W, Xiu D. Parasympathetic neurogenesis is strongly associated with tumor budding and correlates with an adverse prognosis in pancreatic ductal adenocarcinoma. Chin J Cancer Res 2016, 28: 180–186.
Zhang L, Xiu D, Zhan J, He X, Guo L, Wang J, et al. High expression of muscarinic acetylcholine receptor 3 predicts poor prognosis in patients with pancreatic ductal adenocarcinoma. Onco Targets Ther 2016, 9: 6719–6726.
Yang MW, Tao LY, Jiang YS, Yang JY, Huo YM, Liu DJ, et al. Perineural invasion reprograms the immune microenvironment through cholinergic signaling in pancreatic ductal adenocarcinoma. Cancer Res 2020, 80: 1991–2003.
Momi N, Ponnusamy MP, Kaur S, Rachagani S, Kunigal SS, Chellappan S, et al. Nicotine/cigarette smoke promotes metastasis of pancreatic cancer through α7nAChR-mediated MUC4 upregulation. Oncogene 2013, 32: 1384–1395.
Renz BW, Tanaka T, Sunagawa M, Takahashi R, Jiang Z, Macchini M, et al. Cholinergic signaling via muscarinic receptors directly and indirectly suppresses pancreatic tumorigenesis and cancer stemness. Cancer Discov 2018, 8: 1458–1473.
Zhao CM, Hayakawa Y, Kodama Y, Muthupalani S, Westphalen CB, Andersen GT, et al. Denervation suppresses gastric tumorigenesis. Sci Transl Med 2014, 6: 250ra115.
Wang N, Yao M, Xu J, Quan Y, Zhang K, Yang R, et al. Autocrine activation of CHRM3 promotes prostate cancer growth and castration resistance via CaM/CaMKK-mediated phosphorylation of Akt. Clin Cancer Res 2015, 21: 4676–4685.
Amit M, Takahashi H, Dragomir MP, Lindemann A, Gleber-Netto FO, Pickering CR, et al. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature 2020, 578: 449–454.
Balood M, Ahmadi M, Eichwald T, Ahmadi A, Majdoubi A, Roversi K, et al. Nociceptor neurons affect cancer immunosurveillance. Nature 2022, 611: 405–412.
Zhang Y, Lin C, Liu Z, Sun Y, Chen M, Guo Y, et al. Cancer cells co-opt nociceptive nerves to thrive in nutrient-poor environments and upon nutrient-starvation therapies. Cell Metab 2022, 34: 1999-2017.e10.
Assas BM, Pennock JI, Miyan JA. Calcitonin gene-related peptide is a key neurotransmitter in the neuro-immune axis. Front Neurosci 2014, 8: 23.
Wallrapp A, Burkett PR, Riesenfeld SJ, Kim SJ, Christian E, Abdulnour REE, et al. Calcitonin gene-related peptide negatively regulates alarmin-driven type 2 innate lymphoid cell responses. Immunity 2019, 51: 709-723.e6.
Baral P, Umans BD, Li L, Wallrapp A, Bist M, Kirschbaum T, et al. Nociceptor sensory neurons suppress neutrophil and γδ T cell responses in bacterial lung infections and lethal pneumonia. Nat Med 2018, 24: 417–426.
Bapat AA, Munoz RM, Von Hoff DD, Han H. Blocking nerve growth factor signaling reduces the neural invasion potential of pancreatic cancer cells. PLoS ONE 2016, 11: e0165586.
Wakabayashi H, Wakisaka S, Hiraga T, Hata K, Nishimura R, Tominaga M, et al. Decreased sensory nerve excitation and bone pain associated with mouse Lewis lung cancer in TRPV1-deficient mice. J Bone Miner Metab 2018, 36: 274–285.
Huang C, Li Y, Guo Y, Zhang Z, Lian G, Chen Y, et al. MMP1/PAR1/SP/NK1R paracrine loop modulates early perineural invasion of pancreatic cancer cells. Theranostics 2018, 8: 3074–3086.
Li X, Ma G, Ma Q, Li W, Liu J, Han L, et al. Neurotransmitter substance P mediates pancreatic cancer perineural invasion via NK-1R in cancer cells. Mol Cancer Res 2013, 11: 294–302.
Sinha S, Fu YY, Grimont A, Ketcham M, Lafaro K, Saglimbeni JA, et al. PanIN neuroendocrine cells promote tumorigenesis via neuronal cross-talk. Cancer Res 2017, 77: 1868–1879.
Gluexam T, Grandits AM, Schlerka A, Nguyen CH, Etzler J, Finkes T, et al. CGRP signaling via CALCRL increases chemotherapy resistance and stem cell properties in acute myeloid leukemia. Int J Mol Sci 2019, 20: 5826.
Angenendt L, Bormann E, Pabst C, Alla V, Görlich D, Braun L, et al. The neuropeptide receptor calcitonin receptor-like (CALCRL) is a potential therapeutic target in acute myeloid leukemia. Leukemia 2019, 33: 2830–2841.
Dallmayer M, Li J, Ohmura S, Alba Rubio R, Baldauf MC, Hölting TLB, et al. Targeting the CALCB/RAMP1 axis inhibits growth of Ewing sarcoma. Cell Death Dis 2019, 10: 116.
Fukushima A, Mamada K, Iimura A, Ono H. Supraspinal-selective TRPV1 desensitization induced by intracerebroventricular treatment with resiniferatoxin. Sci Rep 2017, 7: 12452.
Fattori V, Hohmann MSN, Rossaneis AC, Pinho-Ribeiro FA, Verri WA. Capsaicin: Current understanding of its mechanisms and therapy of pain and other pre-clinical and clinical uses. Molecules 2016, 21: 844.
Iadarola MJ, Mannes AJ. The vanilloid agonist resiniferatoxin for interventional-based pain control. Curr Top Med Chem 2011, 11: 2171–2179.
Holzer P. Transient receptor potential (TRP) channels as drug targets for diseases of the digestive system. Pharmacol Ther 2011, 131: 142–170.
Sharkey KA, Williams RG, Dockray GJ. Sensory substance P innervation of the stomach and pancreas. Demonstration of capsaicin-sensitive sensory neurons in the rat by combined immunohistochemistry and retrograde tracing. Gastroenterology 1984, 87: 914–921.
Kissin I, Szallasi A. Therapeutic targeting of TRPV1 by resiniferatoxin, from preclinical studies to clinical trials. Curr Top Med Chem 2011, 11: 2159–2170.
Chu X, Zhuang H, Liu Y, Li J, Wang Y, Jiang Y, et al. Blocking cancer-nerve crosstalk for treatment of metastatic bone cancer pain. Adv Mater 2022, 34: e2108653.
Li F, Li F. Cancer cells don’t live alone: Metabolic communication within tumor microenvironments. Dev Cell 2020, 54: 183–195.
Cervantes-Villagrana RD, Albores-García D, Cervantes-Villagrana AR, García-Acevez SJ. Tumor-induced neurogenesis and immune evasion as targets of innovative anti-cancer therapies. Signal Transduct Target Ther 2020, 5: 99.
Levy MJ, Topazian MD, Wiersema MJ, Clain JE, Rajan E, Wang KK, et al. Initial evaluation of the efficacy and safety of endoscopic ultrasound-guided direct Ganglia neurolysis and block. Am J Gastroenterol 2008, 103: 98–103.
Kaufman M, Singh G, Das S, Concha-Parra R, Erber J, Micames C, et al. Efficacy of endoscopic ultrasound-guided celiac plexus block and celiac plexus neurolysis for managing abdominal pain associated with chronic pancreatitis and pancreatic cancer. J Clin Gastroenterol 2010, 44: 127–134.
Doi S, Yasuda I, Kawakami H, Hayashi T, Hisai H, Irisawa A, et al. Endoscopic ultrasound-guided celiac Ganglia neurolysis vs. celiac plexus neurolysis: A randomized multicenter trial. Endoscopy 2013, 45: 362–369.
Cornman-Homonoff J, Holzwanger DJ, Lee KS, Madoff DC, Li D. Celiac plexus block and neurolysis in the management of chronic upper abdominal pain. Semin Intervent Radiol 2017, 34: 376–386.
Coarfa C, Florentin D, Putluri N, Ding Y, Au J, He D, et al. Influence of the neural microenvironment on prostate cancer. Prostate 2018, 78: 128–139.
Yan BM, Myers RP. Neurolytic celiac plexus block for pain control in unresectable pancreatic cancer. Am J Gastroenterol 2007, 102: 430–438.
Teo ZHT, Tey BLJ, Foo CW, Wong WY, Low JK. Intraoperative celiac plexus block with preperitoneal infusion reduces opioid usage in major hepato-pancreato-biliary surgery: A pilot study. Ann Surg 2021, 274: e97–e99.
Urits I, Jones MR, Orhurhu V, Peck J, Corrigan D, Hubble A, et al. A comprehensive review of the celiac plexus block for the management of chronic abdominal pain. Curr Pain Headache Rep 2020, 24: 42.
Levy MJ, Gleeson FC, Topazian MD, Fujii-Lau LL, Enders FT, Larson JJ, et al. Combined celiac Ganglia and plexus neurolysis shortens survival, without benefit, vs plexus neurolysis alone. Clin Gastroenterol Hepatol 2019, 17: 728-738.e9.
Jiang SH, Hu LP, Wang X, Li J, Zhang ZG. Neurotransmitters: Emerging targets in cancer. Oncogene 2020, 39: 503–515.
Kamiya A, Hiyama T, Fujimura A, Yoshikawa S. Sympathetic and parasympathetic innervation in cancer: Therapeutic implications. Clin Auton Res 2021, 31: 165–178.
Martin LJ, Piltonen MH, Gauthier J, Convertino M, Acland EL, Dokholyan NV, et al. Differences in the antinociceptive effects and binding properties of propranolol and bupranolol enantiomers. J Pain 2015, 16: 1321–1333.
Kaduri M, Sela M, Kagan S, Poley M, Abumanhal-Masarweh H, Mora-Raimundo P, et al. Targeting neurons in the tumor microenvironment with bupivacaine nanoparticles reduces breast cancer progression and metastases. Sci Adv 2021, 7: eabj5435.
Li B, Xu H, He C, Zou W, Tu Y. Lidocaine prevents breast cancer growth by targeting neuronatin to inhibit nerve fibers formation. J Toxicol Sci 2021, 46: 329–339.
Di Donato M, Galasso G, Giovannelli P, Sinisi AA, Migliaccio A, Castoria G. Targeting the nerve growth factor signaling impairs the proliferative and migratory phenotype of triple-negative breast cancer cells. Front Cell Dev Biol 2021, 9: 676568.
Shi DD, Guo JA, Hoffman HI, Su J, Mino-Kenudson M, Barth JL, et al. Therapeutic avenues for cancer neuroscience: Translational frontiers and clinical opportunities. Lancet Oncol 2022, 23: e62–e74.
Besikcioglu HE, Yurteri Ü, Munkhbaatar E, Ye L, Zhang F, Moretti A, et al. Innervated mouse pancreas organoids as an ex vivo model to study pancreatic neuropathy in pancreatic cancer. STAR Protoc 2021, 2: 100935.
Zhao Q, Gu X, Zhang C, Lu Q, Chen H, Xu L. Blocking M2 muscarinic receptor signaling inhibits tumor growth and reverses epithelial-mesenchymal transition (EMT) in non-small cell lung cancer (NSCLC). Cancer Biol Ther 2015, 16: 634–643.
Song P, Sekhon HS, Jia Y, Keller JA, Blusztajn JK, Mark GP, et al. Acetylcholine is synthesized by and acts as an autocrine growth factor for small cell lung carcinoma. Cancer Res 2003, 63: 214–221.
Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, et al. Author Correction: Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med 2021, 27: 2246.
Almasi S, Sterea AM, Fernando W, Clements DR, Marcato P, Hoskin DW, et al. TRPM2 ion channel promotes gastric cancer migration, invasion and tumor growth through the AKT signaling pathway. Sci Rep 2019, 9: 4182.
Guo K, Ma Q, Wang L, Hu H, Li J, Zhang D, et al. Norepinephrine-induced invasion by pancreatic cancer cells is inhibited by propranolol. Oncol Rep 2009, 22: 825–830.
Acknowledgements
This review was supported by a grant from the Tianjin “Project + Team” Key Cultivation Program (XC202034) and a Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-009A). The figures were created with BioRender.com.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ni, B., Yin, Y., Li, Z. et al. Crosstalk Between Peripheral Innervation and Pancreatic Ductal Adenocarcinoma. Neurosci. Bull. 39, 1717–1731 (2023). https://doi.org/10.1007/s12264-023-01082-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-023-01082-1