
Abstract
Understanding the fundamental processes of human brain development and diseases is of great importance for our health. However, existing research models such as non-human primate and mouse models remain limited due to their developmental discrepancies compared with humans. Over the past years, an emerging model, the “brain organoid” integrated from human pluripotent stem cells, has been developed to mimic developmental processes of the human brain and disease-associated phenotypes to some extent, making it possible to better understand the complex structures and functions of the human brain. In this review, we summarize recent advances in brain organoid technologies and their applications in brain development and diseases, including neurodevelopmental, neurodegenerative, psychiatric diseases, and brain tumors. Finally, we also discuss current limitations and the potential of brain organoids.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The development of the nervous system is a strictly orchestrated spatial and temporal process that generates an immense diversity of cell types. Understanding the basic mechanisms of human brain formation and disorder is important because of the widespread health burden of neurological disorders. The human brain differs significantly from model animals like mice in terms of size, surface area, and the complexity of cytoarchitecture [1, 2]. Due to the limited accessibility of live human brains and inconsistent treatment of post-mortem or surgically removed human brain samples, it is difficult to replicate data and apply it to clinical treatment.
Human pluripotent stem cells (hPSCs), including human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs), which possess a unique ability for self-renewal and broad plasticity for differentiation, have emerged as invaluable tools for exploring the human brain. By providing different levels of inhibition of bone morphogenic protein (BMP) and transforming growth factor-β/NODAL signaling, known as “Dual SMAD” inhibition, hPSCs can be induced into neural stem cells and cortical pyramidal neurons, among others [3]. This method obtains highly consistent neural cells in vitro. However, monolayer cultures lack cell-type diversity and spatial complexity, and cannot recapitulate cell-cell interactions and certain important cellular properties, such as cell polarity and guided cell migration. In 2008, in vitro neural differentiation was further improved using more reproducible serum-free methods, known as SFEBq (serum-free, floating culture of embryoid body (EB)-like aggregates) [4]. Aggregates plated on coated dishes would efficiently differentiate into multiple small rosettes of neural precursors surrounding and growing around apical lumens. Afterward, aggregates embedded in Matrigel formed an organized architecture, referred to as a brain organoid, mimicking the cellular and structural complexity of the prenatal developing brain and the radial migration of later-born neurons from the ventricular zone in the center of the brain out to the superficial layers [5]. The advent of brain organoid technology has brought a new era of human brain research. Brain organoids have been used to model human-specific developmental processes and recapitulate disease-specific pathologies associated with neurodevelopmental, neuropsychiatric, and neurodegenerative disorders [2, 6,7,8]. In this review, we discuss the development of brain organoid culture systems and their applications to various neurological disorders, including neurodevelopmental disorders, neurodegenerative diseases, major psychiatric diseases, and brain tumors.
Development of the Brain Organoid System
Human brain development begins around the third week of gestation, when the neural tube forms, and differentiation occurs along the anterior-posterior axis. By the sixth gestational week, the difference in the rates of proliferation of cells in rostral regions of the neural tube results in the formation of three brain vesicles, the prospective forebrain, midbrain, and hindbrain [9]. These basic structures provide the basis for the progressive development of regionally more defined brain regions such as the cerebral cortex and thalamus (forebrain), parts of the brainstem (midbrain), and the pons and cerebellum (hindbrain) [9]. To reconstruct the developmental processes of the human brain in vitro, researchers have established various culture systems for the whole brain and specialized regions of the mature central nervous system, such as the forebrain organoid, midbrain organoid, and hindbrain/spinal cord organoid [10].
The method of forming the whole brain is referred to as the unguided method (Fig. 1). This protocol was developed by Lancaster et al [5], who embedded EBs in Matrigel and cultured them in a neural induction medium without the use of patterning growth factors, focusing instead on improving growth conditions and providing the environment necessary for intrinsic cues to help self-organization. Taking advantage of this method, embryonic stem cells or human induced pluripotent stem cells (hiPSCs) can self-assemble to form cerebral organoids, including various discrete, but interdependent brain regions (Fig. 1). This can be used to study the interactions of multi-regional brain areas, but is less subject to external control and more stochastic [5, 11]. Other techniques for developing specialized brain regions are known as guided approaches, which involve the addition of small molecules to organoids to steer them in a certain direction [11]. To promote specific neural fates that generate organoids with varied identities from the forebrain to the midbrain to the hindbrain, defined developmental patterning cues are used, among which some have previously been effectively used in 2D differentiation procedures [3]. Dorsal forebrain organoids can be developed after continuous “Dual SMAD” inhibitor induction, and a sonic hedgehog antagonist is added after the SMAD inhibitor to induce ventral forebrain [12,13,14,15]. Functional hippocampal granule- and pyramidal-like neurons have been generated via long-term dissociation culture of the self-organizing dorsomedial telencephalic tissues derived from hESCs treated with Wnt agonist and BMP ligand, under optimized culture and treatment conditions [16]. After receiving initial dual SMAD inhibitors, EBs have been preprogrammed to have a neuroectodermal fate. Insulin and MAPK/ERK inhibitors were then used to prevent over-causation to a midbrain fate, and the addition of BMP7 guided toward thalamus tissue development [17]. The forebrain organoids are similar to the human cerebral cortex in the aspects of cell types and structures, containing several ventricular structures, each with a defined ventricular zone enriched for FOXG1+ forebrain progenitors and also producing astrocytes at a later stage of development [13]. Many cortical organoid techniques have poor lamination and layer distinction, but some have demonstrated the sequential production of neurons expressing layer-specific markers in the right order, including RELN, TBR1, CTIP2, and SATB2 [5]. The ability of brain organoids to mimic fetal brain development in utero has been tested by transcriptome sequencing, single-cell sequencing, and epitranscriptome analysis of multi-period organoids [18, 19]. Several studies have examined the physiological characteristics of neurons produced using organoid methods and demonstrated how their functional development proceeds through time [5, 20, 21]. Electrophysiological recordings and Ca2+ surges have shown that neurons produced in brain organoids functionally mature gradually and fire spontaneously [5, 13]. The frequency of this firing is sensitive to the application of glutamate and glutamate receptor antagonists, indicating the presence of glutamatergic neurons [13].

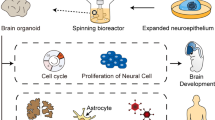
Advances in brain organoid methods. Human stem cell/pluripotent stem cells can be differentiated in self-organizing 3D cultures to derive unguided neural organoids (cerebral organoids) or brain region-specific organoids resembling various regions of the nervous system. Brain region-specific organoids can be combined to generate assemblies to model complex cell-cell interactions and neural circuit formation in the human nervous system. Brain organoids also can be fused with non-neuronal cellular components such as vessels and microglia or transplanted into animals to vascularize brain organoids. The figure was created with BioRender.com.
Although spinning bioreactors have been widely used to help increase nutrient and oxygen diffusion within organoids by agitating and better circulating the medium, the small contact surface of the organoid with the culture medium has the disadvantage that the inside cells die at a later stage of culture. Different techniques such as sectioning, adding vessel-like structures, and orthotopic xenotransplantation, have been used to extend the culture time of the organoid [21,22,23,24,25] (Fig. 1). Organoids cultured at the air-liquid interface were able to show a great improvement in the survival and maturation of neurons [21, 22]. Some studies co-cultured hiPSC-derived endothelial cells or umbilical vein endothelial cells with brain organoids and reported robust engraftment with the formation of capillary-like structures [23,24,25]. Recently, we generated a human brain vessel organoid, which possesses multiple cell types including pericytes, endothelial cells, and microglia, and has a vascular lumen structure [25]. By encapsulating them with brain organoids, the fused organoids not only provided a variety of cell types but also reduced the apoptosis, and increased the neural progenitor proliferation and cortical thickness [25]. An alternate strategy engineered hESCs to ectopically express human ETV2 to create endothelial cells within cortical organoids, resulting in the appearance of vascular network-like structures along with enhanced neuron maturity [26]. In addition, some studies have transplanted organoids into rodent brains so that the implanted organoids can integrate into the host brain circuits to extend the cultural life of organoids with the production of more mature functional neurons [27,28,29,30,31] (Fig. 1).
Organoids that are patterned to particular brain regions contain more homogeneous populations of progenitor cells and neurons, which minimizes inter-organoid variation. However, this enhancement also reduces the possibility of investigating the interactions between various brain regions. To overcome this limitation, several studies have created assembled organoids, named assemblies. which are formed by fusing organoids from different regions. Ventral forebrain-like organoids containing gamma-aminobutyric acid (GABA)-ergic cortical interneurons can be combined with dorsal forebrain-like organoids in order to capture the cell-cell interactions in vitro [32, 33] (Fig. 1). The construction of a thalamus-cortex assembly has modeled the connection loops between the cortex and the thalamus [17]. Recently, Jimena et al. have created a cortico-spinal-muscle assembly to model multi-organ connections with functional regulatory effects [34]. In this system, glutamate uncaging or optogenetic stimulation of cortical spheroids can trigger strong muscle contraction, and these assemblies are morphologically and functionally intact for up to 10 weeks post-fusion [34].
The nervous system contains not only cells of neuroectodermal origin but also microglia from the yolk sac and vascular cells from the mesoderm. Microglia are the brain’s resident immune cells that play crucial roles in regulating neuronal circuits, preserving homeostasis, and monitoring the surrounding area [35]. Due to the diverse germ layer origins, it has been challenging to generate brain organoids containing blood vessels and microglia. Some studies have tried to add in vitro cultured microglia-like cells to brain organoids [36,37,38]. Recently, we developed a new strategy for vascularizing brain organoids [25]. First, brain vessel organoids were generated by sequential mesodermal and endothelial cell induction and then fused with brain organoids at an early stage. The fused organoids formed brain-blood barrier (BBB)-like structures and contained extensive amounts of microglia, which responded to immune stimuli and engulfed synapses [25]. The current induction of blood vessels and microglia in vitro does not fully mimic the in vivo situation, and more work is needed to further optimize the induction conditions of the organoid to make its development more stable and mature.
Neurodevelopmental Disorders
Most neurodevelopmental disorders occur with complex conditions in childhood and remain incurable and irreversible [39, 40]. Limited research models and poor diagnostic conditions and standards render neurodevelopmental disorder studies at a bottleneck stage. The advent of brain organoid models offers an opportunity to uncover unknowns and develop new intervention strategies [41].
Autism spectrum disorder (ASD) is considered to result from overall brain developmental defects, especially at the synapse level, and genetic and environmental factors can both lead to the pathologies of ASD [42] (Fig. 2). As a genetically heterogeneous syndrome, ASD is found in patients with fragile X syndrome, tuberous sclerosis, Joubert syndrome, and Rett syndrome, among others. Besides, genomic copy-number variants are also closely associated with ASD such as deletions and duplications of chromosome 16p11.2 and duplications of maternal 15q11-q13 [43].

Applications of brain organoids. A Neurodevelopmental disorders. Brain organoids have been used to study neurodevelopmental disorders such as autism spectrum disorder, microcephaly, and Rett syndrome. B Neurodegenerative disorders. iPSC-induced or CRISPR-Cas9 gene-edited brain organoids have been successfully established for studying aging-dependent Alzheimer’s disease (AD), Parkinson’s disease (PD), and Huntington’s disease (HD). C Psychiatric disorders. Mental diseases mainly include depressive disorder, schizophrenia, and bipolar disorder. D Brain tumors. They also provide a unique opportunity to model brain tumors such as glioblastoma, medulloblastoma, and meningioma. The figure was created with BioRender.com.
The induced neural cells from hiPSCs of ASD patients in a 2D culture system have been used to study the molecular and cellular mechanisms of ASD. For example, hiPSC-derived neuronal progenitor cells (NPCs) from ASD patients with megalencephaly have been found to have a greater ability of proliferation regulated by β-catenin/BRN2 transcriptional activity [44]. Furthermore, ASD-derived neurons display decreased synaptogenesis and defects in neuronal networks [44]. Co-culture of ASD-derived astrocytes and neurons has shown that astrocytes play a negative role in neuronal morphology and synaptogenesis via the production of reactive oxygen species and the cytokine interleukin-6 [45]. These 2D culture models provide some clues about the relationships between neuronal cell types in ASD. Nevertheless, as a heterogeneous disorder, limited cell types and a lack of brain environment restrict the usage of 2D culture systems. The emergence of organoid models brings a new opportunity for deeper mechanistic understanding.
The hiPSC-derived telencephalic organoid model has been developed to study severe idiopathic ASD [46]. Using this model, it has been revealed that the transcription factor FOXG1 dysregulates the proliferation and differentiation of GABAergic inhibitory neurons, and the imbalance of GABA/glutamate neuronal fates can be reversed by the knockdown intervention of FOXG1 [47]. The single-nucleotide mutation of the CDH8 gene has been associated with ASD [43]. The cerebral organoids derived from CDH8+/− iPSCs exhibit large-scale overlapped differentially-expressed genes (DEGs) with the transcriptome of idiopathic ASD organoids [48]. Non-coding RNA DLX6-AS1 is dramatically upregulated in these ASD organoids and essential for GABAergic interneuron differentiation [48]. ASD forebrain organoids have also revealed the heterochronicity of developmental gene networks, which are associated with morphological growth acceleration [49]. The cortical organoid with haploinsufficiency of three ASD risk genes SUV420H1, ARID1B, and CHD8 displays phenotypic convergence with the asynchronous development of GABAergic neurons and deep-layer excitatory projection neurons through distinct molecular pathways, which leads to abnormal circuit activity [50].
Microcephaly is a kind of neurodevelopmental disorder with a smaller brain size in patients [51] (Fig. 2). Several genes have been shown to be linked with microcephaly, such as MCPH1, WDR62, CDK5RAP2, CEP152, and ASPM [52,53,54]. Knockdown of the CDK5RAP2 gene in cerebral organoids causes premature neural differentiation and leads to a marked reduction in organoid size, and over-expression of CDK5RAP2 can rescue these defects. Mutation of Centrosomal-P4.1-associated protein (CPAP) leads to Seckel syndrome with microcephaly [55]. Cerebral organoids with the natural CPAP microcephaly mutation show smaller size caused by depletion of the cortical neural progenitor radial glial cells (RGCs), and early neuronal differentiation, probably via the action of the cilium disassembly complex [55]. Asparaginyl-tRNA synthetase1 (NARS1) has also been reported to be a risk gene associated with microcephaly [56]. Cortical organoids induced by patients with the NARS1 mutation exhibit reduced proliferation of RGCs, impaired differentiation, and smaller sizes, indicating that NARS1 is important for normal human brain development [56]. By using brain organoid models, several studies have investigated the effects of Zika virus (ZIKV) infection, which is associated with the occurrence of microcephaly in newborns, on brain development and neural precursor cell proliferation [20, 57]. Human neural progenitors, neurospheres, and brain organoids infected with ZIKV exhibit reduced growth and increased cell death [58]. The immune receptor Toll-like-receptor 3 and downstream pathways are activated by ZIKV, thus affecting neurogenesis and resulting in apoptosis [59]. Taken together, brain organoid models have faithfully mimicked brain developmental phenotypes and offer a platform for exploring the molecular basis of microcephaly.
Rett Syndrome (RTT) is another type of neurodevelopmental disorder caused by the mutation of X-linked methyl-CpG binding protein 2 (MECP2), leading to mental retardation primarily in females [60] (Fig. 2). Before the appearance of brain organoid models, it was difficult to study the dynamic molecular and cellular processes due to limited patient samples and unreliable mouse models. Benefiting from 3D brain organoids, more molecular features of RTT have been revealed [61]. For example, MECP2-deficient or mutant cerebral organoids show defects in early neurogenesis and increased expression of miR-199 and miR-214 [61]. Interestingly, the defects in neural development can be rescued by the down-regulation of these miRNAs. Another study showed that MECP2 mutant human interneurons (INs) are abnormal and identified an epigenetic reader BRD4 as a trigger of IN dysfunction [62]. Thus, brain organoids provide an opportunity for the identification of targets for RTT therapy.
Fragile X syndrome (FXS) is an X-linked dominant disorder caused by the low expression of the FMR1 gene due to excessive CGG repeats in its 5′ untranslated region [63, 64]. The encoded protein FMRP inhibits the translation of specific mRNAs and thus decreased FMRP protein incurs an excess of translated products, which further affects neuronal maturation and synaptic plasticity [63, 64]. The 2D culture system has been applied to the study of FXS and found that decreased expression of FMR1 leads to poor neuronal maturation [65, 66]. Brain organoids derived from FMRP-KO iPSCs showed bigger sizes and an increased number of glial cells [67]. In addition, FXS forebrain organoids from patient-derived iPSCs exhibit reduced neural progenitor proliferation, dysregulated differentiation, increased synapse formation, hyperexcitability, and a deficit in the production of GABAergic neurons [68]. Interestingly, pharmacological inhibition of the phosphoinositide 3-kinase pathway rescues the neurodevelopmental and synapse formation defects in FXS forebrain organoids [68].
Down syndrome (DS) is caused by the presence of an additional copy of Homo sapiens chromosome 21 (HSA21), termed trisomy 21 [69, 70]. The prevalence of DS is around 0.125% worldwide [69]. DS patients usually have intellectual disabilities and other defects of bodily systems such as the musculoskeletal and cardiovascular systems [69, 70]. By adding a partial trisomy of Mus musculus chromosome 16 (MMU16) that is orthologous to HSA21 in humans, several DS mouse models have been established [70, 71]. However, these models are artificial and cannot reflect the true DS pathologies because of the discrepancies between humans and mice. Xu et al. [72] have established DS human brain organoids and found overproduction of OLIGO2+ neural progenitors, which leads to excessive GABAergic interneuron production. Another study focused on microglia in DS organoids and found that tau protein triggers microglial interferon (IFN)-I signaling, which causes increased synaptic pruning, and the microglia dysfunction and senescence can be rescued by inhibiting the IFN-I receptor [73]. Interestingly, DS cerebral organoids show increased DSCAM/PAK1 pathway activity and down-regulation or inhibition of this pathway reverses the abnormal neurogenesis in DS organoids [74].
Angelman syndrome (AS) is a rare neurodevelopmental disorder with 0.4% prevalence, caused by the loss of UBE3A protein, an E3 ubiquitin ligase in neurons [75, 76]. Notably, AS human brain organoids exhibit early silencing of paternal UBE3A and abnormal neuronal activity, which are partially rescued by topoisomerase inhibitors [77]. Using organoid systems, it has been revealed that UBE3A suppresses neuronal excitability via ubiquitin-mediated degradation of calcium- and voltage-dependent big potassium (BK) channels, providing mechanistic insights into AS occurrence [78].
Besides, brain organoids have also been used to model other neural developmental disorders, such as macrocephaly [79, 80], neuronal heterotopia [81, 82], tuberous sclerosis [83, 84], and Timothy syndrome [85]. These studies have revealed the molecular basis of these syndromic disorders, leading to the identification of intervention targets or the development of strategies to restore deficits in the context of disease.
Neurodegenerative Disorders
Neurodegenerative diseases (NDDs) are major threats to human health caused by the progressive death of selectively vulnerable populations of neurons and the loss of normal brain functions [86]. Major types of NDD include Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), spinal muscular atrophy, Batten disease, multiple sclerosis, and Huntington’s disease (HD) (Fig. 2). Aging, genetics, protein misfolding, and programmed cell death are the main causal factors of NDDs [87,88,89]. Patients with NDDs have various clinical manifestations including memory loss, cognitive dysfunction, and abnormalities in behavior, language, and respiration, which severely affect the normal life of patients and endanger their safety [90,91,92]. NDDs are in urgent need of effective treatment programs and drugs, which require a deep understanding of the pathogenesis.
Various animal models have been used to investigate the pathogenesis of NDDs [93,94,95,96]. However, due to the large inherent differences between human and animal models, the value of the results based on model systems is suboptimal. Because of the rarity and difficulty in obtaining human tissues, some studies have used stem cell induction technology in vitro to induce neurons and glial cells for the mechanistic investigation of NDDs [97, 98]. However, it is difficult to simulate the complex environment inside the real human brain for the induced single cell types, so some studies are beginning to use organoid technology to study related NDDs by establishing organoids representing different brain regions, such as the whole brain, forebrain, midbrain, striatum, and sensorimotor cortex, derived from patient iPSCs [99,100,101,102,103,104].
AD is the most common neurodegenerative disease [90], with pathological features mainly including amyloid plaques formed by the accumulation of extracellular amyloid-beta (Aβ) and intracellular neurofibrillary tangles formed by the accumulation of phosphorylated tau protein [105]. Brain organoids can be induced from hiPSCs from patients with familial AD (APP, PSEN1, and PSEN2 mutations) or patients with sporadic AD, leading to the identification of modulators of the tau interactome, reproduction of AD pathology, and findings of cell fate changes in AD organoids [99,100,101]. Apolipoprotein E4 (APOE4) is the strongest genetic risk factor associated with late-onset AD among the three polymorphic alleles (APOE2, APOE3, and APOE4), and has recently been proposed to impair myelination via cholesterol dysregulation in oligodendrocytes [106, 107]. Interestingly, APOE4 organoids exhibit more severe synaptic loss and neurodegeneration phenotypes [108]. Cerebral organoids increase Aβ and p-Tau by inducing beta-secretase 1 and glycogen synthase kinase-3 alpha/beta levels after being exposed to serum from AD patients [109]. By integrating mathematical modeling and the pathological features of AD in iPSC-derived cerebral organoids, a high-content screening platform has been established for drug screening and testing [110].
PD is the most common movement disorder; it is characterized pathologically by the accumulation of Lewy Bodies consisting of insoluble aggregates of α-synuclein and reduced dopamine levels due to degeneration of the substantia nigra [91, 111]. Previous studies have identified a number of PD risk genes including SNCA, PARK2, PINK2, and LRRK2 [112]. In 2D cell culture, inducing hiPSCs into neurons and astrocytes, the degeneration of PD neurons was found to be associated with the accumulation of toxic α-synuclein in astrocytes [113, 114]. In cultured midbrain organoids from PD patients with the LRRK2-G2019S mutation, high-content imaging data has shown decreased dopaminergic differentiation, altered mitochondrial morphology, and increased cell death compared to the organoids from isogenic lines [102]. PD organoids can also be established by CRISPR-Cas9-mediated gene editing in human embryonic stem cells. After introducing the DNAJC6 mutation in human midbrain-like organoids, PD pathologic features such as midbrain-type dopamine neuron degeneration, pathological α-synuclein aggregation, an increase in intrinsic neuronal firing frequency, and mitochondrial and lysosomal dysfunctions were detected [115].
ALS is a neurodegenerative disease specifically affecting motor neurons; it typically results in progressive muscle atrophy and usually death from respiratory failure [92, 116]. Cultured motor neurons from ALS patients show degeneration, abnormal protein aggregation, and increased cell death [117, 118]. Given the unfeasibility of obtaining presymptomatic samples, the development of brain organoids may help elucidate initial molecular events. By using a combination of single-cell RNA sequencing and biological assays, it has been revealed that the cortical organoids from patients with ALS overlapping with frontotemporal dementia harboring the C9ORF72 hexanucleotide repeat expansion mutation exhibit distinct transcriptional, proteostasis, and DNA repair disturbances in astroglia and neurons [119]. Apart from the traditional method of culturing brain organoids, ALS can be modeled by sensorimotor organoids, which contain sensory neurons, astrocytes, microglia, and vasculature, and include functional neuromuscular junctions (NMJs), and this ALS organoid displays impaired NMJs [103].
HD is the most frequent autosomal-dominant neurodegenerative disease; it is caused by somatic expansion of CAG repeats in the Huntington (HTT) gene, which results in neurodegeneration in the striatum and cortex [120, 121]. Patients with HD show motor, cognitive, and mental abnormalities in mid-life [122]. Fetal brains of HD patients and mutant mouse models display mislocalization of mutant huntingtin and junctional complex proteins, defects in neural progenitor cell polarity and differentiation, abnormal ciliogenesis, and changes in mitosis and cell-cycle progression [123]. By comparing HD organoids with controls at the transcriptome level, HD organoids had a more immature transcription profile as well as disrupted cortical cytoarchitecture, indicating a possible connection between mutant huntingtin and abnormal neural development [124]. Another study found that heat shock transcription factor 1 (HSF1) accumulates in the mitochondria of HD cell models, a mouse model, and human striatal organoids derived from induced HD iPSCs, and suppressing the mitochondrial localization of HSF1 by interfering with its binding to dynamin-related protein 1 can rescue the pathological HD changes in striatal organoids [104]. Taken together, organoids have become a powerful model in which to explore the pathogenesis and develop potential treatments for NDDs.
To date, there is no satisfactory treatment for NDDs. Many drugs are effective in laboratory animals but do not achieve the desired therapeutic effect in humans, most likely due to inherent differences between humans and other species. Thus, human brain organoids can be used as a compensatory approach for target identification and drug development. Previous studies have built a complete experimental system for drug screening for cancer using organoid technology [125,126,127], but the application of organoids in NDDs is more challenging because of the complexity of the origin of neuronal cell types, cell-type specific pathogenesis, and complicated cell-cell interactions, as well as the involvement of environmental and immune factors. Nevertheless, brain organoids resemble some key features of NDDs and thus can be used to test the effects of potential modulators. For example, treatment with the LRRK2 inhibitor 2 has shown some rescue effects on LRRK2-G2019S-dependent dopaminergic phenotypes in PD organoids [102]. The BBB is another element that should be considered because most NDDs at late stages are accompanied by disruption of the BBB, which fails to prevent the entrance of toxic substances from the circulatory system into the brain [25]. However, the current brain organoid models lack BBB structure, which limits their applications.
Psychiatric Disorders
Because the clinical manifestations of psychiatric disease are very subjective, most being diagnosed by doctors through oral communications with patients, and objectively quantifiable phenotypes [128]. Thus, simulating mental diseases using animal models is extremely challenging. Owing to the research on the family pedigrees of patients with psychiatric diseases and the development of gene technology, many risk genes for psychiatric diseases have been identified, and various experimental models based on these genes have been established [128].
Mental diseases mainly include depressive disorder, schizophrenia, and bipolar disorder (Fig. 2). Major depression is one of the most common mental diseases; patients often present with loss of interest or pleasure, insomnia or hypersomnia, and mental disorders [129, 130]. However, the current understanding of the mechanism of depressive disorder is still incomplete, and some symptoms and etiology have great heterogeneity. In postmortem brain scans of people with depressive disorder, it has been found that GABA receptor-mediated inhibition is dysregulated in depressed individuals with a history of suicidal behavior, but the molecular mechanism underlying this abnormality is not clear [131, 132]. Recent studies have used hiPSCs from patients with a depressive disorder to induce the ventral forebrain organoids and GABAergic neurons in vitro [133]. Through transcriptome sequencing and single-cell technology, it has been found that the decreased expression of serotonergic receptor 2C in neurons under the condition of depressive disorder may lead to defective neuronal activity, and targeting the 5-HT2C receptor by adding small molecule agonists and genetic methods can effectively restore neural activity [133].
Schizophrenia is a chronic brain disease that occurs mostly in early adulthood. It is polygenic and is hypothesized to be a neurodevelopmental disorder with an as-yet-unknown molecular origin [134,135,136]. Disordered neurogenesis, impaired synaptic transmission, and dysfunction of mitochondria have been identified in neurons derived from hiPSCs from patients with schizophrenia [137,138,139,140,141]. The astrocytes induced by schizophrenic hiPSCs show DEGs related to inflammation and synaptic function, and transplanting schizophrenic astrocytes to mouse brains results in behavioral changes in cognitive and olfactory functions [142]. The reduction in synaptic density in schizophrenic patients is caused by the excessive synaptic pruning by microglia [143]. In an in vitro model of microglia-mediated synapse engulfment, schizophrenic microglia increase synapse elimination [144]. Recent studies have used hiPSCs from patients with schizophrenia to construct whole-brain organoids [145, 146]. Using transcriptome sequencing analysis, the authors found that the genes related to mitochondrial function showed marked differences [145]. By applying the Seahorse Mito Stress test of organoids, it has been found that the level of oxygen consumption of schizophrenic organoids decreases significantly [145]. These organoids also show neuronal dysfunction as reflected by weakened responses to electrical stimulation and KCl depolarization measured with a microelectrode array [145]. By analyzing the single-cell RNA sequence profile of schizophrenic organoids, decreased progenitor survival and disrupted neurogenesis were detected [147]. Transcription factor BRN2 and growth factor PTN have been identified as mechanistic substrates of neurogenesis and cellular survival, respectively, in schizophrenic organoids [147].
Bipolar patients experience recurrent episodes of depression and mania that affect perception, emotion, thought, and social behavior [148]. Due to the complexity of the symptoms, the diagnosis and treatment of bipolar cognitive disorder are still subjective. In cultured neurons derived from bipolar hiPSCs, mitochondrial abnormalities, hyperexcitability, anomalous calcium signaling, impaired neural differentiation, and decreased proliferation have been reported [149,150,151]. By transcriptome analysis of brain organoids derived from the hiPSCs of bipolar patients, it has been found that the expression of genes related to cell adhesion, neurodevelopment, and synaptic regulation is decreased, while the expression of genes related to immune signaling is increased in bipolar organoids [152]. In another study, bipolar organoids showed specific deficits in response to stimulation and depolarization, as reflected by neuronal activity measured by microelectrode arrays, and enrichment of endoplasmic reticulum pathways as analyzed by Gene Ontology analysis for DEGs [152]. In conclusion, organoids provide an in vitro model in which to study psychiatric disorders and help explore the causes and develop treatment strategies.
Brain Tumors
Brain tumors are a type of neoplasm that arises from brain tissue or systemic cancer metastases, classified as malignant and benign tumors [153, 154]. Malignant brain tumors cause high morbidity and mortality and thus are considered one of the most devastating neoplasms due to the complexity of cell types and structures in the human brain. In 2021, the World Health Organization published the fifth edition of the classification of brain tumors, which recapitulated their characteristics and linked the molecular mechanisms of a set of brain tumors including gliomas, glioneuronal tumors, and neuronal tumors [155].
For a better understanding of brain tumors and the associated therapies, genetically engineered mouse models (GEMMs) have been established and used for mimicking the pathological phenotypes and elucidating the innate mechanisms of brain tumors [156,157,158]. However, GEMMs cannot fully simulate the phenotypes because of the species differences. Patient-derived xenografts (PDXs) are a model that transplants dissected tumor tissue from cancer patients into immunodeficient mice to mimic tumor growth in vivo [159, 160], and have been used for anti-tumor drug screening [161]. Nevertheless, PDXs lack the procedures for tumor origination and formation, which limits their application. Recently, the production of organoids has enabled further advances in brain tumor research [162]. Brain tumor organoids are classified by the origin of tumor cells such as genetically-engineered stem cells or dissected tumor tissues or cancer stem cells. For example, Bian et al. [163] established a neoplastic cerebral organoid from hESCs using genome-editing technologies to introduce mutation of tumor-suppressor genes combined with Sleeping Beauty transposon gene insertion. Tumor overgrowth occurs in induced cerebral organoids and mimics brain tumor formation. The MYC gene, a proto-oncogene, is over-expressed in cerebral organoids; it is exhibited in brain tumors such as glioblastoma (GBM), central nervous system primitive neuroectodermal tumor, atypical teratoid/rhabdoid tumor, and medulloblastoma. Another human cerebral organoid modeling GBM has been developed by targeting an HRasG12V-IRES-tdTomato sequence into the TP53 locus using the CRISPR-Cas9 system [164], which exhibits an increased fraction of tumor cells, accompanied by the expression of the GBM stem cell markers OLIG2, GFAP, and SOX2, and the proliferation marker KI67, indicating the GBM identity. Transplantation of brain tumor organoids into immunodeficient mice shows the invasiveness of the brain tumor and higher mortality of mice [164].
To overcome the limitation of the lack of a “normal” human brain microenvironment, Linkous et al. [165] have established a cerebral organoid glioma (GLICO) model system in which they introduced patient-derived glioma stem cells (GSCs) into hESC-derived cerebral organoids. In the GLICO system, GSCs invade, proliferate, and form tumors within the host organoids, and these processes faithfully phenocopy patient GBMs. Furthermore, the sensitivity to chemotherapeutic agents and ionizing radiation of GLICO tumors also confirmed it as a suitable model resembling in vivo tumors compared with 2D cultured tumor cells. Considering the cellular and genetic heterogeneity in inter- and intra-GBM samples, a comprehensive study has established patient-derived glioblastoma organoids (GBOs) and transplanted them into immunodeficient mouse brains [166]. These GBOs have been used experimentally for personalized drug testing, in particular T cell immunotherapy. This offers a possible approach to the development of patient-specific treatment strategies. By combining single-cell transcriptomics and live imaging of primary tumor resections, multiple GSC subtypes have been identified, leading to the finding of an invasive population similar to outer radial glia (oRG), a type of cortical neural progenitor which is believed to contribute to cortical expansion and folding [167]. Transplanting GFP-labelled oRG-like tumor cells into hPSC-derived cortical organoids confirmed the tumorigenic and invasive properties of this tumor cell population [167]. Thus, the analysis of GSC heterogeneity may provide an optimized strategy for an individual patient.
Brain tumor organoids have been applied to studies of GBM, medulloblastoma (MB), and meningioma [168] (Fig. 2). MB is the most common malignant brain tumor in the cerebellum that occurs mostly in childhood [169]. Transcriptional profiling has revealed four main subgroups of MB, and group 3 is characterized by c-MYC upregulation and has the worst outcome [170]. Based on the previous protocol of cerebellar organoid generation [171], a group 3 MB organoid model has been developed by overexpression of the Otx2 and c-MYC genes, which promote tumor cell over-proliferation [172]. Using the MB organoids, the authors found that up-regulation of the on-co-suppressor SMARC4 or treatment with Tazemetostat, an EZH2-specific inhibitor, reduces Otx2/c-MYC-induced tumorigenesis [172]. In another study, overexpression of the MB driver genes MYC and Gfi1 in human cerebellar organoids induces group 3 MB with an epigenetic profile similar to human patients in vivo [173]. Moreover, activation of the Notch1 pathway fosters group 3 MB formation [173]. Meningiomas are the most common intracranial tumors, but the molecular drivers are poorly understood and effective treatments are lacking because of the shortage of research models. Recently, several studies have established meningioma organoid models from patient-derived tumor cells or tissues [174,175,176]. The histological features and innate molecular profiles of meningioma organoids are similar to corresponding parental tumors, and this advantage has allowed the model to be applied to the identification of potential targets for meningioma therapy [174].
Outlook
Improvements in organoid technology in recent years have contributed greatly to our understanding of the mechanisms of human brain development and the pathogenesis of neurological diseases. However, in vitro culture conditions limit the size of the organoid, neuronal maturation, and subsequent production of more complete cell types, such as astrocytes and oligodendrocytes. Furthermore, the lack of blood vessels and immune cells also limits the applications of brain organoids. Although some advances have been made, the innermost parts of an organoid eventually die due to the lack of oxygen and nutrients. Several studies have attempted to establish assembled organoids to resemble inter-regional interactions in the brain and brain periphery interactions, but they only partially mimic the counterparts of the real human body. Nevertheless, partial simulation has already demonstrated broad prospects in disease modeling and the clarification of mechanisms, as well as drug screening or testing. Transplantation of specific types of brain organoids into injured or degenerative regions provides another avenue for the repair of the related neural circuits under disease contexts. A combination of multidisciplinary strategies would help optimize the brain organoid system and broaden its applications.
References
Rakic P. Evolution of the neocortex: A perspective from developmental biology. Nat Rev Neurosci 2009, 10: 724–735.
Giandomenico SL, Lancaster MA. Probing human brain evolution and development in organoids. Curr Opin Cell Biol 2017, 44: 36–43.
Mertens J, Marchetto MC, Bardy C, Gage FH. Evaluating cell reprogramming, differentiation and conversion technologies in neuroscience. Nat Rev Neurosci 2016, 17: 424–437.
Eiraku M, Watanabe K, Matsuo-Takasaki M, Kawada M, Yonemura S, Matsumura M. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 2008, 3: 519–532.
Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, et al. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501: 373–379.
Benito-Kwiecinski S, Lancaster MA. Brain organoids: Human neurodevelopment in a dish. Cold Spring Harb Perspect Biol 2020, 12: a035709.
Clevers H. Modeling development and disease with organoids. Cell 2016, 165: 1586–1597.
Luo X, Liu Y, Dang D, Hu T, Hou Y, Meng X, et al. 3D Genome of macaque fetal brain reveals evolutionary innovations during primate corticogenesis. Cell 2021, 184: 723-740.e21.
Stiles J, Jernigan TL. The basics of brain development. Neuropsychol Rev 2010, 20: 327–348.
Mansour AA, Schafer ST, Gage FH. Cellular complexity in brain organoids: Current progress and unsolved issues. Semin Cell Dev Biol 2021, 111: 32–39.
Jacob F, Schnoll JG, Song H, Ming GL. Building the brain from scratch: Engineering region-specific brain organoids from human stem cells to study neural development and disease. Curr Top Dev Biol 2021, 142: 477–530.
Kadoshima T, Sakaguchi H, Nakano T, Soen M, Ando S, Eiraku M, et al. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc Natl Acad Sci U S A 2013, 110: 20284–20289.
Paşca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods 2015, 12: 671–678.
Birey F, Andersen J, Makinson CD, Islam S, Wei W, Huber N, et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545: 54–59.
Xiang Y, Tanaka Y, Patterson B, Kang YJ, Govindaiah G, Roselaar N, et al. Fusion of regionally specified hPSC-derived organoids models human brain development and interneuron migration. Cell Stem Cell 2017, 21: 383-398.e7.
Sakaguchi H, Kadoshima T, Soen M, Narii N, Ishida Y, Ohgushi M, et al. Generation of functional hippocampal neurons from self-organizing human embryonic stem cell-derived dorsomedial telencephalic tissue. Nat Commun 2015, 6: 8896.
Xiang Y, Tanaka Y, Cakir B, Patterson B, Kim KY, Sun P, et al. hESC-derived thalamic organoids form reciprocal projections when fused with cortical organoids. Cell Stem Cell 2019, 24: 487-497.e7.
Kanton S, Boyle MJ, He Z, Santel M, Weigert A, Sanchís-Calleja F, et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 2019, 574: 418–422.
Uzquiano A, Kedaigle AJ, Pigoni M, Paulsen B, Adiconis X, Kim K, et al. Proper acquisition of cell class identity in organoids allows definition of fate specification programs of the human cerebral cortex. Cell 2022, 185: 3770-3788.e27.
Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, et al. Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 2016, 165: 1238–1254.
Giandomenico SL, Mierau SB, Gibbons GM, Wenger LMD, Masullo L, Sit T, et al. Cerebral organoids at the air-liquid interface generate diverse nerve tracts with functional output. Nat Neurosci 2019, 22: 669–679.
Qian X, Su Y, Adam CD, Deutschmann AU, Pather SR, Goldberg EM, et al. Sliced human cortical organoids for modeling distinct cortical layer formation. Cell Stem Cell 2020, 26: 766–781.
Pham MT, Pollock KM, Rose MD, Cary WA, Stewart HR, Zhou P, et al. Generation of human vascularized brain organoids. Neuroreport 2018, 29: 588–593.
Shi Y, Sun L, Wang M, Liu J, Zhong S, Li R, et al. Vascularized human cortical organoids (vOrganoids) model cortical development in vivo. PLoS Biol 2020, 18: e3000705.
Sun XY, Ju XC, Li Y, Zeng PM, Wu J, Zhou YY, et al. Generation of vascularized brain organoids to study neurovascular interactions. Elife 2022, 11: e76707.
Cakir B, Xiang Y, Tanaka Y, Kural MH, Parent M, Kang YJ, et al. Engineering of human brain organoids with a functional vascular-like system. Nat Methods 2019, 16: 1169–1175.
Mansour AA, Gonçalves JT, Bloyd CW, Li H, Fernandes S, Quang D, et al. An in vivo model of functional and vascularized human brain organoids. Nat Biotechnol 2018, 36: 432–441.
Lancaster MA. Brain organoids get vascularized. Nat Biotechnol 2018, 36: 407–408.
Dong X, Xu SB, Chen X, Tao M, Tang XY, Fang KH, et al. Human cerebral organoids establish subcortical projections in the mouse brain after transplantation. Mol Psychiatry 2021, 26: 2964–2976.
Revah O, Gore F, Kelley KW, Andersen J, Sakai N, Chen X, et al. Maturation and circuit integration of transplanted human cortical organoids. Nature 2022, 610: 319–326.
Jgamadze D, Lim JT, Zhang Z, Harary PM, Germi J, Mensah-Brown K, et al. Structural and functional integration of human forebrain organoids with the injured adult rat visual system. Cell Stem Cell 2023, 30: 137-152.e7.
Bagley JA, Reumann D, Bian S, Levi-Strauss J, Knoblich JA. Fused cerebral organoids model interactions between brain regions. Nat Methods 2017, 14: 743–751.
Paşca SP. Assembling human brain organoids. Science 2019, 363: 126–127.
Lee JH, Shin H, Shaker MR, Kim HJ, Park SH, Kim JH, et al. Production of human spinal-cord organoids recapitulating neural-tube morphogenesis. Nat Biomed Eng 2022, 6: 435–448.
Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med 2017, 23: 1018–1027.
Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, et al. iPSC-derived human microglia-like cells to study neurological diseases. Neuron 2017, 94: 278-293.e9.
Douvaras P, Sun B, Wang M, Kruglikov I, Lallos G, Zimmer M, et al. Directed differentiation of human pluripotent stem cells to microglia. Stem Cell Rep 2017, 8: 1516–1524.
Zhang Y, Cui D. Evolving models and tools for microglial studies in the central nervous system. Neurosci Bull 2021, 37: 1218–1233.
Thapar A, Cooper M, Rutter M. Neurodevelopmental disorders. Lancet. Psychiatry 2017, 4: 339–346.
D’Souza H, Karmiloff-Smith A. Neurodevelopmental disorders. Wiley Interdiscip Rev Cogn Sci 2017, https://doi.org/10.1002/wcs.1398.
Adams JW, Cugola FR, Muotri AR. Brain organoids as tools for modeling human neurodevelopmental disorders. Physiology (Bethesda) 2019, 34: 365–375.
Lord C, Elsabbagh M, Baird G, Veenstra-Vanderweele J. Autism spectrum disorder. Lancet 2018, 392: 508–520.
Geschwind DH. Autism: Many genes, common pathways? Cell 2008, 135: 391–395.
Marchetto MC, Belinson H, Tian Y, Freitas BC, Fu C, Vadodaria K, et al. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry 2017, 22: 820–835.
Russo FB, Freitas BC, Pignatari GC, Fernandes IR, Sebat J, Muotri AR, et al. Modeling the interplay between neurons and astrocytes in autism using human induced pluripotent stem cells. Biol Psychiatry 2018, 83: 569–578.
Yin F, Zhang X, Wang L, Wang Y, Zhu Y, Li Z, et al. HiPSC-derived multi-organoids-on-chip system for safety assessment of antidepressant drugs. Lab Chip 2021, 21: 571–581.
Mariani J, Coppola G, Zhang P, Abyzov A, Provini L, Tomasini L, et al. FOXG1-dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 2015, 162: 375–390.
Wang P, Mokhtari R, Pedrosa E, Kirschenbaum M, Bayrak C, Zheng D, et al. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in cerebral organoids derived from iPS cells. Mol Autism 2017, 8: 11.
Schafer ST, Paquola ACM, Stern S, Gosselin D, Ku M, Pena M, et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat Neurosci 2019, 22: 243–255.
Paulsen B, Velasco S, Kedaigle AJ, Pigoni M, Quadrato G, Deo AJ, et al. Autism genes converge on asynchronous development of shared neuron classes. Nature 2022, 602: 268–273.
Woods CG. Human microcephaly. Curr Opin Neurobiol 2004, 14: 112–117.
Jayaraman D, Bae BI, Walsh CA. The genetics of primary microcephaly. Annu Rev Genomics Hum Genet 2018, 19: 177–200.
Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S, Askham JM, et al. ASPM is a major determinant of cerebral cortical size. Nat Genet 2002, 32: 316–320.
Bond J, Roberts E, Springell K, Lizarraga SB, Scott S, Higgins J, et al. A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet 2005, 37: 353–355.
Gabriel E, Wason A, Ramani A, Gooi LM, Keller P, Pozniakovsky A, et al. CPAP promotes timely cilium disassembly to maintain neural progenitor pool. EMBO J 2016, 35: 803–819.
Wang L, Li Z, Sievert D, Smith DEC, Mendes MI, Chen DY, et al. Loss of NARS1 impairs progenitor proliferation in cortical brain organoids and leads to microcephaly. Nat Commun 2020, 11: 4038.
Garcez PP, Loiola EC, Madeiro da Costa R, Higa LM, Trindade P, Delvecchio R, et al. Zika virus impairs growth in human neurospheres and brain organoids. Science 2016, 352: 816–818.
Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 2016, 18: 587–590.
Dang J, Tiwari SK, Lichinchi G, Qin Y, Patil VS, Eroshkin AM, et al. Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell 2016, 19: 258–265.
Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet 1999, 23: 185–188.
Mellios N, Feldman DA, Sheridan SD, Ip JK, Kwok S, Amoah SK, et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol Psychiatry 2018, 23: 1051–1065.
Xiang Y, Tanaka Y, Patterson B, Hwang SM, Hysolli E, Cakir B, et al. Dysregulation of BRD4 function underlies the functional abnormalities of MeCP2 mutant neurons. Mol Cell 2020, 79: 84-98.e9.
Hagerman RJ, Berry-Kravis E, Hazlett HC, Bailey DB Jr, Moine H, Kooy RF, et al. Fragile X syndrome. Nat Rev Dis Primers 2017, 3: 17065.
Garber KB, Visootsak J, Warren ST. Fragile X syndrome. Eur J Hum Genet 2008, 16: 666–672.
Urbach A, Bar-Nur O, Daley GQ, Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 2010, 6: 407–411.
Lee A, Xu J, Wen Z, Jin P. Across dimensions: Developing 2D and 3D human iPSC-based models of fragile X syndrome. Cells 2022, 11: 1725.
Brighi C, Salaris F, Soloperto A, Cordella F, Ghirga S, de Turris V, et al. Novel fragile X syndrome 2D and 3D brain models based on human isogenic FMRP-KO iPSCs. Cell Death Dis 2021, 12: 498.
Kang Y, Zhou Y, Li Y, Han Y, Xu J, Niu W, et al. A human forebrain organoid model of fragile X syndrome exhibits altered neurogenesis and highlights new treatment strategies. Nat Neurosci 2021, 24: 1377–1391.
Bull MJ. Down syndrome. N Engl J Med 2020, 382: 2344–2352.
Antonarakis SE, Skotko BG, Rafii MS, Strydom A, Pape SE, Bianchi DW, et al. Down syndrome. Nat Rev Dis Primers 2020, 6: 9.
Aziz NM, Guedj F, Pennings JLA, Olmos-Serrano JL, Siegel A, Haydar TF, et al. Lifespan analysis of brain development, gene expression and behavioral phenotypes in the Ts1Cje, Ts65Dn and Dp(16)1/Yey mouse models of Down syndrome. Dis Model Mech 2018, 11: dmm031013.
Xu R, Brawner AT, Li S, Liu JJ, Kim H, Xue H, et al. OLIG2 drives abnormal neurodevelopmental phenotypes in human iPSC-based organoid and chimeric mouse models of down syndrome. Cell Stem Cell 2019, 24: 908-926.e8.
Jin M, Xu R, Wang L, Alam MM, Ma Z, Zhu S, et al. Type-I-interferon signaling drives microglial dysfunction and senescence in human iPSC models of Down syndrome and Alzheimer’s disease. Cell Stem Cell 2022, 29: 1135-1153.e8.
Tang XY, Xu L, Wang J, Hong Y, Wang Y, Zhu Q, et al. DSCAM/PAK1 pathway suppression reverses neurogenesis deficits in iPSC-derived cerebral organoids from patients with Down syndrome. J Clin Invest 2021, 131: e135763.
Margolis SS, Sell GL, Zbinden MA, Bird LM. Angelman syndrome. Neurotherapeutics 2015, 12: 641–650.
Jiang Y, Lev-Lehman E, Bressler J, Tsai TF, Beaudet AL. Genetics of angelman syndrome. Am J Hum Genet 1999, 65: 1–6.
Sen D, Voulgaropoulos A, Drobna Z, Keung AJ. Human cerebral organoids reveal early spatiotemporal dynamics and pharmacological responses of UBE3A. Stem Cell Reports 2020, 15: 845–854.
Sun AX, Yuan Q, Fukuda M, Yu W, Yan H, Lim GGY, et al. Potassium channel dysfunction in human neuronal models of Angelman syndrome. Science 2019, 366: 1486–1492.
Zhang W, Ma L, Yang M, Shao Q, Xu J, Lu Z, et al. Cerebral organoid and mouse models reveal a RAB39b-PI3K-mTOR pathway-dependent dysregulation of cortical development leading to macrocephaly/autism phenotypes. Genes Dev 2020, 34: 580–597.
Dang LT, Vaid S, Lin G, Swaminathan P, Safran J, Loughman A, et al. STRADA-mutant human cortical organoids model megalencephaly and exhibit delayed neuronal differentiation. Dev Neurobiol 2021, 81: 696–709.
Klaus J, Kanton S, Kyrousi C, Ayo-Martin AC, Di Giaimo R, Riesenberg S, et al. Altered neuronal migratory trajectories in human cerebral organoids derived from individuals with neuronal heterotopia. Nat Med 2019, 25: 561–568.
O’Neill AC, Kyrousi C, Klaus J, Leventer RJ, Kirk EP, Fry A, et al. A primate-specific isoform of PLEKHG6 regulates neurogenesis and neuronal migration. Cell Rep 2018, 25: 2729-2741.e6.
Blair JD, Hockemeyer D, Bateup HS. Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat Med 2018, 24: 1568–1578.
Eichmüller OL, Corsini NS, Vértesy Á, Morassut I, Scholl T, Gruber VE, et al. Amplification of human interneuron progenitors promotes brain tumors and neurological defects. Science 2022, 375: eabf5546.
Paşca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Paşca AM, et al. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med 2011, 17: 1657–1662.
Wang J, Hu WW, Jiang Z, Feng MJ. Advances in treatment of neurodegenerative diseases: Perspectives for combination of stem cells with neurotrophic factors. World J Stem Cells 2020, 12: 323–338.
Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol 2019, 15: 565–581.
Soto C, Pritzkow S. Protein misfolding, aggregation, and conformational strains in neurodegenerative diseases. Nat Neurosci 2018, 21: 1332–1340.
Chi H, Chang HY, Sang TK. Neuronal cell death mechanisms in major neurodegenerative diseases. Int J Mol Sci 2018, 19: 3082.
Livingston G, Huntley J, Sommerlad A, Ames D, Ballard C, Banerjee S. Dementia prevention, intervention, and care: 2020 report of the lancet commission. Lancet 2020, 396: 413–446.
Yemula N, Njoku P, Takyi J. The second brain in Parkinson’s disease: Fact or fantasy? Neural Regen Res 2022, 17: 1737–1738.
Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, et al. Amyotrophic lateral sclerosis. Nat Rev Dis Primers 2017, 3: 17085.
Yan Y, Wang X, Chaput D, Shin MK, Koh Y, Gan L, et al. X-linked ubiquitin-specific peptidase 11 increases tauopathy vulnerability in women. Cell 2022, 185: 3913-3930.e19.
Sanz FJ, Solana-Manrique C, Muñoz-Soriano V, Calap-Quintana P, Moltó MD, Paricio N. Identification of potential therapeutic compounds for Parkinson’s disease using Drosophila and human cell models. Free Radic Biol Med 2017, 108: 683–691.
Elovsson G, Bergkvist L, Brorsson AC. Exploring aβ proteotoxicity and therapeutic candidates using Drosophila melanogaster. Int J Mol Sci 2021, 22: 10448.
Wie J, Liu Z, Song H, Tropea TF, Yang L, Wang H, et al. A growth-factor-activated lysosomal K+ channel regulates Parkinson’s pathology. Nature 2021, 591: 431–437.
Kikuchi T, Morizane A, Doi D, Magotani H, Onoe H, Hayashi T, et al. Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 2017, 548: 592–596.
McAlpine CS, Park J, Griciuc A, Kim E, Choi SH, Iwamoto Y, et al. Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature 2021, 595: 701–706.
Choi H, Kim HJ, Yang J, Chae S, Lee W, Chung S, et al. Acetylation changes tau interactome to degrade tau in Alzheimer’s disease animal and organoid models. Aging Cell 2020, 19: e13081.
Fan W, Sun Y, Shi Z, Wang H, Deng J. Mouse induced pluripotent stem cells-derived Alzheimer’s disease cerebral organoid culture and neural differentiation disorders. Neurosci Lett 2019, 711: 134433.
Arber C, Lovejoy C, Harris L, Willumsen N, Alatza A, Casey JM, et al. Familial alzheimer’s disease mutations in PSEN1 lead to premature human stem cell neurogenesis. Cell Rep 2021, 34: 108615.
Bolognin S, Fossépré M, Qing X, Jarazo J, Ščančar J, Moreno EL, et al. 3D cultures of parkinson’s disease-specific dopaminergic neurons for high content phenotyping and drug testing. Adv Sci (Weinh) 2019, 6: 1800927.
Pereira JD, DuBreuil DM, Devlin AC, Held A, Sapir Y, Berezovski E, et al. Human sensorimotor organoids derived from healthy and amyotrophic lateral sclerosis stem cells form neuromuscular junctions. Nat Commun 2021, 12: 4744.
Liu C, Fu Z, Wu S, Wang X, Zhang S, Chu C, et al. Mitochondrial HSF1 triggers mitochondrial dysfunction and neurodegeneration in Huntington’s disease. EMBO Mol Med 2022, 14: e15851.
Hanseeuw BJ, Betensky RA, Jacobs HIL, Schultz AP, Sepulcre J, et al. Association of amyloid and tau with cognition in preclinical alzheimer disease: A longitudinal study. JAMA Neurol 2019, 76: 915–924.
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261: 921–923.
Blanchard JW, Akay LA, Davila-Velderrain J, von Maydell D, Mathys H, Davidson SM, et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 2022, 611: 769–779.
Zhao J, Fu Y, Yamazaki Y, Ren Y, Davis MD, Liu CC, et al. APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer’s disease patient iPSC-derived cerebral organoids. Nat Commun 2020, 11: 5540.
Chen X, Sun G, Tian E, Zhang M, Davtyan H, Beach TG, et al. Modeling sporadic alzheimer’s disease in human brain organoids under serum exposure. Adv Sci (Weinh) 2021, 8: e2101462.
Park JC, Jang SY, Lee D, Lee J, Kang U, Chang H, et al. A logical network-based drug-screening platform for Alzheimer’s disease representing pathological features of human brain organoids. Nat Commun 2021, 12: 280.
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in lewy bodies. Nature 1997, 388: 839–840.
Singleton AB, Farrer MJ, Bonifati V. The genetics of Parkinson’s disease: Progress and therapeutic implications. Mov Disord 2013, 28: 14–23.
di Domenico A, Carola G, Calatayud C, Pons-Espinal M, Muñoz JP, Richaud-Patin Y, et al. Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in parkinson’s disease. Stem Cell Reports 2019, 12: 213–229.
de Rus Jacquet A, Tancredi JL, Lemire AL, DeSantis MC, Li WP, O'Shea EK. The LRRK2 G2019S mutation alters astrocyte-to-neuron communication via extracellular vesicles and induces neuron atrophy in a human iPSC-derived model of Parkinson’s disease. eLife 2021, 10: e73062.
Wulansari N, Darsono WHW, Woo HJ, Chang MY, Kim J, Bae EJ, et al. Neurodevelopmental defects and neurodegenerative phenotypes in human brain organoids carrying Parkinson’s disease-linked DNAJC6 mutations. Sci Adv 2021, 7: eabb1540.
van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, et al. Amyotrophic lateral sclerosis. Lancet 2017, 390: 2084–2098.
Ho R, Workman MJ, Mathkar P, Wu K, Kim KJ, O’Rourke JG, et al. Cross-comparison of human iPSC motor neuron models of familial and sporadic ALS reveals early and convergent transcriptomic disease signatures. Cell Syst 2021, 12: 159-175.e9.
Sances S, Bruijn LI, Chandran S, Eggan K, Ho R, Klim JR, et al. Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat Neurosci 2016, 19: 542–553.
Szebényi K, Wenger LMD, Sun Y, Dunn AWE, Limegrover CA, Gibbons GM, et al. Human ALS/FTD brain organoid slice cultures display distinct early astrocyte and targetable neuronal pathology. Nat Neurosci 2021, 24: 1542–1554.
Walker FO. Huntington’s disease. Semin Neurol 2007, 27: 143–150.
Tabrizi SJ, Estevez-Fraga C, van Roon-Mom WMC, Flower MD, Scahill RI, Wild EJ, et al. Potential disease-modifying therapies for Huntington’s disease: Lessons learned and future opportunities. Lancet Neurol 2022, 21: 645–658.
Estevez-Fraga C, Tabrizi SJ, Wild EJ. Huntington’s disease clinical trials corner: November 2022. J Huntingtons Dis 2022, 11: 351–367.
Barnat M, Capizzi M, Aparicio E, Boluda S, Wennagel D, Kacher R, et al. Huntington’s disease alters human neurodevelopment. Science 2020, 369: 787–793.
Conforti P, Besusso D, Bocchi VD, Faedo A, Cesana E, Rossetti G, et al. Faulty neuronal determination and cell polarization are reverted by modulating HD early phenotypes. Proc Natl Acad Sci U S A 2018, 115: E762–E771.
Yan HHN, Siu HC, Law S, Ho SL, Yue SSK, Tsui WY, et al. A comprehensive human gastric cancer organoid biobank captures tumor subtype heterogeneity and enables therapeutic screening. Cell Stem Cell 2018, 23: 882-897.e11.
Driehuis E, Kretzschmar K, Clevers H. Establishment of patient-derived cancer organoids for drug-screening applications. Nat Protoc 2020, 15: 3380–3409.
Broutier L, Mastrogiovanni G, Verstegen MM, Francies HE, Gavarró LM, Bradshaw CR, et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat Med 2017, 23: 1424–1435.
Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci 2010, 13: 1161–1169.
First MB, Reed GM, Hyman SE, Saxena S. The development of the ICD-11 clinical descriptions and diagnostic guidelines for mental and behavioural disorders. World Psychiatry 2015, 14: 82–90.
Li Z, Ruan M, Chen J, Fang Y. Major depressive disorder: Advances in neuroscience research and translational applications. Neurosci Bull 2021, 37: 863–880.
Bernstein HG, Tausch A, Wagner R, Steiner J, Seeleke P, Walter M, et al. Disruption of glutamate-glutamine-GABA cycle significantly impacts on suicidal behaviour: Survey of the literature and own findings on glutamine synthetase. CNS Neurol Disord Drug Targets 2013, 12: 900–913.
Lewis CP, Nakonezny PA, Blacker CJ, Vande Voort JL, Port JD, Worrell GA, et al. Cortical inhibitory markers of lifetime suicidal behavior in depressed adolescents. Neuropsychopharmacology 2018, 43: 1822–1831.
Lu K, Hong Y, Tao M, Shen L, Zheng Z, Fang K, et al. Depressive patient-derived GABA interneurons reveal abnormal neural activity associated with HTR2C. EMBO Mol Med 2023, 15: e16364.
Sayed Y, Garrison JM. The dopamine hypothesis of schizophrenia and the antagonistic action of neuroleptic drugs—a review. Psychopharmacol Bull 1983, 19: 283–288.
Stahl SM. Beyond the dopamine hypothesis to the NMDA glutamate receptor hypofunction hypothesis of schizophrenia. CNS Spectr 2007, 12: 265–268.
Wu Q, Wang X, Wang Y, Long YJ, Zhao JP, Wu RR. Developments in biological mechanisms and treatments for negative symptoms and cognitive dysfunction of schizophrenia. Neurosci Bull 2021, 37: 1609–1624.
Grunwald LM, Stock R, Haag K, Buckenmaier S, Eberle MC, Wildgruber D, et al. Comparative characterization of human induced pluripotent stem cells (hiPSC) derived from patients with schizophrenia and autism. Transl Psychiatry 2019, 9: 179.
Hoffman GE, Hartley BJ, Flaherty E, Ladran I, Gochman P, Ruderfer DM, et al. Transcriptional signatures of schizophrenia in hiPSC-derived NPCs and neurons are concordant with post-mortem adult brains. Nat Commun 2017, 8: 2225.
Ishii T, Ishikawa M, Fujimori K, Maeda T, Kushima I, Arioka Y, et al. In vitro modeling of the bipolar disorder and schizophrenia using patient-derived induced pluripotent stem cells with copy number variations of PCDH15 and RELN. eNeuro 2019, 6: ENEURO.0403–ENEURO.0418.2019.
Kathuria A, Lopez-Lengowski K, Watmuff B, McPhie D, Cohen BM, Karmacharya R. Synaptic deficits in iPSC-derived cortical interneurons in schizophrenia are mediated by NLGN2 and rescued by N-acetylcysteine. Transl Psychiatry 2019, 9: 321.
Li J, Ryan SK, Deboer E, Cook K, Fitzgerald S, Lachman HM, et al. Mitochondrial deficits in human iPSC-derived neurons from patients with 22q11.2 deletion syndrome and schizophrenia. Transl Psychiatry 2019, 9: 302.
Koskuvi M, Lehtonen Š, Trontti K, Keuters M, Wu YC, Koivisto H, et al. Contribution of astrocytes to familial risk and clinical manifiestation of schizophrenia. Glia 2022, 70: 650–660.
Cannon TD. How schizophrenia develops: Cognitive and brain mechanisms underlying onset of psychosis. Trends Cogn Sci 2015, 19: 744–756.
Sellgren CM, Gracias J, Watmuff B, Biag JD, Thanos JM, Whittredge PB, et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat Neurosci 2019, 22: 374–385.
Kathuria A, Lopez-Lengowski K, Jagtap SS, McPhie D, Perlis RH, Cohen BM, et al. Transcriptomic landscape and functional characterization of induced pluripotent stem cell-derived cerebral organoids in schizophrenia. JAMA Psychiatry 2020, 77: 745–754.
Schnepp PM, Ahmed A, Escara-Wilke J, Dai J, Shelley G, Keller J, et al. Transcription factor network analysis based on single cell RNA-seq identifies that Trichostatin-a reverses docetaxel resistance in prostate Cancer. BMC Cancer 2021, 21: 1316.
Notaras M, Lodhi A, Dündar F, Collier P, Sayles NM, Tilgner H, et al. Schizophrenia is defined by cell-specific neuropathology and multiple neurodevelopmental mechanisms in patient-derived cerebral organoids. Mol Psychiatry 2022, 27: 1416–1434.
Bessonova L, Ogden K, Doane MJ, O’Sullivan AK, Tohen M. The economic burden of bipolar disorder in the United States: A systematic literature review. Clin Outcomes Res 2020, 12: 481–497.
Mertens J, Wang QW, Kim Y, Yu DX, Pham S, Yang B, et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 2015, 527: 95–99.
Madison JM, Zhou F, Nigam A, Hussain A, Barker DD, Nehme R, et al. Characterization of bipolar disorder patient-specific induced pluripotent stem cells from a family reveals neurodevelopmental and mRNA expression abnormalities. Mol Psychiatry 2015, 20: 703–717.
Chen HM, DeLong CJ, Bame M, Rajapakse I, Herron TJ, McInnis MG, et al. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients. Transl Psychiatry 2014, 4: e375.
Kathuria A, Lopez-Lengowski K, Vater M, McPhie D, Cohen BM, Karmacharya R. Transcriptome analysis and functional characterization of cerebral organoids in bipolar disorder. Genome Med 2020, 12: 34.
McFaline-Figueroa JR, Lee EQ. Brain tumors. Am J Med 2018, 131: 874–882.
DeAngelis LM. Brain tumors. N Engl J Med 2001, 344: 114–123.
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro-oncology 2021, 23: 1231–1251.
Goodrich LV, Milenković L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997, 277: 1109–1113.
Jacques TS, Swales A, Brzozowski MJ, Henriquez NV, Linehan JM, Mirzadeh Z, et al. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumour phenotypes. EMBO J 2010, 29: 222–235.
Xiao A, Yin C, Yang C, Di Cristofano A, Pandolfi PP, Van Dyke T. Somatic induction of Pten loss in a preclinical astrocytoma model reveals major roles in disease progression and avenues for target discovery and validation. Cancer Res 2005, 65: 5172–5180.
Yoshida GJ. Applications of patient-derived tumor xenograft models and tumor organoids. J Hematol Oncol 2020, 13: 4.
Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, et al. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol 2012, 9: 338–350.
Huszthy PC, Daphu I, Niclou SP, Stieber D, Nigro JM, Sakariassen PØ, et al. In vivo models of primary brain tumors: Pitfalls and perspectives. Neuro-oncology 2012, 14: 979–993.
Qian X, Song H, Ming GL. Brain organoids: Advances, applications and challenges. Development 2019, 146: dev166074.
Bian S, Repic M, Guo Z, Kavirayani A, Burkard T, Bagley JA, et al. Genetically engineered cerebral organoids model brain tumor formation. Nat Methods 2018, 15: 631–639.
Ogawa J, Pao GM, Shokhirev MN, Verma IM. Glioblastoma model using human cerebral organoids. Cell Rep 2018, 23: 1220–1229.
Linkous A, Balamatsias D, Snuderl M, Edwards L, Miyaguchi K, Milner T, et al. Modeling patient-derived glioblastoma with cerebral organoids. Cell Rep 2019, 26: 3203-3211.e5.
Jacob F, Salinas RD, Zhang DY, Nguyen PTT, Schnoll JG, Wong SZH, et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell 2020, 180: 188-204.e22.
Bhaduri A, Di Lullo E, Jung D, Müller S, Crouch EE, Espinosa CS, et al. Outer radial Glia-like cancer stem cells contribute to heterogeneity of glioblastoma. Cell Stem Cell 2020, 26: 48-63.e6.
Antonica F, Aiello G, Soldano A, Abballe L, Miele E, Tiberi L. Modeling brain tumors: A perspective overview of in vivo and organoid models. Front Mol Neurosci 2022, 15: 818696.
Rutkowski S, von Hoff K, Emser A, Zwiener I, Pietsch T, Figarella-Branger D, et al. Survival and prognostic factors of early childhood medulloblastoma: An international meta-analysis. J Clin Oncol 2010, 28: 4961–4968.
Northcott PA, Jones DTW, Kool M, Robinson GW, Gilbertson RJ, Cho YJ, et al. Medulloblastomics: The end of the beginning. Nat Rev Cancer 2012, 12: 818–834.
Muguruma K, Nishiyama A, Kawakami H, Hashimoto K, Sasai Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep 2015, 10: 537–550.
Ballabio C, Anderle M, Gianesello M, Lago C, Miele E, Cardano M, et al. Modeling medulloblastoma in vivo and with human cerebellar organoids. Nat Commun 2020, 11: 583.
Ballabio C, Gianesello M, Lago C, Okonechnikov K, Anderle M, Aiello G, et al. Notch1 switches progenitor competence in inducing medulloblastoma. Sci Adv 2021, 7: eabd2781.
Magill ST, Vasudevan HN, Seo K, Villanueva-Meyer JE, Choudhury A, John Liu S, et al. Multiplatform genomic profiling and magnetic resonance imaging identify mechanisms underlying intratumor heterogeneity in meningioma. Nat Commun 2020, 11: 4803.
Chan HSC, Ng HK, Chan AKY, Cheng SH, Chow C, Wong N, et al. Establishment and characterization of meningioma patient-derived organoid. J Clin Neurosci 2021, 94: 192–199.
Yamazaki S, Ohka F, Hirano M, Shiraki Y, Motomura K, Tanahashi K, et al. Newly established patient-derived organoid model of intracranial meningioma. Neuro-oncology 2021, 23: 1936–1948.
Acknowledgements
This review was supported by the National Natural Science Foundation of China (32130035 and 92168107), STI2030-Major Projects (2021ZD0202500), the Frontier Key Project of the Chinese Academy of Sciences (QYZDJ-SSW-SMC025), Shanghai Municipal Science and Technology Projects (2018SHZDZX05), and Shanghai Frontiers Science Center for Biomacromolecules and Precision Medicine at ShanghaiTech University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Y., Zeng, PM., Wu, J. et al. Advances and Applications of Brain Organoids. Neurosci. Bull. 39, 1703–1716 (2023). https://doi.org/10.1007/s12264-023-01065-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-023-01065-2