
Abstract
Chronic pain is challenging to treat due to the limited therapeutic options and adverse side-effects of therapies. Astrocytes are the most abundant glial cells in the central nervous system and play important roles in different pathological conditions, including chronic pain. Astrocytes regulate nociceptive synaptic transmission and network function via neuron–glia and glia–glia interactions to exaggerate pain signals under chronic pain conditions. It is also becoming clear that astrocytes play active roles in brain regions important for the emotional and memory-related aspects of chronic pain. Therefore, this review presents our current understanding of the roles of astrocytes in chronic pain, how they regulate nociceptive responses, and their cellular and molecular mechanisms of action.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pain, as defined by the International Association for the Study of Pain, is an unpleasant sensory and emotional experience associated with, or resembling that associated with actual or potential tissue damage. Under physiological conditions, pain plays a protective role to warn the organism to evade noxious stimuli (such as heat, chemical irritants, and cold) and avoid them in future. However, under injury or disease conditions, pain can persist for months to years, and this type of pain is called pathological or chronic pain. Chronic pain is characterized by spontaneous pain, allodynia (pain evoked by a normally innocuous stimulus), and hyperalgesia (enhanced pain evoked by a noxious stimulus). Changes in neuronal plasticity are the major mechanisms of chronic pain [1]. Thus, several neuron-targeting drugs such as NMDA receptor antagonists, opioids (such as morphine, oxycodone, and codeine), and Na+ channel blockers (such as lidocaine, oxcarbazepine, and carbamazepine) are used for the treatment of chronic pain. Although these drugs have therapeutic effects, they also have different degrees of side-effects [2]. Therefore, the development of new types of analgesic with few adverse reactions and better targeting are urgently needed. In the last two decades, non-neuronal cells, especially glial cells, have attracted increasing attention. Targeting the function of glial cells is likely to be a new direction for chronic pain treatment.
In the central nervous system (CNS), more than half of the cells are glia (including astrocytes, microglia, and oligodendrocytes), ~20%–40% of which are astrocytes [3, 4]. Astrocytes not only provide structural and nutritional support for neurons, they also play important roles in many neural processes [5]. Under normal conditions, astrocytes are mostly in a resting state; however, when tissue injury or disease occurs, they transform into a reactive state and contribute to the development of neurological disorders. One of the important features of astrocytes, different from other glial cells, is that they directly communicate with each other by forming gap-junction protein complexes, which allow adjoining cells to freely exchange ions and small cytosolic components [6]. In addition, when CNS neurons are activated, astrocytes regulate blood flow through their extensive contact with cerebral blood vessels [7]. Astrocytes also widely connect with neuronal synapses: a single cortical astrocyte can contact 4–6 neuronal somata, almost 140,000 synapses, and 300–600 neuronal dendrites [3, 4]. Close contact with neurons and synapses makes it possible for astrocytes to support neurons and regulate the physiological/pathological environment during synaptic transmission. These features show that astrocytes play important roles in signal transmission and processing.
In this review, we provide an overview of the roles of astrocytes in the pathogenesis of chronic pain and the interactions between astrocytes and microglia/neurons. We discuss recent neurobiological mechanisms and possible downstream molecular pathways of the astrocytic control of chronic pain. Finally, we discuss how they can be targeted as an alternative strategy for the treatment of chronic pain.
Spinal Astrocytes in Chronic Pain
Classically, chronic pain is classified into two main categories: nociceptive and neuropathic. Nociceptive pain is associated with an ongoing input from real or threatened tissue injury, such as arthritis, trauma, and visceral inflammation. Neuropathic pain is caused by a direct consequence of a lesion or disease affecting the somatosensory system, such as nerve or nerve root compression, toxins, and ischemia. In 2016, the term nociplastic pain was proposed to describe pain that arises from the abnormal processing of pain signals without any clear evidence of tissue damage or discrete pathology involving the somatosensory system, such as fibromyalgia, irritable bowel syndrome, and temporomandibular disorder [8]. In the past two decades, the role of astrocytes in nociceptive pain and neuropathic pain has been widely studied. Under various disease conditions, astrocytes are activated and change to a reactive state characterized by morphological, molecular, and functional changes. Reactive astrocytes are identified by glial fibrillary acidic protein (GFAP) upregulation and hypertrophy after nerve injuries such as spinal nerve ligation (SNL) [9,10,11,12], chronic constriction injury (CCI) [13,14,15], and spinal cord injury (SCI) [16,17,18]. In addition, this process has also been reported in other pain models like tissue inflammation [19, 20], chemotherapy-induced pain [21, 22], arthritic pain [23, 24], and chronic post-cast pain [25, 26].
Reactive astrocytes can display various states. Some reactive states happen within minutes, such as a change in the phosphorylation of signaling molecules or an increase in intracellular Ca2+. Others occur after hours or days (e.g., astrocyte hypertrophy or translational regulation). When a peripheral nerve is injured, astrocyte hypertrophy occurs 3 days later and lasts for several months [27]. Different from astrocytes, microglia immediately respond to a stimulus and proliferate [28], and this process grows to maximal levels in the first week following nerve injury. Raghavendra et al. reported that in a neuropathic pain model, when minocycline (a microglia activation inhibitor) administration is started at the time of nerve transection (pre-emptive treatment), it reduces allodynia and hyperalgesia, which is associated with its ability to suppress microgliosis. However, administration on day 5 after surgery (treatment of existing hypersensitivity) fails to attenuate the behavioral hyperalgesia and allodynia, although it inhibits microglial activation [29]. Meanwhile, astrocyte inhibitors work in both the early and late phases of neuropathic pain [30], indicating that microglia and astrocytes play different roles in the induction and persistence of chronic pain. Evidence shows that microglial reaction may lead to astrocyte reaction [31,32,33,34]; however, astrocyte reaction can also cause microglial reaction [33, 35]. A recent study showed that direct activation of astrocytes using an optogenetic approach induces microglial reactivity and pain hypersensitivity [36]. In addition, inhibition of astrocyte reaction by deletion of astrocyte-expressing CXCR5 reduces the microglial reaction under neuropathic pain conditions [37]. These reports indicate that there is a bilateral interaction between microglia and astrocytes in pain states. Thus, we discuss some of the signaling molecules and related signaling pathways in astrocyte-mediated chronic pain, as well as how astrocytes contribute to chronic pain via communicating with microglia and neurons.
Signaling Molecules Related to Astrocyte-mediated Chronic Pain
As noted above, reactive astrocytes not only change in shape, size, and number, but also change in the expression of various molecules. An increasing list of signaling molecules in astrocytes have been implicated in persistent pain, such as cytokines, chemokines, ion channels, enzymes, and structural proteins. Meanwhile, astrocyte reactivity can be triggered by various molecules. Here, we introduce some classical and important signaling molecules in astrocyte-mediated chronic pain (Table 1).
Inflammatory Cytokines and Chemokines
Cytokines and chemokines are secreted proteins that regulate the immune responses and control immune cell trafficking. It is well known that cytokines and chemokines in peripheral tissues, dorsal root ganglia (DRG), spinal cord, and even the brain play a certain role in the pathogenesis of chronic pain.
Tumor necrosis factor-α (TNF-α) belongs to a superfamily of ligand/receptor proteins called TNF superfamily proteins. It is a cytokine that is usually produced by activated microglia and astrocytes and has pleiotropic effects on normal and malignant cells [38]. When a peripheral nerve is injured, TNF-α is released from activated microglia and stimulates astrocytes [39,40,41,42]. Accordingly, inhibition of TNF-α signaling by neutralizing antibodies or inhibitors alleviates chronic pain [43,44,45]. Our previous study indicated that spinal injection of TNF-α-activated astrocytes induce persistent pain by releasing CCL2 (C-C Motif Chemokine Ligand 2) [46]. Similar to TNF-α, IL-1β is also expressed in microglia and astrocytes. IL-1β is upregulated in astrocytes in different types of chronic pain such as inflammatory pain [47], neuropathic pain [48], and bone cancer pain [49]. IL-1β activates the IL-1 receptor, which is expressed on nociceptive neurons and activates the mitogen-activated protein kinase (MAPK) pathway and sensitizes the neurons. IL-1β and TNF-α regulate the phosphorylation of the NR2B and NR1 subunit of the NMDA (N-methyl D-aspartate) receptor [50] and enhance NMDA-induced currents [51, 52], which suggest a potentiation of glutamatergic synaptic transmission. TNF-α and IL-1β also increase the frequency and amplitude of spontaneous excitatory postsynaptic currents (sEPSCs) [53]. IL-1 receptor-knockout mice show decreased nociceptive responses after SNL [54]. In addition, optogenetic activation of astrocytes causes an increase of IL-1β and TNF-α secretion [36]. In human studies, TNF-α and IL-1β are also increased in the spinal astrocytes of patients with HIV-associated chronic pain [55]. These results suggest the important role of TNF-α and IL-1β in regulating neuropathic pain.
In addition to cytokines, several chemokines such as CCL1, CCL2, CCL3, CCL4, CCL7, CXCL10, CXCL12, CXCL13, and CX3CL1 and their receptors contribute to the pathogenesis of neuropathic pain [56]. Here, we focus on the chemokines or chemokine receptors associated with astrocytes. CCL1 was initially identified in T cells and stimulates the migration of human monocytes through binding to its receptor CCR8. In the SNL model, CCL1 is mainly produced in the DRG and transported to the spinal cord [57]. Meanwhile, CCR8 is increased in astrocytes of the ipsilateral superficial dorsal horn. Inhibition of CCL1 by intrathecal injection of a neutralizing antibody reduces nerve ligation-induced tactile allodynia [57]. Also, oral administration of RAP-103, a peptide inhibitor of CCR8, fully prevents mechanical allodynia and inhibits the development of thermal hyperalgesia after SNL, suggesting the involvement of CCR8 in the initiation and maintenance of nerve injury-induced neuropathic pain [58, 59].
CCL2 is highly expressed by spinal astrocytes and is upregulated in the SNL model [46]. Meanwhile, CCL2 is produced in cultured astrocytes after stimulation with lipopolysaccharide (LPS), TNF-α, or IL-1β [60]. CCR2 is the major receptor of CCL2 and is expressed in primary afferents and neurons in the spinal cord [61]. Mice overexpressing CCL2 in astrocytes display enhanced nociceptive responses in the CFA (complete Freund’s adjuvant) model [62]. Our previous study demonstrated that CCL2 induces rapid phosphorylation of ERK (extracellular signal-activated kinase) in spinal cord neurons. In addition, when lamina II neurons in the spinal cord slice are recorded, the application of CCL2 immediately enhances NMDA- and AMPA-induced inward currents and causes an increase in the frequency and amplitude of sEPSCs [46]. CCL2 also modulates inhibitory synaptic transmission since it inhibits GABA-induced currents in spinal neurons [63].
CXCL10 belongs to the CXC chemokine family and is also known as keratinocyte-derived chemokine-10. CXCL10 is the major ligand of CXCR3 and is increased in neurons and astrocytes of the spinal cord after SNL or spinal cord ischemia reperfusion [64, 65]. Inhibition of CXCL10 by spinal injection of shRNA lentivirus attenuates SNL-induced mechanical allodynia and heat hyperalgesia [64, 66]. CXCL9 and CXCL11 belong to the same subfamily as CXCL10 [67]. However, the roles of these chemokines in pain hypersensitivity are different from CXCL10. Intrathecal injection of CXCL9 or CXCL11 does not induce hyperalgesia or allodynia behaviors, and their inhibition does not inhibit neuropathic pain either [68], suggesting different roles of these chemokines in pain regulation. Other chemokines and their receptors such as CX3CL1/CX3CR1, CXCL1/CXCR2, and CXCL12/CXCR4 are also involved in chronic pain and have been introduced in our previous and others’ reviews [56, 69, 70], so they are not discussed in detail here.
Channel Proteins
Different types of cationic or anionic channels are located on astrocytic membranes to regulate ions for the resting membrane potential or conductance and intracellular signaling. The ion channels on astrocytes are also involved in regulating the release of various gliotransmitters associated with several physiological processes. Here, we introduce some typical water and ion channels that are expressed on astrocytes involved in chronic pain regulation.
Aquaporin-4 (AQP4) is a major water channel expressed in the central nervous system, primarily in astrocytes. The role of AQP4 has been widely studied in a range of pathological conditions [71]. In the spinal cord, AQP4 exhibits a graded decline in distribution from the dorsal to the ventral horn, with abundant expression in laminae I and II [72]. The function of AQP4 is to regulate water influx or efflux driven by osmotic pressure to maintain water homeostasis. AQP4 is increased in spinal cord astrocytes after SCI and nerve injury in humans [73]. In addition, AQP4-knock-out mice show reduced pain sensitivity and dorsal horn sensitivity to noxious stimulation [74].
Sulfonylurea receptor 1 (SUR1), encoded by the Abcc8 gene, is a regulatory subunit that co-assembles with the inward rectifier K+-selective channel to form the KATP channel [75]. SUR1 also co-assembles with the non-selective cation channel, transient receptor potential melastatin 4 (TRPM4) to form the SUR1-TRPM4 complex. SUR1-TRPM4 is upregulated in dorsal horn astrocytes, and global or astrocytes-targeted deletion of SUR1-TRPM4 relieves mechanical allodynia and thermal hyperalgesia in a sciatic nerve cuffing mouse model [76]. Meanwhile, chronic administration of glibenclamide (an SUR1 antagonist) to mice with neuropathic pain causes a reduction of pain behaviors and the expression of IL-6, CCL2, and CXCL1 in astrocytes. Thus, glibenclamide may be an astrocyte-targeted candidate drug for the treatment of some kinds of neuropathic pain.
P2X3 is a non-selective ligand-gated ion channel that belongs to the purinergic receptor family [77]. P2X3 is activated by adenosine triphosphate (ATP) and is selectively permeable to Na+, K+, and Ca2+, especially Ca2+, which plays an important role in the generation and transmission of nociceptive information [78]. Nerve injury causes the release of a large amount of ATP, which activates the P2X3 receptor in the presynaptic membrane and causes Ca2+ influx, resulting in phosphorylation of PKA and PKC and the release of glutamate. This process further activates the NMDA receptors on neurons and causes EPSC generation and central sensitization [79]. It is well known that activation of P2X3 in the DRG causes abnormal nerve discharge, strengthens the transmission of sensory information, and induces visceral hyperalgesia [80, 81]. P2X3 is also expressed on astrocytes in the spinal cord and is increased in a rat model of neuropathic pain [82]. Inhibition of P2X3 in the spinal cord reduces hypersensitivity after nerve injury. In addition, administration of MPEP (2-methyl-6-(phenylethynyl)pyridine; an mGluR5 antagonist) reduces the mechanical allodynia and abolishes the increase in the density of P2X3 in astrocytes induced by nerve injury [82].
Enzymes
Metalloproteases (MMPs) have been suggested to act in the cleavage of extracellular matrix proteins, cytokines, and chemokines to control the inflammation and tissue remodeling associated with various neurodegenerative diseases [83]. MMP-2 and MMP-9 are members of the MMP family involved in IL-1β cleavage [83, 84]. Spinal astrocytes continuously secrete MMP2 after SNL. Downregulation of MMP-2 through intrathecal injection of MMP-2 siRNA reduces mechanical allodynia and the level of spinal GFAP and phosphorylated c-Jun N-terminal kinase 1/2 (JNK1/2), an astrocyte-expressing kinase, in a neuropathic pain model. Local inhibition of MMP-9 inhibits the early phase of neuropathic pain, whereas inhibition of MMP-2 suppresses the late phase of neuropathic pain [83].
Transforming growth factor-β-activated kinase 1 (TAK-1), also known as MAPK kinase kinase 7, is an enzyme regulating innate immunity and pro-inflammatory signaling [85]. TAK-1 mediates the activation of the nuclear factor-kB (NF-kB), JNK, and p38 pathways. Soto-Diaz et al. found that both the production of chemokines and neutrophil migration caused by astrocyte reaction are dependent on TAK1 signaling [86]. Another study reported by Katura et al. showed that peripheral nerve injury induces an increase in TAK1 expression in astrocytes in the spinal dorsal horn, and this TAK1 upregulation increases JNK1 phosphorylation in spinal astrocytes and contributes to the development and maintenance of mechanical hypersensitivity [87].
Tissue type plasminogen activator (tPA) is a well-known extracellular serine protease that converts zymogen plasminogen into an active serine protease. tPA is found on the endothelial cells of blood vessels and is involved in the degradation of blood clots [88]. In addition, tPA participates in modification of the extracellular matrix that leads to long-term potentiation in the hippocampus [89]. Kozai et al. reported that tPA is upregulated in spinal astrocytes following root injury [90]. Moreover, continuous intrathecal administration of a tPA inhibitor suppresses root ligation-induced mechanical allodynia. These data suggest that astrocyte-derived tPA in the dorsal horn is essential for the mechanical hypersensitivity following root injury.
Other Molecules
N-myc downstream-regulated gene 2 (NDRG2) is a member of the NDRG family and is widely distributed in the CNS but only expressed in astrocytes. NDRG2 members have different functions in cell differentiation, proliferation, and maintenance of cell morphology [91]. Ma et al. found that down-regulation of NDRG2 in spinal astrocytes inhibits their reactivity and reduces nociceptive behaviors in a rat model of spared nerve injury (SNI) [92]. Another study reported by Li et al. also indicated that inhibition of NDRG2 contributes to astrocyte-specific neuroprotection [93].
FGFR3 is a member of the fibroblast growth factor receptor (FGFR) family, which contains four members (FGFR1–4) that mediate FGF signal transduction. FGF/FGFR signaling plays an important role in cell differentiation, neuronal survival, and cell development [94]. Previous studies have shown that activated FGFR3 promotes the proliferation and development of astrocytes [95]. Xie et al. showed that FGFR3 upregulates GFAP and TNF-α expression in astrocytes in vivo and in vitro. Inhibition of FGFR3 leads to reduced GFAP and TNF-α and increases the withdrawal threshold in SNI model [95]. These results suggest that FGFR3 induces production of the inflammatory mediator TNF-α and astrocyte reactivity to cause hyperpathia in neuropathic pain.
TRAF6 belongs to the TNF receptor-associated factor (TRAF) protein family which has 6 members (TRAF1–TRAF6). A growing body of literature has established the important role of TRAF6 in the development and maintenance of chronic pain. Our previous study showed that TNF-α, IL-1β, and TLR4 mediate TRAF6 upregulation in spinal astrocytes after SNL in mice [96]. However, TRAF6 is increased in spinal microglia in CFA-induced chronic inflammatory pain [97]. Direct inhibition of TRAF6 by siRNA or indirect inhibition by docosahexaenoic acid has therapeutic effects on neuropathic pain and inflammatory pain [97]. Another study showed that TRAF6 is increased in spinal astrocytes in the chronic visceral pain model [98]. Knockdown of TRAF6 remarkably reduces the amplitude of EPSCs of spinal dorsal horn neurons and relieves visceral hypersensitivity [98].
Long non-coding RNAs (lncRNAs) have hundreds of nucleotides with no protein-coding potential. Currently, there is growing evidence that lncRNAs play an important role in regulating chronic pain. For example, lncRNA PVT1 is up-regulated in spinal astrocytes after SCI [99]. Depletion of PVT1 in the spinal cord reduces nociceptive responses such as thermal hyperalgesia and mechanical allodia as well as the expression of neuroinflammatory factors and proteins. Furthermore, it has been confirmed that PVT1 is a competitive endogenous RNA of miR-186-5p, while miR-186-5p targets CXCL13 [99]. Inhibition of lncRNA PVT1 alleviates neuropathic pain in SCI rats by upregulating miR-186-5p and down-regulating CXCL13/CXCR5 [99, 100]. Another similar study showed that lncRNA MEG3 aggravates neuropathic pain and astrocyte reactivity by mediating the miR-130a-5p/CXCL12/CXCR4 axis [101].
Interaction of Astrocytes, Microglia, and Neurons Under Chronic Pain Conditions
It is well-accepted that neuronal plasticity is a key mechanism for the development and maintenance of chronic pain. Astrocytes and microglia are important players in the regulation of neuronal functions [102]. The communication between astrocytes, microglia, and neurons in the spinal cord and brain facilitates central sensitization, which is manifested as an increased responsiveness of neurons to normal or subthreshold afferent inputs.
Microglia are the resident immune cells of the CNS. They are activated by tissue damage or nerve injury [28] and their morphology changes to an amoeboid shape, accompanied by enhanced secretion of numerous inflammatory factors and microglial phagocytosis [3]. Evidence has already demonstrated that microglia play an important role in the pathogenesis of chronic pain [28]. With different stimuli (usually extracellular/intracellular signals like inflammatory mediators or anti-inflammatory mediators), microglia display two phenotypes, M1 and M2 [33]. The M1-like phenotype is induced by TNF-α, IL-1β, or other inflammatory mediators, and then microglia secrete pro-inflammatory cytokines such as IL-6, IL-23, or chemokines (CCL2 and CCL5) to induce neuroinflammation, resulting in the maintenance of chronic pain [28]. The M2-like phenotype is induced by IL-4 or IL-13. Microglia with M2-like phenotypes have increased phagocytosis and produce growth factors such as insulin-like growth factor-1 and anti-inflammatory cytokines such as IL-10 [103]. The cross-talk between astrocytes and microglia is maintained in part via secreted mediators, such as growth factors, neurotransmitters, cytokines, chemokines, innate-immunity mediators, tissue damage molecules (e.g., ATP), mitogenic factors, NO, ROS, and metabolic mediators such as amino-acids, that can be used for cell metabolism and may also mediate tissue changes [104]. The production and release of these mediators are normally controlled by key intracellular signaling pathways, such as the MAPK pathway. Many chemokines, such as CXCL12, CXCL10, CXCL1, and CCL2 are mainly stored in astrocytes, and microglia express the corresponding chemokine receptors like CXCR4 or CCR2 [105]. This suggests a strong association between microglia and astrocytes [6]. IL-18, one member of the IL-1 family, is an important regulator of innate and acquired immune responses. Kan et al. reported that IL-18 and IL-18 receptors are expressed in microglia and astrocytes, respectively, mediate microglia-astrocyte interaction in the spinal cord, and enhance neuropathic pain processing after nerve injury [106]. These results suggest that activated microglia in the spinal dorsal horn are directly responsible for the induction of astrocyte reactivity after nerve injury. However, reactive astrocytes sometimes are not correlated with reactive microglia. Hald et al. found that bone cancer markedly induces spinal astrocyte reactivity, not microglial reactivity [107]. In addition, Robinson et al. reported that astrocyte but not microglial reactivity is induced in oxaliplatin- and bortezomib-induced peripheral neuropathy in rats [108].
In addition to the interaction with microglia, astrocytes also directly interact with neurons. We found that CXCL13-CXCR5 mediates neuron-astrocyte interaction in the spinal cord after SNL [37]. SNL increases the expression of CXCL13 and its receptor CXCR5 in neurons and astrocytes in mice. In Cxcr5-KO mice, the reactivity of astrocytes and microglia in the dorsal horn is remarkably reduced after nerve injury [37]. Furthermore, when astrocytes are activated, they release CCL2 and CXCL1 to act on their receptors CCR2 and CXCR2 on spinal neurons, thus causing an enhancement of excitatory synaptic transmission and chronic pain [46, 56, 109, 110]. Recent work has shown that CXCL2 secreted by astrocytes interacts with CXCR2 expressed on neurons in the spinal cord and this contributes to carrageenan-induced prostatitis pain [111]. Meanwhile, the CXCL1/CXCR2-mediated interaction between astrocytes and neurons in the periaqueductal gray (PAG) also facilitates chronic pain [112]. A recent study showed that Wnt5a from neurons is crucial for reactive astrogliosis in an animal model of HIV-associated pain [113]. Wnt5a from neurons targets its receptor ROR2, which is expressed in astrocytes. Furthermore, conditional knockout of either Wnt5a in neurons or its receptor ROR2 abolishes not only gp120-induced astrocyte reactivity but also hyperalgesia. These results show that astrocytes, microglia, and neurons form a loop to interact with each other and regulate chronic pain.
The Intracellular Signaling Pathway in Reactive Astrocytes in Chronic Pain
Several intricate roles are played by astrocytes in the pathogenesis of chronic pain, not only in the means of intercellular communication but also in the alteration of intracellular downstream signaling pathways as well as the variation in metabolic patterns. Nerve injury or pathogen invasion can be the initial factor for astrocyte activation in chronic pain. Here, we introduce astrocytic intracellular signaling and the actions of astrocyte-released neuromodulators in chronic pain. Some typical pathways such as the p38 and JNK pathways have been described in previous reviews [4, 6], thus we do not discuss them in detail.
Janus kinase (JAK) signal transducers and activators of the transcription 3 (STAT3) signaling pathway is involved in the restricted proliferation of dorsal horn astrocytes after peripheral nerve injury [114]. Inhibition of JAK-STAT3 suppresses both the proliferation of dorsal horn astrocytes and the maintenance of tactile allodynia. It has been shown that the dimerization of gp130 participates in the conduction pathway of activated JAK1 and JAK2, followed by phosphorylating STAT3 [115]. Gp130 cooperates with other receptors such as IL-6, IL-11, and IL-27 to constitute receptor complexes [116,117,118]. Considering the pleiotropic effects and widespread expression of these cytokines, the gp130-JAK-STAT3 signaling pathway may provide new directions in the research on reactive astrogliosis concerned with chronic pain.
NF-κB plays important roles in many aspects of cell regulation such as proliferation, apoptosis, and differentiation [119]. NF-κB can regulate neuropathic pain by mediating neuroinflammation, neuron apoptosis, and synaptic plasticity [120]. NF-κB is also strongly activated in reactive astrocytes and contributes to the development of neuropathic pain. During the progression of SCI, the inactivation of NF-κB in transgenic GFAP-IκBα-dn mice causes a reduction of lesion volume, an increase of white matter preservation, and a reduction in the expression of proteoglycan and chemokines CXCL10 and CCL2, and improves functional recovery [121]. During the inflammatory pain process, NF-κB plays an important role in gene transcription induced by cytokines. IL-1β and TNF-α activate NF-κB and lead to the transcription of several genes including pro-inflammatory factors and chemokines. This is a positive feedback process that further activates NF-κB and leads to more expression of downstream genes associated with neuroinflammation. In addition, Toll-like receptors (TLRs) are key regulators of the NF-κB signaling pathway. TLR4 regulates neuropathic pain through its activation of microglia and astrocytes in the spinal cord. When TLR4 is activated by LPS or other stimuli, NF-κB acting as its main downstream pathway is activated to begin the next process to develop chronic pain [122]. Using different methods including blockers or siRNA to inhibit the NF-κB signaling pathway can relieve different types of chronic pain. Thus, regulation of the NF-κB signaling pathway could be a potential therapeutic strategy [121, 123, 124].
The transcription factor Olig2 is considered to play an essential role in the differentiation of oligodendrocytes and motor neurons in the embryonic spinal cord. However, recent findings suggest that Olig2-lineage astrocytes are a different subgroup from GFAP-lineage astrocytes, both of which frequently occupy mutually exclusive territories and have distinct mRNA expression patterns [125,126,127]. In addition, Olig2-lineage astrocytes tend to express GABA transporter-3 and are involved in inhibitory neuronal transmission [126]. Ablation of Olig2 decreases the proliferation of reactive astrocytes in response to injury [128]. Olig2 is a direct target of Notch signaling [129], which upregulates Olig2 expression and promotes Olig2 localization in the nucleus in reactive astrogliogenesis in the brain. Inhibitor of the Notch-activating enzyme reduces the number of reactive astrocytes [130]. Given the fact that Olig2 influences Wnt signaling in gliomas and neural stem cells and participates in the interaction between Wnt signaling and Notch signaling, further research on the Notch-Olig2-Wnt pathway is important in the area of astrogliogenesis and chronic pain [131, 132].
TGF-β is rapidly and chronically elevated in response to CNS injury. Intrathecal injection of TGF-β inhibits neuropathy-induced hyperalgesia as well as spinal microglial and astrocytic reactivity [133, 134]. TGF-β attenuates the upregulation of pp38 and pERK in spinal microglia and astrocytes of mice with CCI. Despite the neuroprotective role of TGF-β, it also stimulates astrocytes to a reactive state with up-regulated GFAP [135]. Schachtrup et al. emphasized that fibrinogen is a carrier of latent TGF-β and induces phosphorylation of Smad2 in astrocytes after leakage through the disrupted blood–brain barrier or vascular damage [136]. Nuclear Smads interact with a large number of transcription factors to activate target genes [137]. TGF-β regulates adenine nucleotide translocator 1 gene expression, which is responsible for the removal of extracellular glutamate through a cooperative interaction of both Smad and Sp1 binding elements located immediately upstream of the transcriptional start site [138, 139].
Supraspinal Astrocytes in Chronic Pain
Chronic pain results from peripheral and central sensitization [1]. Central sensitization occurs not only in the spinal cord but also in supraspinal areas. Noxious stimuli are detected and transduced into electrical signals and further transmitted from the DRG to the dorsal horn. The signal is then sent up through the spinothalamic tract to the thalamus and then to the primary somatosensory cortex (S1) [140]. Several areas such as the PAG, parabrachial nucleus, nucleus accumbens, and anterior cingulate cortex (ACC) are important for pain regulation [141]. Under nerve injury or inflammatory conditions, astrocytes undergo varied changes in the brainstem, thalamus, ACC, and S1 [142, 143], and these changes contribute to central sensitization and chronic pain.
The spinal trigeminal nucleus is similar to the spinal dorsal horn and plays an essential role in trigeminal pain transmission. Reactive astrocytes in the spinal trigeminal nucleus contribute to the pathogenesis of chronic orofacial pain [144]. In reactive astrocytes, the TNF signaling pathway is activated and increases the expression of GFAP, CX43, and IL-1β, leading to neuropathic pain in the trigeminal system [48, 145]. This activation process of astrocytes is initiated by upregulation of several marker genes, including complement 3, complement factor B, MX dynamin-like GTPase 1, and S100a10 [32]. Neurons and glial cells regulate this astrogliosis process through various signaling pathways, including the JAK-STAT3, Notch-OLIG2, and TGFβ-SMAD pathways [114, 128, 146,147,148]. In addition, after infraorbital nerve injury, reactive astrocytes are detected in the rostral ventromedial medulla (RVM), a major component of the brainstem for descending pain modulatory circuitry [48]. The expression levels of the pro-inflammatory factors TNF-α and IL-1β are increased in RVM astrocytes. Intra-RVM administration of astrocytic inhibitors attenuates mechanical hyperalgesia and allodynia behaviors. Moreover, TNFR1 and IL-1R are expressed in RVM neurons that express the NMDA receptor subunit NR1. Injection of recombinant TNF-α or IL-1β upregulates NR1 phosphorylation and causes an NMDAR-dependent allodynia [48]. Supraspinal astrocytes also participate in descending nociceptive modulation in a cancer-induced bone pain model. Ni et al. showed that activation of astrocytes in the ventrolateral PAG is implicated in facilitating bone cancer pain in rats via the JNK signaling pathway [149]. Intrathecal administration of astrocytic cytotoxin or a JNK inhibitor reduces the expression of GFAP and mechanical allodynia. All the above suggest a contribution of supraspinal astrocytes and central glia-neuronal interactions to the descending facilitation of chronic pain.
Astrocytes in other regions of the brain have also been reported to be involved in the pain matrix such as the ACC and S1. Astrocytes in the ACC play a role in the affective component of pain, including unpleasantness or aversion [142]. SNL activates astrocytes in S1 and increases mGluR5 expression [143]. This leads to an increase in S1 astrocytic Ca2+ transients and, thus, the release of thrombospondin 1 from astrocytes, which promotes synapse formation as well as mechanical allodynia [143]. In addition, Wahis et al. reported a new function of astrocytes in the central nucleus of the amygdala (CeA) under neuropathic pain conditions [150]. They found that activation of oxytocin receptors (OTRs) in the CeA reduces neuropathic pain-induced anxiety behavior. Meanwhile, the deletion of OTRs from the astrocytes in the lateral part of the CeA abolishes the anxiolytic effects of OTRs agonists [150]. These data highlight the central role of astrocyte-mediated oxytocin signaling in the regulation of emotional states under chronic pain conditions.
It is important to note that the research above was mostly based on animal studies. Human astrocytes have several distinct properties that are quite different from those of rodents. The human brain contains subtypes of GFAP-positive astrocytes that are not expressed in rodents [135]. Also, the size of human cortical astrocytes is almost double that in rodents [151]. Real-time imagining studies on the brains of patients with chronic lower back pain have demonstrated glial activation in multiple regions including the thalamus and S1 [152]. This research suggests possible astrocyte activation in higher brain regions in humans. Thus, determining whether there is concomitant neuronal activation with astrocytes activation in chronic pain may be of major importance in future research.
Targeting Astrocytes as an Alternative Strategy for the Treatment of Chronic Pain
Long-term unendurable and severe chronic pain impairs patients’ quality of life and imposes a heavy economic burden on society. Non-steroidal anti-inflammatory drugs, opioids, and adjuvant analgesics like antidepressants and antiepileptics are common pharmacological treatments for managing chronic pain. However, most patients face the problem of receiving inadequate analgesic therapy and the prescription is limited by the side-effects including gastrointestinal hemorrhage, thrombotic cardiovascular events, and addiction [153]. Hence, there is an urgent need for developing new therapeutic strategies for chronic pain.
With the improving understanding of the roles of astrocytes in chronic pain, therapeutic strategies may target their underlying mechanisms in order to reduce pain or enhance the recovery process. For example, matrix metalloproteinases (MMPs), particularly MMP-2, are known to participate in neuropathic pain after nerve injury [83, 84]. MMP-2 is released by astrocytes after the injury and induces activation of IL-1β [83]. Therefore, it would be ideal to target MMP-2 and reduce inflammation after injury. Unfortunately, most available drugs have a non-specific affinity for MMPs and thus induce various side-effects or minimal desired effects. MMP-1 and MMP-9 inhibitors have recently been developed, and yet not much progress has been made in the development of MMP inhibitors [154]. TNF-α is a cytokine that is usually produced by activated microglia and astrocytes. Administration of TNF-α antibody effectively alleviates hyperalgesia [155], indicating the prospect of anti-TNFα therapy in the treatment of chronic pain. An ongoing phase III clinical trial is aimed to test the efficacy of infliximab (a TNFα antagonist) in treating lower-back pain in patients (NCT03704363). Multiple compounds and medicines targeting astrocytes alleviate chronic pain by regulating pro-inflammatory mediators such as TNFα, IL-1β, and CCL2 [156,157,158,159,160]. Of note, besides the neuronal expression of TNFR1 and IL-1R, they are also expressed in astrocytes and microglia and contribute to glial activation [45, 96, 161]. Pharmacological inhibition of TNF-α also attenuates glial activation then relieves chronic pain. However, since a certain amount of TNFR1 or IL-1R is expressed on neurons, blocking TNF-α also affects neuronal function.
In addition, pharmacological inhibition of P2X3 in rats following CCI of the trigeminal infraorbital nerve attenuates facial pain [82]. The purinergic receptor P2X3 is found in astrocytes in the spinal trigeminal nucleus, and blocking P2X3 inhibits reactive astrogliosis and the release of downstream inflammatory factors [82]. Thus, it might also be possible to target downstream molecules of reactive astrogliosis to reduce its effect on pain behaviors. For example, patients with neuromyelitis optica have been treated with tocilizumab, an IL-6 antibody, which was found to be safe and effective [162]. Although some of these compounds have antinociceptive effects in animal models and inhibit the reactivity of astrocytes, more designs for clinical trials to test their analgesic efficacy on humans are needed.
Another potential approach is to block gap junction proteins, such as connexin-43 (Cx43) and pannexin 1 (Panx1). Cx43 is specifically upregulated in spinal astrocytes after CCI [30]. Once upregulated, Cx43 enhances ATP release from astrocytes and finally leads to microglial activation and allodynia [163, 164]. Besides, Cx43 also contributes to the release of glutamate and chemokines, and blocking this protein remarkably attenuates neuropathic pain sensitization [30, 165]. The main mechanism is that, in chronic pain states, Cx43 modulates hemichannel function, leading to an increase in the permeability to various cytokines and chemokines [30, 166]. In addition, glial Panx1 contributes to the tactile hypersensitivity in chronic orofacial pain by inducing hyper-responsiveness to ATP [167]. Some Panx1 blockers (including mefloquine and probenecid) have been reported to improve morphine withdrawal syndrome [168]. However, further studies are needed to clarify their effects on humans.
Conclusion and Future Perspectives
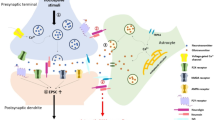
In summary, we have reviewed different kinds of evidence to demonstrate the necessity and sufficiency of astrocytes in chronic pain. We also explain how astrocytes promote chronic pain through astrocyte-microglia or astrocyte-neuron interactions (Fig. 1). When peripheral nerve injury or tissue damage occurs, astrocytes change to a reactive state in response to different neurotransmitters or neuromodulators in the spinal cord or brain. Reactive astrocytes are usually accompanied by the activation of a variety of intracellular signaling pathways. Therefore, the molecular mechanisms of astrocyte-microglia-neuron crosstalk in the spinal cord and brain under chronic pain conditions deserve further study.

Astrocytic, microglial, and neuronal interaction in chronic pain. Nerve injury induces the release of CXCL13, which activates astrocytes via the CXCR5 receptor. The activation of astrocytes results in the upregulation of CX43 expression and a switch in CX43-mediated function from gap-junction communication to CX43 hemichannel-mediated paracrine signaling, resulting in the increased release of pro-inflammatory cytokines, chemokines, glutamate, and ATP, which activate microglia through P2RX4, P2RX7, and other receptors. The activation of these microglial receptors induces the release of pro-inflammatory cytokines (including TNF-α and IL-1β) and further amplifies neuronal excitability. These cytokines also result in the upregulation of the transcriptional regulators TRAF6, STAT3, and subsequent activation of the JNK and ERK pathways in astrocytes, further increasing their production and release of chemokines and facilitating neuropathic pain. The figure was created with BioRender.com.
Given the important role of astrocytes in the facilitation of chronic pain, targeting them may provide novel prevention and treatment strategies. However, because astrocytes play an essential supportive and protective role in the CNS, it is important to target specific signaling events in astrocytes without disrupting their overall well-being. The recent identification of astrocyte-expressing genes by transcriptome analyses suggests that astrocytes display inter- or intra-regional heterogeneity and act as a gate for descending noradrenergic control of mechanosensory behavior, which indicates the diverse functions and phenotypes of astrocytes for chronic pain regulation [169]. Akdemir et al. also found that subpopulations of Lfng-labeled astrocytes in laminae III and IV of the dorsal horn are involved in the regulation of neuronal activity and maintaining sensory-processing circuity associated with light touch [170]. Ablation of Lfng+ astrocytes reduces glutamatergic synapses and mechanosensory responses. In addition, compared to the classical neuronal gate control theory of pain, Xu et al. reported a new function of astrocytes in the gating of nociceptive signals in the spinal cord [171]. Spinal astrocytes are activated by electrical stimulation of peripheral Aβ fibers, which induces long-term depression in NK1R+ neurons and antinociception. Meanwhile, suppression of reactive astrocytes by different methods blocks such processes. Their results demonstrate astrocytes as a new and important component of pain gating by activation of Aβ fibers that exert non-neuronal control of pain. Thus, these recent discoveries may provide a new research direction for astrocyte regulation of chronic pain.
References
Ji RR, Nackley A, Huh Y, Terrando N, Maixner W. Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology 2018, 129: 343–366.
Bacchi S, Palumbo P, Sponta A, Coppolino MF. Clinical pharmacology of non-steroidal anti-inflammatory drugs: A review. Antiinflamm Antiallergy Agents Med Chem 2012, 11: 52–64.
Donnelly CR, Andriessen AS, Chen G, Wang K, Jiang C, Maixner W. Central nervous system targets: Glial cell mechanisms in chronic pain. Neurotherapeutics 2020, 17: 846–860.
Gao YJ, Ji RR. Targeting astrocyte signaling for chronic pain. Neurotherapeutics 2010, 7: 482–493.
Haim LB, Rowitch DH. Functional diversity of astrocytes in neural circuit regulation. Nat Rev Neurosci 2017, 18: 31–41.
Ji RR, Donnelly CR, Nedergaard M. Astrocytes in chronic pain and itch. Nat Rev Neurosci 2019, 20: 667–685.
Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci 2007, 10: 1369–1376.
Fitzcharles MA, Cohen SP, Clauw DJ, Littlejohn G, Usui C, Häuser W. Nociplastic pain: Towards an understanding of prevalent pain conditions. Lancet 2021, 397: 2098–2110.
Colburn RW, Rickman AJ, DeLeo JA. The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exp Neurol 1999, 157: 289–304.
Guo S, Song Z, He J, Yin G, Zhu J, Liu H, et al. Akt/aquaporin-4 signaling aggravates neuropathic pain by activating astrocytes after spinal nerve ligation in rats. Neuroscience 2022, 482: 116–131.
Tsuda M. Modulation of pain and itch by spinal glia. Neurosci Bull 2018, 34: 178–185.
Chen G, Luo X, Qadri MY, Berta T, Ji RR. Sex-dependent glial signaling in pathological pain: Distinct roles of spinal microglia and astrocytes. Neurosci Bull 2018, 34: 98–108.
Garrison CJ, Dougherty PM, Kajander KC, Carlton SM. Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res 1991, 565: 1–7.
Xue P, Chen L, Lu X, Zhang J, Bao G, Xu G, et al. Vimentin promotes astrocyte activation after chronic constriction injury. J Mol Neurosci 2017, 63: 91–99.
Ono T, Kohro Y, Kohno K, Tozaki-Saitoh H, Nakashima Y, Tsuda M. Mechanical pain of the lower extremity after compression of the upper spinal cord involves signal transducer and activator of transcription 3-dependent reactive astrocytes and interleukin-6. Brain Behav Immun 2020, 89: 389–399.
Okada S, Hara M, Kobayakawa K, Matsumoto Y, Nakashima Y. Astrocyte reactivity and astrogliosis after spinal cord injury. Neurosci Res 2018, 126: 39–43.
Allahyari RV, Heinsinger NM, Hwang D, Jaffe DA, Rasouli J, Shiers S, et al. Response of astrocyte subpopulations following spinal cord injury. Cells 2022, 11: 721.
Miranpuri GS, Bali P, Nguyen J, Kim JJ, Modgil S, Mehra P, et al. Role of microglia and astrocytes in spinal cord injury induced neuropathic pain. Ann Neurosci 2021, 28: 219–228.
Wei X, Jin XH, Meng XW, Hua J, Ji FH, Wang LN, et al. Platelet-rich plasma improves chronic inflammatory pain by inhibiting PKM2-mediated aerobic glycolysis in astrocytes. Ann Transl Med 2020, 8: 1456.
Ducza L, Szücs P, Hegedűs K, Bakk E, Gajtkó A, Wéber I, et al. NLRP2 is overexpressed in spinal astrocytes at the peak of mechanical pain sensitivity during complete Freund adjuvant-induced persistent pain. Int J Mol Sci 2021, 22: 11408.
Luo H, Liu HZ, Zhang WW, Matsuda M, Lv N, Chen G, et al. Interleukin-17 regulates neuron-glial communications, synaptic transmission, and neuropathic pain after chemotherapy. Cell Rep 2019, 29: 2384-2397.e5.
Sheng HY, Zhang YQ. Emerging molecular targets for the management of cancer pain. Neurosci Bull 2020, 36: 1225–1228.
Old EA, Clark AK, Malcangio M. The role of glia in the spinal cord in neuropathic and inflammatory pain. Handb Exp Pharmacol 2015, 227: 145–170.
Sagar DR, Burston JJ, Hathway GJ, Woodhams SG, Pearson RG, Bennett AJ, et al. The contribution of spinal glial cells to chronic pain behaviour in the monosodium iodoacetate model of osteoarthritic pain. Mol Pain 2011, 7: 88.
Li T, Liu T, Chen X, Li L, Feng M, Zhang Y, et al. Microglia induce the transformation of A1/A2 reactive astrocytes via the CXCR7/PI3K/Akt pathway in chronic post-surgical pain. J Neuroinflammation 2020, 17: 211.
Ohmichi M, Ohmichi Y, Ohishi H, Yoshimoto T, Morimoto A, Li Y, et al. Activated spinal astrocytes are involved in the maintenance of chronic widespread mechanical hyperalgesia after cast immobilization. Mol Pain 2014, 10: 6.
Li X, Li M, Tian L, Chen J, Liu R, Ning B. Reactive astrogliosis: Implications in spinal cord injury progression and therapy. Oxid Med Cell Longev 2020, 2020: 9494352.
Inoue K, Tsuda M. Microglia in neuropathic pain: Cellular and molecular mechanisms and therapeutic potential. Nat Rev Neurosci 2018, 19: 138–152.
Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther 2003, 306: 624–630.
Chen G, Park CK, Xie RG, Berta T, Nedergaard M, Ji RR. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain 2014, 137: 2193–2209.
Yang J, Wang T, Jin X, Wang G, Zhao F, Jin Y. Roles of crosstalk between astrocytes and microglia in triggering neuroinflammation and brain edema formation in 1, 2-dichloroethane-intoxicated mice. Cells 2021, 10: 2647.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541: 481–487.
Liu LR, Liu JC, Bao JS, Bai QQ, Wang GQ. Interaction of microglia and astrocytes in the neurovascular unit. Front Immunol 2020, 11: 1024.
Sano F, Shigetomi E, Shinozaki Y, Tsuzukiyama H, Saito K, Mikoshiba K, et al. Reactive astrocyte-driven epileptogenesis is induced by microglia initially activated following status epilepticus. JCI Insight 2021, 6: 135391.
Ovanesov MV, Ayhan Y, Wolbert C, Moldovan K, Sauder C, Pletnikov MV. Astrocytes play a key role in activation of microglia by persistent Borna disease virus infection. J Neuroinflammation 2008, 5: 50.
Nam Y, Kim JH, Kim JH, Jha MK, Jung JY, Lee MG, et al. Reversible induction of pain hypersensitivity following optogenetic stimulation of spinal astrocytes. Cell Rep 2016, 17: 3049–3061.
Jiang BC, Cao DL, Zhang X, Zhang ZJ, He LN, Li CH, et al. CXCL13 drives spinal astrocyte activation and neuropathic pain via CXCR5. J Clin Investig 2016, 126: 745–761.
Sawada M, Kondo N, Suzumura A, Marunouchi T. Production of tumor necrosis factor-alpha by microglia and astrocytes in culture. Brain Res 1989, 491: 394–397.
McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J Neuroinflammation 2008, 5: 45.
Probert L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302: 2–22.
Gao YJ, Zhang L, Ji RR. Spinal injection of TNF-α-activated astrocytes produces persistent pain symptom mechanical allodynia by releasing monocyte chemoattractant protein-1. Glia 2010, 58: 1871–1880.
Hyvärinen T, Hagman S, Ristola M, Sukki L, Veijula K, Kreutzer J, et al. Co-stimulation with IL-1β and TNF-α induces an inflammatory reactive astrocyte phenotype with neurosupportive characteristics in a human pluripotent stem cell model system. Sci Rep 2019, 9: 16944.
Mata M, Hao S, Fink DJ. Gene therapy directed at the neuroimmune component of chronic pain with particular attention to the role of TNFα. Neurosci Lett 2008, 437: 209–213.
Zheng X, Ouyang H, Liu S, Mata M, Fink DJ, Hao S. TNFα is involved in neuropathic pain induced by nucleoside reverse transcriptase inhibitor in rats. Brain Behav Immun 2011, 25: 1668–1676.
Xu JT, Xin WJ, Zang Y, Wu CY, Liu XG. The role of tumor necrosis factor-alpha in the neuropathic pain induced by Lumbar 5 ventral root transection in rat. Pain 2006, 123: 306–321.
Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, et al. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci 2009, 29: 4096–4108.
Weyerbacher AR, Xu Q, Tamasdan C, Shin SJ, Inturrisi CE. N-Methyl-D-aspartate receptor (NMDAR) independent maintenance of inflammatory pain. Pain 2010, 148: 237–246.
Wei F, Guo W, Zou S, Ren K, Dubner R. Supraspinal glial-neuronal interactions contribute to descending pain facilitation. J Neurosci 2008, 28: 10482–10495.
Zhang RX, Liu B, Wang L, Ren K, Qiao JT, Berman BM, et al. Spinal glial activation in a new rat model of bone cancer pain produced by prostate cancer cell inoculation of the tibia. Pain 2005, 118: 125–136.
Zhang RX, Li A, Liu B, Wang L, Ren K, Zhang H, et al. IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. Pain 2008, 135: 232–239.
Zhang L, Berta T, Xu ZZ, Liu T, Park JY, Ji RR. TNF-alpha contributes to spinal cord synaptic plasticity and inflammatory pain: Distinct role of TNF receptor subtypes 1 and 2. Pain 2011, 152: 419–427.
Wheeler D, Knapp E, Bandaru VVR, Wang Y, Knorr D, Poirier C, et al. Tumor necrosis factor-alpha-induced neutral sphingomyelinase-2 modulates synaptic plasticity by controlling the membrane insertion of NMDA receptors. J Neurochem 2009, 109: 1237–1249.
Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: Distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci 2008, 28: 5189–5194.
Wolf G, Gabay E, Tal M, Yirmiya R, Shavit Y. Genetic impairment of interleukin-1 signaling attenuates neuropathic pain, autotomy, and spontaneous ectopic neuronal activity, following nerve injury in mice. Pain 2006, 120: 315–324.
Shi Y, Gelman BB, Lisinicchia JG, Tang SJ. Chronic-pain-associated astrocytic reaction in the spinal cord dorsal horn of human immunodeficiency virus-infected patients. J Neurosci 2012, 32: 10833–10840.
Jiang BC, Liu T, Gao YJ. Chemokines in chronic pain: Cellular and molecular mechanisms and therapeutic potential. Pharmacol Ther 2020, 212: 107581.
Akimoto N, Honda K, Uta D, Beppu K, Ushijima Y, Matsuzaki Y, et al. CCL-1 in the spinal cord contributes to neuropathic pain induced by nerve injury. Cell Death Dis 2013, 4: e679.
Padi SSV, Shi XQ, Zhao YQ, Ruff MR, Baichoo N, Pert CB, et al. Attenuation of rodent neuropathic pain by an orally active peptide, RAP-103, which potently blocks CCR2- and CCR5-mediated monocyte chemotaxis and inflammation. Pain 2012, 153: 95–106.
Noda M, Tomonaga D, Kitazono K, Yoshioka Y, Liu J, Rousseau JP, et al. Neuropathic pain inhibitor, RAP-103, is a potent inhibitor of microglial CCL1/CCR8. Neurochem Int 2018, 119: 184–189.
Croitoru-Lamoury J, Guillemin GJ, Boussin FD, Mognetti B, Gigout LI, Chéret A, et al. Expression of chemokines and their receptors in human and Simian astrocytes: Evidence for a central role of TNFα and IFNγ in CXCR4 and CCR5 modulation. Glia 2003, 41: 354–370.
Illias AM, Gist AC, Zhang H, Kosturakis AK, Dougherty PM. Chemokine CCL2 and its receptor CCR2 in the dorsal root ganglion contribute to oxaliplatin-induced mechanical hypersensitivity. Pain 2018, 159: 1308–1316.
Menetski J, Mistry S, Lu M, Mudgett JS, Ransohoff RM, DeMartino JA, et al. Mice overexpressing chemokine ligand 2 (CCL2) in astrocytes display enhanced nociceptive responses. Neuroscience 2007, 149: 706–714.
Gosselin RD, Varela C, Banisadr G, Mechighel P, Rostene W, Kitabgi P, et al. Constitutive expression of CCR2 chemokine receptor and inhibition by MCP-1/CCL2 of GABA-induced currents in spinal cord neurones. J Neurochem 2005, 95: 1023–1034.
Jiang BC, He LN, Wu XB, Shi H, Zhang WW, Zhang ZJ, et al. Promoted interaction of C/EBPα with demethylated Cxcr3 gene promoter contributes to neuropathic pain in mice. J Neurosci 2017, 37: 685–700.
Yu Q, Tian DL, Tian Y, Zhao XT, Yang XY. Elevation of the chemokine pair CXCL10/CXCR3 initiates sequential glial activation and crosstalk during the development of bimodal inflammatory pain after spinal cord ischemia reperfusion. Cell Physiol Biochem 2018, 49: 2214–2228.
Kong YF, Sha WL, Wu XB, Zhao LX, Ma LJ, Gao YJ. CXCL10/CXCR3 signaling in the DRG exacerbates neuropathic pain in mice. Neurosci Bull 2021, 37: 339–352.
Loetscher M, Gerber B, Loetscher P, Jones SA, Piali L, Clark-Lewis I, et al. Chemokine receptor specific for IP10 and mig: Structure, function, and expression in activated T-lymphocytes. J Exp Med 1996, 184: 963–969.
Wu XB, He LN, Jiang BC, Shi H, Bai XQ, Zhang WW, et al. Spinal CXCL9 and CXCL11 are not involved in neuropathic pain despite an upregulation in the spinal cord following spinal nerve injury. Mol Pain 2018, 14: 174480691877740.
Sommer C, Leinders M, Üçeyler N. Inflammation in the pathophysiology of neuropathic pain. Pain 2018, 159: 595–602.
Montague K, Malcangio M. The therapeutic potential of targeting chemokine signalling in the treatment of chronic pain. J Neurochem 2017, 141: 520–531.
Nagelhus EA, Ottersen OP. Physiological roles of aquaporin-4 in brain. Physiol Rev 2013, 93: 1543–1562.
Nesic O, Lee J, Johnson KM, Ye Z, Xu GY, Unabia GC, et al. Transcriptional profiling of spinal cord injury-induced central neuropathic pain. J Neurochem 2005, 95: 998–1014.
Tackley G, Vecchio D, Hamid S, Jurynczyk M, Kong Y, Gore R, et al. Chronic neuropathic pain severity is determined by lesion level in aquaporin 4-antibody-positive myelitis. J Neurol Neurosurg Psychiatry 2017, 88: 165–169.
Bao F, Chen M, Zhang Y, Zhao Z. Hypoalgesia in mice lacking aquaporin-4 water channels. Brain Res Bull 2010, 83: 298–303.
Jha RM, Rani A, Desai SM, Raikwar S, Mihaljevic S, Munoz-Casabella A, et al. Sulfonylurea receptor 1 in central nervous system injury: An updated review. Int J Mol Sci 2021, 22: 11899.
Tsymbalyuk O, Gerzanich V, Mumtaz A, Andhavarapu S, Ivanova S, Makar TK, et al. SUR1, newly expressed in astrocytes, mediates neuropathic pain in a mouse model of peripheral nerve injury. Mol Pain 2021, 17: 17448069211006604.
Chen CC, Akopian AN, Sivilottit L, Colquhoun D, Burnstock G, Wood JN. A P2X purinoceptor expressed by a subset of sensory neurons. Nature 1995, 377: 428–431.
Chizh BA, Illes P. P2X receptors and nociception. Pharmacol Rev 2001, 53: 553–568.
McGaraughty S, Wismer CT, Zhu CZ, Mikusa J, Honore P, Chu KL, et al. Effects of A-317491, a novel and selective P2X3/P2X2/3 receptor antagonist, on neuropathic, inflammatory and chemogenic nociception following intrathecal and intraplantar administration. Br J Pharmacol 2003, 140: 1381–1388.
He YQ, Lang XQ, Lin L, Ji L, Yuan XY, Chen Q, et al. P2X3 receptor-mediated visceral hyperalgesia and neuronal sensitization following exposure to PTSD-like stress in the dorsal root Ganglia of rats. Neurogastroenterol Motil 2017, https://doi.org/10.1111/nmo.12976.
Hu S, Sun Q, Du WJ, Song J, Li X, Zhang PA, et al. Adult stress promotes purinergic signaling to induce visceral pain in rats with neonatal maternal deprivation. Neurosci Bull 2020, 36: 1271–1280.
Mah W, Lee SM, Lee J, Bae JY, Ju JS, Lee CJ, et al. A role for the purinergic receptor P2X3 in astrocytes in the mechanism of craniofacial neuropathic pain. Sci Rep 2017, 7: 13627.
Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH, et al. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med 2008, 14: 331–336.
Parks WC, Wilson CL, López-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nat Rev Immunol 2004, 4: 617–629.
Wang W, Gao W, Zhu Q, Alasbahi A, Seki E, Yang L. TAK1: A molecular link between liver inflammation, fibrosis, steatosis, and carcinogenesis. Front Cell Dev Biol 2021, 9: 734749.
Soto-Díaz K, Juda MB, Blackmore S, Walsh C, Steelman AJ. TAK1 inhibition in mouse astrocyte cultures ameliorates cytokine-induced chemokine production and neutrophil migration. J Neurochem 2020, 152: 697–709.
Katsura H, Obata K, Miyoshi K, Kondo T, Yamanaka H, Kobayashi K, et al. Transforming growth factor-activated kinase 1 induced in spinal astrocytes contributes to mechanical hypersensitivity after nerve injury. Glia 2008, 56: 723–733.
Suzuki Y, Nagai N, Yamakawa K, Kawakami J, Lijnen HR, Umemura K. Tissue-type plasminogen activator (t-PA) induces stromelysin-1 (MMP-3) in endothelial cells through activation of lipoprotein receptor-related protein. Blood 2009, 114: 3352–3358.
Echeverry R, Wu J, Haile WB, Guzman J, Yepes M. Tissue-type plasminogen activator is a neuroprotectant in the mouse hippocampus. J Clin Invest 2010, 120: 2194–2205.
Kozai T, Yamanaka H, Dai Y, Obata K, Kobayashi K, Mashimo T, et al. Tissue type plasminogen activator induced in rat dorsal horn astrocytes contributes to mechanical hypersensitivity following dorsal root injury. Glia 2007, 55: 595–603.
Kim G, Lim S, Kim KD. N-myc downstream-regulated gene 2 (NDRG2) function as a positive regulator of apoptosis: A new insight into NDRG2 as a tumor suppressor. Cells 2021, 10: 2649.
Zuo ZF, Liao YH, Ding T, Dong YL, Qu J, Wang J, et al. Astrocytic NDRG2 is involved in glucocorticoid-mediated diabetic mechanical allodynia. Diabetes Res Clin Pract 2015, 108: 128–136.
Li X, Luo P, Wang F, Yang Q, Li Y, Zhao M, et al. Inhibition of N-myc downstream-regulated gene-2 is involved in an astrocyte-specific neuroprotection induced by sevoflurane preconditioning. Anesthesiology 2014, 121: 549–562.
Xie Y, Su N, Yang J, Tan Q, Huang S, Jin M, et al. FGF/FGFR signaling in health and disease. Signal Transduct Target Ther 2020, 5: 181.
Xie KY, Wang Q, Cao DJ, Liu J, Xie XF. Spinal astrocytic FGFR3 activation leads to mechanical hypersensitivity by increased TNF-α in spared nerve injury. Int J Clin Exp Pathol 2019, 12: 2898–2908.
Lu Y, Jiang BC, Cao DL, Zhang ZJ, Zhang X, Ji RR, et al. TRAF6 upregulation in spinal astrocytes maintains neuropathic pain by integrating TNF-α and IL-1β signaling. Pain 2014, 155: 2618–2629.
Lu Y, Cao DL, Ma LJ, Gao YJ. TRAF6 contributes to CFA-induced spinal microglial activation and chronic inflammatory pain in mice. Cell Mol Neurobiol 2022, 42: 1543–1555.
Weng RX, Chen W, Tang JN, Sun Q, Li M, Xu X, et al. Targeting spinal TRAF6 expression attenuates chronic visceral pain in adult rats with neonatal colonic inflammation. Mol Pain 2020, 16: 1744806920918059.
Zhang P, Sun H, Ji Z. Downregulating lncRNA PVT1 relieves astrocyte overactivation induced neuropathic pain through targeting miR-186-5p/CXCL13/CXCR5 axis. Neurochem Res 2021, 46: 1457–1469.
Xu S, Dong H, Zhao Y, Feng W. Differential expression of long non-coding RNAs and their role in rodent neuropathic pain models. J Pain Res 2021, 14: 3935–3950.
Dong J, Xia R, Zhang Z, Xu C. lncRNA MEG3 aggravated neuropathic pain and astrocyte overaction through mediating miR-130a-5p/CXCL12/CXCR4 axis. Aging 2021, 13: 23004–23019.
Allen NJ, Lyons DA. Glia as architects of central nervous system formation and function. Science 2018, 362: 181–185.
Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J Neuroinflammation 2014, 11: 98.
Bezzi P, Domercq M, Vesce S, Volterra A. Neuron-astrocyte cross-talk during synaptic transmission: Physiological and neuropathological implications. Prog Brain Res 2001, 132: 255–265.
Hughes CE, Nibbs RJB. A guide to chemokines and their receptors. FEBS J 2018, 285: 2944–2971.
Miyoshi K, Obata K, Kondo T, Okamura H, Noguchi K. Interleukin-18-mediated microglia/astrocyte interaction in the spinal cord enhances neuropathic pain processing after nerve injury. J Neurosci 2008, 28: 12775–12787.
Hald A, Nedergaard S, Hansen RR, Ding M, Heegaard AM. Differential activation of spinal cord glial cells in murine models of neuropathic and cancer pain. Eur J Pain 2009, 13: 138–145.
Robinson CR, Zhang H, Dougherty PM. Astrocytes, but not microglia, are activated in oxaliplatin and bortezomib-induced peripheral neuropathy in the rat. Neuroscience 2014, 274: 308–317.
Zhang ZJ, Cao DL, Zhang X, Ji RR, Gao YJ. Chemokine contribution to neuropathic pain: Respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. Pain® 2013, 154: 2185–2197.
Cao DL, Zhang ZJ, Xie RG, Jiang BC, Ji RR, Gao YJ. Chemokine CXCL1 enhances inflammatory pain and increases NMDA receptor activity and COX-2 expression in spinal cord neurons via activation of CXCR2. Exp Neurol 2014, 261: 328–336.
Deng GC, Lu M, Zhao YY, Yuan Y, Chen G. Activated spinal astrocytes contribute to the later phase of carrageenan-induced prostatitis pain. J Neuroinflammation 2019, 16: 189.
Ni H, Wang Y, An K, Liu Q, Xu L, Zhu C, et al. Crosstalk between NFκB-dependent astrocytic CXCL1 and neuron CXCR2 plays a role in descending pain facilitation. J Neuroinflammation 2019, 16: 1–16.
Liu X, Bae C, Gelman BB, Chung JM, Tang SJ. A neuron-to-astrocyte Wnt5a signal governs astrogliosis during HIV-associated pain pathogenesis. Brain 2022: awac015.
Tsuda M, Kohro Y, Yano T, Tsujikawa T, Kitano J, Tozaki-Saitoh H, et al. JAK-STAT3 pathway regulates spinal astrocyte proliferation and neuropathic pain maintenance in rats. Brain 2011, 134: 1127–1139.
Oliva AA Jr, Kang Y, Sanchez-Molano J, Furones C, Atkins CM. STAT3 signaling after traumatic brain injury. J Neurochem 2012, 120: 710–720.
Simpson RJ, Hammacher A, Smith DK, Matthews JM, Ward LD. Interleukin-6: Structure-function relationships. Protein Sci 1997, 6: 929–955.
Metcalfe RD, Aizel K, Zlatic CO, Nguyen PM, Morton CJ, Lio DSS, et al. The structure of the extracellular domains of human interleukin 11α receptor reveals mechanisms of cytokine engagement. J Biol Chem 2020, 295: 8285–8301.
Pflanz S, Hibbert L, Mattson J, Rosales R, Vaisberg E, Bazan JF, et al. WSX-1 and glycoprotein 130 constitute a signal-transducing receptor for IL-27. J Immunol 2004, 172: 2225–2231.
Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 2009, 1: a000034.
Miao J, Zhou X, Ji T, Chen G. NF-κB p65-dependent transcriptional regulation of histone deacetylase 2 contributes to the chronic constriction injury-induced neuropathic pain via the microRNA-183/TXNIP/NLRP3 axis. J Neuroinflammation 2020, 17: 225.
Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, et al. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med 2005, 202: 145–156.
Liu T, Gao YJ, Ji RR. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci Bull 2012, 28: 131–144.
Song ZP, Xiong BR, Guan XH, Cao F, Manyande A, Zhou YQ, et al. Minocycline attenuates bone cancer pain in rats by inhibiting NF-κB in spinal astrocytes. Acta Pharmacol Sin 2016, 37: 753–762.
Kuno R, Yoshida Y, Nitta A, Nabeshima T, Wang J, Sonobe Y, et al. The role of TNF-alpha and its receptors in the production of NGF and GDNF by astrocytes. Brain Res 2006, 1116: 12–18.
Takebayashi H, Nabeshima Y, Yoshida S, Chisaka O, Ikenaka K, Nabeshima YI. The basic Helix-loop-Helix factor Olig2 is essential for the development of motoneuron and oligodendrocyte lineages. Curr Biol 2002, 12: 1157–1163.
Tatsumi K, Isonishi A, Yamasaki M, Kawabe Y, Morita-Takemura S, Nakahara K, et al. Olig2-lineage astrocytes: A distinct subtype of astrocytes that differs from GFAP astrocytes. Front Neuroanat 2018, 12: 8.
Tatsumi K, Kinugawa K, Isonishi A, Kitabatake M, Okuda H, Takemura S, et al. Olig2-astrocytes express neutral amino acid transporter SLC7A10 (Asc-1) in the adult brain. Mol Brain 2021, 14: 163.
Chen Y, Miles DK, Hoang T, Shi J, Hurlock E, Kernie SG, et al. The basic helix-loop-helix transcription factor olig2 is critical for reactive astrocyte proliferation after cortical injury. J Neurosci 2008, 28: 10983–10989.
Fujimoto M, Takagi Y, Muraki K, Nozaki K, Yamamoto N, Tsuji M, et al. RBP-J promotes neuronal differentiation and inhibits oligodendroglial development in adult neurogenesis. Dev Biol 2009, 332: 339–350.
Marumo T, Takagi Y, Muraki K, Hashimoto N, Miyamoto S, Tanigaki K. Notch signaling regulates nucleocytoplasmic Olig2 translocation in reactive astrocytes differentiation after ischemic stroke. Neurosci Res 2013, 75: 204–209.
Bejoy J, Bijonowski B, Marzano M, Jeske R, Ma T, Li Y. Wnt-Notch signaling interactions during neural and astroglial patterning of human stem cells. Tissue Eng Part A 2020, 26: 419–431.
Ahn SM, Byun K, Kim D, Lee K, Yoo JS, Kim SU, et al. Olig2-induced neural stem cell differentiation involves downregulation of Wnt signaling and induction of Dickkopf-1 expression. PLoS One 2008, 3: e3917.
Echeverry S, Shi XQ, Haw A, Liu H, Zhang ZW, Zhang J. Transforming growth factor-beta1 impairs neuropathic pain through pleiotropic effects. Mol Pain 2009, 5: 16.
Chen NF, Chen WF, Sung CS, Lu CH, Chen CL, Hung HC, et al. Contributions of p38 and ERK to the antinociceptive effects of TGF-β1 in chronic constriction injury-induced neuropathic rats. J Headache Pain 2016, 17: 72.
Jurga AM, Paleczna M, Kadluczka J, Kuter KZ. Beyond the GFAP-astrocyte protein markers in the brain. Biomolecules 2021, 11: 1361.
Schachtrup C, Ryu JK, Helmrick MJ, Vagena E, Galanakis DK, Degen JL, et al. Fibrinogen triggers astrocyte scar formation by promoting the availability of active TGF-beta after vascular damage. J Neurosci 2010, 30: 5843–5854.
Tzavlaki K, Moustakas A. TGF-β signaling. Biomolecules 2020, 10: 487.
Law AK, Gupta D, Levy S, Wallace DC, McKeon RJ, Buck CR. TGF-beta1 induction of the adenine nucleotide translocator 1 in astrocytes occurs through Smads and Sp1 transcription factors. BMC Neurosci 2004, 5: 1.
Buck CR, Jurynec MJ, Gupta DK, Law AKT, Bilger J, Wallace DC, et al. Increased adenine nucleotide translocator 1 in reactive astrocytes facilitates glutamate transport. Exp Neurol 2003, 181: 149–158.
Goldberg JS. Chronic opioid therapy and opioid tolerance: A new hypothesis. Pain Res Treat 2013, 2013: 407504.
Kuner R, Kuner T. Cellular circuits in the brain and their modulation in acute and chronic pain. Physiol Rev 2021, 101: 213–258.
Chen FL, Dong YL, Zhang ZJ, Cao DL, Xu J, Hui J, et al. Activation of astrocytes in the anterior cingulate cortex contributes to the affective component of pain in an inflammatory pain model. Brain Res Bull 2012, 87: 60–66.
Kim SK, Hayashi H, Ishikawa T, Shibata K, Shigetomi E, Shinozaki Y, et al. Cortical astrocytes rewire somatosensory cortical circuits for peripheral neuropathic pain. J Clin Invest 2016, 126: 1983–1997.
Okada-Ogawa A, Suzuki I, Sessle BJ, Chiang CY, Salter MW, Dostrovsky JO, et al. Astroglia in medullary dorsal horn (trigeminal spinal subnucleus caudalis) are involved in trigeminal neuropathic pain mechanisms. J Neurosci 2009, 29: 11161–11171.
Guo W, Wang H, Watanabe M, Shimizu K, Zou S, LaGraize SC, et al. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci 2007, 27: 6006–6018.
Koyama Y. Signaling molecules regulating phenotypic conversions of astrocytes and glial scar formation in damaged nerve tissues. Neurochem Int 2014, 78: 35–42.
Wang Y, Moges H, Bharucha Y, Symes A. Smad3 null mice display more rapid wound closure and reduced scar formation after a stab wound to the cerebral cortex. Exp Neurol 2007, 203: 168–184.
Racchetti G, D’Alessandro R, Meldolesi J. Astrocyte stellation, a process dependent on Rac1 is sustained by the regulated exocytosis of enlargeosomes. Glia 2012, 60: 465–475.
Ni HD, Xu LS, Wang Y, Li H, An K, Liu M, et al. Astrocyte activation in the periaqueductal gray promotes descending facilitation to cancer-induced bone pain through the JNK MAPK signaling pathway. Mol Pain 2019, 15: 1744806919831909.
Wahis J, Baudon A, Althammer F, Kerspern D, Goyon S, Hagiwara D, et al. Astrocytes mediate the effect of oxytocin in the central amygdala on neuronal activity and affective states in rodents. Nat Neurosci 2021, 24: 529–541.
Oberheim NA, Takano T, Han X, He W, Lin JHC, Wang F, et al. Uniquely hominid features of adult human astrocytes. J Neurosci 2009, 29: 3276–3287.
Loggia ML, Chonde DB, Akeju O, Arabasz G, Catana C, Edwards RR, et al. Evidence for brain glial activation in chronic pain patients. Brain 2015, 138: 604–615.
Hylands-White N, Duarte RV, Raphael JH. An overview of treatment approaches for chronic pain management. Rheumatol Int 2017, 37: 29–42.
Winer A, Adams S, Mignatti P. Matrix metalloproteinase inhibitors in cancer therapy: Turning past failures into future successes. Mol Cancer Ther 2018, 17: 1147–1155.
Dib P, Zhang Y, Ihnat MA, Gallucci RM, Standifer KM. TNF-alpha as an initiator of allodynia and anxiety-like behaviors in a preclinical model of PTSD and comorbid pain. Front Psychiatry 2021, 12: 721999.
Wang YS, Li YY, Cui W, Li LB, Zhang ZC, Tian BP, et al. Melatonin attenuates pain hypersensitivity and decreases astrocyte-mediated spinal neuroinflammation in a rat model of oxaliplatin-induced pain. Inflammation 2017, 40: 2052–2061.
Wen ZH, Huang SY, Kuo HM, Chen CT, Chen NF, Chen WF, et al. Fumagillin attenuates spinal angiogenesis, neuroinflammation, and pain in neuropathic rats after chronic constriction injury. Biomedicines 2021, 9: 1187.
Jiang W, Wang Y, Sun W, Zhang M. Morin suppresses astrocyte activation and regulates cytokine release in bone cancer pain rat models. Phytother Res 2017, 31: 1298–1304.
Lin YC, Huang SY, Jean YH, Chen WF, Sung CS, Kao ES, et al. Intrathecal lemnalol, a natural marine compound obtained from Formosan soft coral, attenuates nociceptive responses and the activity of spinal glial cells in neuropathic rats. Behav Pharmacol 2011, 22: 739–750.
Chen Z, Doyle TM, Luongo L, Largent-Milnes TM, Giancotti LA, Kolar G, et al. Sphingosine-1-phosphate receptor 1 activation in astrocytes contributes to neuropathic pain. Proc Natl Acad Sci USA 2019, 116: 10557–10562.
Gruber-Schoffnegger D, Drdla-Schutting R, Hönigsperger C, Wunderbaldinger G, Gassner M, Sandkühler J. Induction of thermal hyperalgesia and synaptic long-term potentiation in the spinal cord lamina I by TNF-α and IL-1β is mediated by glial cells. J Neurosci 2013, 33: 6540–6551.
Ringelstein M, Ayzenberg I, Harmel J, Lauenstein AS, Lensch E, Stögbauer F, et al. Long-term therapy with interleukin 6 receptor blockade in highly active neuromyelitis optica spectrum disorder. JAMA Neurol 2015, 72: 756–763.
Koyanagi S, Kusunose N, Taniguchi M, Akamine T, Kanado Y, Ozono Y, et al. Glucocorticoid regulation of ATP release from spinal astrocytes underlies diurnal exacerbation of neuropathic mechanical allodynia. Nat Commun 2016, 7: 13102.
Masuda T, Ozono Y, Mikuriya S, Kohro Y, Tozaki-Saitoh H, Iwatsuki K, et al. Dorsal horn neurons release extracellular ATP in a VNUT-dependent manner that underlies neuropathic pain. Nat Commun 2016, 7: 12529.
Wang H, Cao Y, Chiang CY, Dostrovsky JO, Sessle BJ. The gap junction blocker carbenoxolone attenuates nociceptive behavior and medullary dorsal horn central sensitization induced by partial infraorbital nerve transection in rats. Pain 2014, 155: 429–435.
Ren K, Torres R. Role of interleukin-1β during pain and inflammation. Brain Res Rev 2009, 60: 57–64.
Hanstein R, Hanani M, Scemes E, Spray DC. Glial pannexin1 contributes to tactile hypersensitivity in a mouse model of orofacial pain. Sci Rep 2016, 6: 38266.
Burma NE, Bonin RP, Leduc-Pessah H, Baimel C, Cairncross ZF, Mousseau M, et al. Blocking microglial pannexin-1 channels alleviates morphine withdrawal in rodents. Nat Med 2017, 23: 355–360.
Kohro Y, Matsuda T, Yoshihara K, Kohno K, Koga K, Katsuragi R, et al. Spinal astrocytes in superficial laminae gate brainstem descending control of mechanosensory hypersensitivity. Nat Neurosci 2020, 23: 1376–1387.
Akdemir ES, Woo J, Bosquez Huerta NA, Lozzi B, Groves AK, Harmanci AS, et al. Lunatic fringe-GFP marks Lamina-specific astrocytes that regulate sensory processing. J Neurosci 2022, 42: 567–580.
Xu Q, Ford NC, He S, Huang Q, Anderson M, Chen Z, et al. Astrocytes contribute to pain gating in the spinal cord. Sci Adv 2021, 7: 6287.
Acknowledgements
This review was supported by the Ministry of Science and Technology of China Brain Initiative Grant (2022ZD0204702), the National Natural Science Foundation of China (32030048 and 32100806), and the Natural Science Foundation of the Higher Education Institutions of Jiangsu Province (21KJB310010).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lu, HJ., Gao, YJ. Astrocytes in Chronic Pain: Cellular and Molecular Mechanisms. Neurosci. Bull. 39, 425–439 (2023). https://doi.org/10.1007/s12264-022-00961-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-022-00961-3