
Abstract
Tumor angiogenesis has been identified to play a critical role in tumor growth and tumor progression, and is regulated by a balance of angiogenic and antiangiogenic cytokines. This has been documented for solid tumors, and there is emerging evidence suggesting that tumor progression of hematological malignancies also depends on the induction of new blood vessel formation. Data on angiogenesis in the bone marrow of BCR-ABL1-negative myeloproliferative neoplasm patients suggest an increase of the microvessel density and vascular endothelial growth factor (VEGF) expression, and there is a relation to the JAK2-V617F status. The most important proangiogenic agent is VEGF, activating VEGF receptors 1 and 2. Inhibition of VEGF signaling by monoclonal antibodies or small molecules (kinase inhibitors) has already been successfully established for the treatment of different cancer entities, and multiple new drugs are being tested in clinical trials. Most patients with essential thrombocythemia (ET) or primary myelofibrosis (PMF) that was not associated with a JAK2 or MPL alteration carried a somatic mutation in calreticulin (CALR). Thus, CALR mutations should be included in the next classification system for ET/PMF. This review summarizes recent advances in the basic understanding of the role of angiogenesis in myeloproliferative neoplasms and the translation of such basic findings into clinical studies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
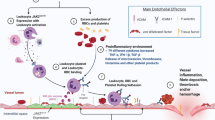
Angiogenesis, new blood vessel formation, is fundamental to tumor growth and spread [1]. Tumors require nutrients and oxygen to grow, and new blood vessels, formed by angiogenesis, provide these substrates [2]. The key mediator of angiogenesis is the vascular endothelial growth factor (VEGF). Its expression is regulated by a plethora of intrinsic and extrinsic factors, with hypoxia and hypoglycemia being the major stimuli [3]. Little is known about the exact trigger mechanisms of VEGF expression in hematolymphoid tumors, but mechanisms analogous to those observed in solid tumors are anticipated [4, 5]. Tight control of angiogenesis is maintained by a balance of endogenous antiangiogenic and proangiogenic factors. VEGF has a key, rate-limiting role in promoting tumor angiogenesis and exerts its effects by binding to one of three tyrosine kinase receptors: VEGF receptor-1 (VEGFR-1; fms-like tyrosine kinase-1, Flt-1), VEGFR-2 (human kinase domain region, KDR/murine fetal liver kinase-1, Flk-1), and VEGFR-3 (Flt-4) [6, 7].
The identification of an acquired somatic mutation in the JAK2 gene, resulting in a valine to phenylalanine substitution at position 617 (JAK2-V617F), has provided new insights into the pathogenesis of BCR-ABL1-negative myeloproliferative neoplasms (MPN), being present in most patients with polycythemia vera (PV) and in approximately 50 % of patients with essential thrombocythemia (ET) and primary myelofibrosis (PMF) [8, 9]. Although the molecular diagnostic gap in JAK2V617F-negative PV was adequately addressed by the discovery of other JAK2 mutations, the same had not been realized for the 35–40 % of patients with ET or PMF who do not express JAK2 mutations. In December 2013, two groups reported the occurrence of novel calreticulin (CALR) mutations in JAK2/MPL-unmutated PMF or ET [10, 11]. Both groups found mutual exclusivity between CALR, JAK2, and MPL mutations. In the study by Klampfl et al., CALR mutations were not seen in 382 cases of PV but were detected in 25 % of patients with ET (n = 311) and 35 % of those with PMF (n = 203).
Pathophysiology of angiogenesis in MPN
We and the others showed that angiogenesis and expression of VEGF and its receptors in the bone marrow of patients with BCR-ABL1-negative MPN is increased, especially in PMF [12–17]. We found significantly increased microvessel density (MVD), particularly that assessed by CD105, and VEGF expression in MPN compared with controls (PMF > PV > MDS/MPN > ET) [12]. We observed stronger association between CD105-MVD and VEGF expression, fibrosis, and JAK2-V617F mutant allele burden, compared with CD34-MVD. CD105-MVD probably better reflects angiogenic activity in MPN than CD34-MVD. CD105/endoglin is a cell transmembrane glycoprotein and a key component of transforming growth factor-b receptor complex [18]. It is mainly expressed by angiogenic endothelial cells and has been correlated with tumor MVD [19]. CD105 is preferentially expressed by newly formed vessels [20, 21], and thus, CD105-based MVD quantification better reveals ongoing proangiogenic activity. MVD was strongly increased in MPN with high JAK2-V617F mutant allele burden. Considering correlations between the JAK2-V617F status and angiogenesis in MPN, we showed that MVD, particularly if assessed by CD105, in PV and PMF patients correlate with a high JAK2-V617F mutant allele burden (≥ 55 % mutant alleles), but not with the diagnostic level for JAK2 mutations of 3 %. These observations implicate that higher activity of JAK2-related pathways, as observed in cases with higher JAK2-V617F mutant allele burdens, may influence angiogenesis in MPN. This assumption is further supported by our observation that VEGF+ cellular amounts did not concurrently rise with increasing JAK2-V617F mutant allele burden regardless of rising MVD [12].
Whether this observed increased neovascularization in neoplastic bone marrow is instrumental for the neoplastic process or rather an “innocent bystander” resulting from growth factors and cytokines being out of balance can only be determined by carefully designed experiments and clinical studies where inhibition of the angiogenic process does or does not inhibit the growth of the neoplasm.
Antiangiogenic therapy in myeloproliferative neoplasms
Antiangiogenic therapies are mostly based on inhibiting the binding of VEGF to VEGFR by neutralizing antibodies to the ligand or to the receptor, soluble receptors, or small molecule inhibitors or are directed against the tyrosine kinase activity of the VEGF receptors (Table 1) [22]. A growing list of antiangiogenics is now available, either in various stages of clinical development or as components of standard clinical regimens. The major classes of antiangiogenic therapy include (1) receptor tyrosine kinase inhibitors (TKI), targeting VEGFR signaling as well as receptors of other (proangiogenic) factors, (2) direct anti-VEGF acting molecules (anti-VEGF antibodies, VEGF-antisense nucleotides), and (3) immunomodulatory drugs (IMiDs) with antiangiogenic properties [22].
Tyrosine kinase inhibitors
Small TKI that target VEGFR are a further important class of antiangiogenic drugs with application to MPN, although their efficacy in hematolymphoid neoplasias might be attributable to inhibition of a variety of pathways. Small-molecule inhibitors targeting VEGFRs and other kinases, e.g., sorafenib (Nexavar®) and sunitinib (Sutent®), have been approved based on their efficacy in treating renal cell and hepatocellular carcinoma [23, 24]. Vatalanib is an oral angiogenesis inhibitor that is active against VEGFR and platelet-derived growth factor receptor tyrosine kinases, thereby offering a novel approach to inhibiting tumor growth [25] by interfering with the ATP-binding sites of VEGFR. In a phase I dose-escalation study, vatalanib, a VEGFR TKI, was administered to 29 PMF patients at doses of 500 or 750 mg bid [26]. One patient (3 %) achieved CR (complete remission) and five (17 %) achieved clinical improvement. Transient potentially vatalanib-related toxicities were mild nausea, vomiting, dizziness, fatigue, or thrombocytopenia. Cumulatively, this study indicated that vatalanib had modest activity in PMF patients.
Anti-VEGF antibodies
The first antiangiogenic agent to be approved in solid tumors was bevacizumab (Avastin®), a humanized anti-VEGF monoclonal antibody. Administration of bevacizumab, in combination with cytotoxic chemotherapy, conferred benefits to patients with several tumor entities like metastatic colorectal cancer, nonsquamous, non-small-cell lung cancer, and metastatic breast cancer [27, 28]. In a phase II study, bevacizumab monotherapy was tested in patients with symptomatic, relapsed/refractory PMF. A total of 13 patients were enrolled in the first stage of this two-stage trial. Among the 11 patients who received therapy, only 3 received more than 4 cycles of therapy; none of the patients achieved an objective response. Furthermore, significant toxicity, not directly related to the vascular or gastrointestinal events typically associated with the anti-VEGF monoclonal antibody preparation in other disease states, was observed. Lack of objective responses coupled with toxicity led to the decision to terminate the study early. The authors concluded that if future studies incorporate bevacizumab in combination therapy for myelofibrosis, more modest doses should be considered [29].
Immunomodulatory drugs
Thalidomide was originally introduced as sedative and withdrawn in the 1960s due to deleterious side effects. Recently, there is increasing evidence for the efficacy of thalidomide in cancer therapy. The surprising effects of thalidomide have led to the development of a series of IMiDs with even higher antiangiogenic potency [30, 31]. The interest on thalidomide as an antineoplastic agent rose after demonstration of its antiangiogenic activity in a rabbit model of corneal neovascularization that was induced in response to bFGF [32]. Thalidomide and the newer IMiDs (e.g., lenalidomide) have been shown to significantly decrease the expression of the proangiogenic factors VEGF and interleukin-6 in multiple myeloma [33]. The newer IMiDs were found to be two to three times more potent compared with thalidomide concerning antiangiogenic activity in various in vivo assays [34]. The antiangiogenic activity of IMiDs has been shown to be independent of their immunomodulatory effects [35]. In a recent study, the antiangiogenic effect of lenalidomide was studied in vitro in bone marrow endothelial cells of patients with multiple myeloma and in vivo using the chorioallantoic membrane assay. Lenalidomide exerted a relevant antiangiogenic effect in vivo at 1.75 μmol/L, a dose reached in interstitial fluids of patients treated with 25 mg/day. In vitro, lenalidomide inhibited angiogenesis and migration of multiple myeloma endothelial cells [36].
In a phase II study of 44 PMF patients, the efficacy of thalidomide monotherapy was assessed [37]. Of 41 evaluable patients receiving treatment for at least 15 days, 17 (41 %) showed a response. CR (complete remission) (without reversal of bone marrow fibrosis) was achieved in four patients (10 %) and partial response in four (10 %), and hematological improvements of anemia, thrombopenia, and/or splenomegaly were observed in nine patients (21 %). In phase II studies with lenalidomide monotherapy in patients with symptomatic PMF, the overall response rates were 22 % for anemia, 33 % for splenomegaly, and 50 % for thrombocytopenia [38]. In a combination study of lenalidomide with prednisone in 40 PMF patients [39], responses were recorded in 12 patients (30 %) and are ongoing in 10 (25 %), with a median time to response of 12 weeks. Three patients (7.5 %) had partial response and nine (22.5 %) had clinical improvement lasting a median of 18 months. Overall response rates were 30 % for anemia and 42 % for splenomegaly. Interestingly, all eight JAK2-V617F-positive responders also experienced a reduction of the baseline mutant allele burden. In a study with pomalidomide, a newer IMiD, in patients with MF, 94 patients were treated with pomalidomide with or without prednisone. Overall anemia response was 27 % and increased to 53 % in JAK2V617F-positive patients. Median duration of anemia response was 16 months. Treatment effect on splenomegaly was negligible [40].
JAK1 and JAK2 inhibitors
Ruxolitinib is a potent and selective inhibitor against both JAK1 and JAK2. It is the first approved JAK2 inhibitor for the treatment of myelofibrosis. The COMFORT-I trial assessed the activity of ruxolitinib at 15 or 20 mg orally twice daily in 309 patients with PMF, or with post-PV or post-ET MF, whereas COMFORT-II trial compared the activity of ruxolitinib in 219 patients with PMF or post-PV or post-ET MF against the best available therapy (BAT). The proportion of patients with at least a 35 % reduction in spleen volume was detected by either magnetic resonance imaging (MRI) or computed tomography at 24 (COMFORT-I) or 48 (COMFORT-II) weeks of therapy. In COMFORT-I, the reduction in spleen volume was observed in 41.9 % of patients taking ruxolitinib compared with 0.7 % taking placebo; the proportion of patients with a reduction of 50 % or more in the total symptom score from baseline to week 24 was 45.9 % in the ruxolitinib group versus 5.3 % in the placebo group [41, 42]. In COMFORT-II, the reduction in spleen volume was observed in 28 % of patients on ruxolitinib compared with 0 % on BAT after a 48-week follow-up period [41, 42]. The 3-year follow-up data with ruxolitinib in patients with myelofibrosis showed, in the ruxolitinib arm, with continued therapy, spleen volume reductions of ≥ 35 % by MRI were sustained for at least 144 weeks, with the probability of 50 %. A total of 45 % of the patients randomized to ruxolitinib remained on treatment, and ruxolitinib continues to be well tolerated. Anemia and thrombocytopenia were the main toxicities, but they were generally manageable, improved over time, and rarely led to treatment discontinuation. Additionally, patients randomized to ruxolitinib showed longer overall survival than those randomized to BAT [43].
Conclusions
Angiogenic and especially VEGF/VEGFR pathways are possibly involved in the pathophysiology of MPN. VEGF/VEGFR interactions can stimulate proliferation, migration, and survival of leukemia/lymphoma cells by autocrinous and paracrinous loops. Novel agents, targeting VEGF, its receptors, and other angiogenic pathways, are in various stages of clinical development and investigation in MPN. As we know from the treatment of solid tumors, combination therapies of different antiangiogenic molecules with chemotherapy or irradiation increases treatment efficacy. However, not all patients treated with antiangiogenic therapies benefit from this kind of therapy, and in most cases, the effect is transient. It has to be kept in mind that most of these studies used anti-VEGF monotherapy to treat heavily pretreated, refractory, or relapsed patients. The molecular diagnostic gap in JAK2/MPL-unmutated ET/PMF is now partially addressed by the recent discovery of CALR mutations in the majority of such cases.
Take-home message
-
Angiogenesis is possibly involved in the pathophysiology of myeloproliferative neoplasms (MPN).
-
New molecular markers like calreticulin provide additional diagnostic and prognostic tools in MPN wild-type for JAK2 and MPL.
Conflict of interest
Michael Medinger and Jakob Passweg declare that they have no conflict of interest.
References
Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1(1):27–31.
Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–6.
Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359(6398):843–5.
Dong X, Han ZC, Yang R. Angiogenesis and antiangiogenic therapy in hematologic malignancies. Crit Rev Oncol Hematol. 2007;62(2):105–18.
Frater JL, Kay NE, Goolsby CL, Crawford SE, Dewald GW, Peterson LC. Dysregulated angiogenesis in B-chronic lymphocytic leukemia: morphologic, immunohistochemical, and flow cytometric evidence. Diagn Pathol. 2008;3:16.
Gille H, Kowalski J, Li B, et al. Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J Biol Chem. 2001;276(5):3222–30.
Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9(6):669–76.
Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–90.
Kralovics R, Teo SS, Li S, Theocharides A, et al. Acquisition of the V617F mutation of JAK2 is a late genetic event in a subset of patients with myeloproliferative disorders. Blood. 2006;108(4):1377–80.
Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–90.
Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med. 2013;369(25):2391–405.
Medinger M, Skoda R, Gratwohl A, et al. Angiogenesis and vascular endothelial growth factor-/receptor expression in myeloproliferative neoplasms: correlation with clinical parameters and JAK2-V617F mutational status. Br J Haematol. 2009;146(2):150–7.
Lundberg LG, Lerner R, Sundelin P, Rogers R, Folkman J, Palmblad J. Bone marrow in polycythemia vera, chronic myelocytic leukemia, and myelofibrosis has an increased vascularity. Am J Pathol. 2000;157(1):15–9.
Wrobel T, Mazur G, Surowiak P, Wolowiec D, Jelen M, Kuliczkowsky K. Increased expression of vascular endothelial growth factor (VEGF) in bone marrow of patients with myeloproliferative disorders (MPD). Pathol Oncol Res. 2003;9(3):170–3.
Boveri E, Passamonti F, Rumi E, et al. Bone marrow microvessel density in chronic myeloproliferative disorders: a study of 115 patients with clinicopathological and molecular correlations. Br J Haematol. 2008;140(2):162–8.
Gianelli U, Vener C, Raviele PR, et al. VEGF expression correlates with microvessel density in Philadelphia chromosome-negative chronic myeloproliferative disorders. Am J Clin Pathol. 2007;128(6):966–73.
Medinger M, Fischer N, Tzankov A. Vascular endothelial growth factor-related pathways in hemato-lymphoid malignancies. J Oncol. 2010;2010:729725.
Gougos A, Letarte M. Primary structure of endoglin, an RGD-containing glycoprotein of human endothelial cells. J Biol Chem. 1990;265(15):8361–4.
Dallas NA, Samuel S, Xia L, et al. Endoglin (CD105): a marker of tumor vasculature and potential target for therapy. Clin Cancer Res. 2008;14(7):1931–7.
Fonsatti E, Del Vecchio L, Altomonte M, et al. Endoglin: an accessory component of the TGF-beta-binding receptor-complex with diagnostic, prognostic, and bioimmunotherapeutic potential in human malignancies. J Cell Physiol. 2001;188(1):1–7.
Warrington K, Hillarby MC, Li C, Letarte M, Kumar S. Functional role of CD105 in TGF-beta1 signalling in murine and human endothelial cells. Anticancer Res. 2005;25(3B):1851–64.
Medinger M, Mross K. Clinical trials with anti-angiogenic agents in hematological malignancies. J Angiogenes Res. 2010;2:10.
Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125–34.
Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356(2):115–24.
Mross K, Drevs J, Müller M, et al. Phase I clinical and pharmacokinetic study of PTK/ZK, a multiple VEGF receptor inhibitor, in patients with liver metastases from solid tumours. Eur J Cancer. 2005;41(9):1291–9.
Giles FJ, List AF, Carroll M, et al. PTK787/ZK 222584, a small molecule tyrosine kinase receptor inhibitor of vascular endothelial growth factor (VEGF), has modest activity in myelofibrosis with myeloid metaplasia. Leuk Res. 2007;31(7):891–7.
Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–42.
Sandler A, Gray R, Perry MC, et al. Paclitaxel-carboplatin alone or with bevacizumab for nonsmall-cell lung cancer. N Engl J Med. 2006;355(24):2542–50.
Mesa RA, Silver RT, Verstovsek S, et al. Single agent bevacizumab for myelofibrosis: results of the myeloproliferative disorders research consortium trial. Haematologica. 2013;98(9):1421–3.
Singhal S, Mehta J, Desikan R, et al. Antitumor activity of thalidomide in refractory multiple myeloma. N Engl J Med. 1999;341(21):1565–71.
Moncada B, Baranda ML, Gonzalez-Amaro R, Urbina R, Loredo CE. Thalidomide-effect on T cell subsets as a possible mechanism of action. Int J Lepr Other Mycobact Dis. 1985;53(2):201–5.
D’Amato RJ, Loughnan MS, Flynn E, Folkman J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci U S A. 1994;91(9):4082–5.
Gupta D, Treon SP, Shima Y, et al. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: therapeutic applications. Leukemia. 2001;15(12):1950–61.
Teo SK. Properties of thalidomide and its analogues: implications for anticancer therapy. AAPS J. 2005;7(1):E14–9.
Dredge K, Marriott JB, Macdonald CD, et al. Novel thalidomide analogues display antiangiogenic activity independently of immunomodulatory effects. Br J Cancer. 2002;87(10):1166–72.
De Luisi AF, Coluccia AM, et al. Lenalidomide restrains motility and overangiogenic potential of bone marrow endothelial cells in patients with active multiple myeloma. Clin Cancer Res. 2011;17(7):1935–46.
Thomas DA, Giles FJ, Albitar M, et al. Thalidomide therapy for myelofibrosis with myeloid metaplasia. Cancer. 2006;106(9):1974–84.
Tefferi A, Cortes J, Verstovsek S, et al. Lenalidomide therapy in myelofibrosis with myeloid metaplasia. Blood. 2006;108(4):1158–64.
Quintás-Cardama A, Kantarjian HM, Manshouri T, et al. Lenalidomide plus prednisone results in durable clinical, histopathologic, and molecular responses in patients with myelofibrosis. J Clin Oncol. 2009;27(28):4760–6.
Begna KH, Pardanani A, Mesa R, et al. Long-term outcome of pomalidomide therapy in myelofibrosis. Am J Hematol. 2012;87(1):66–8.
Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807.
Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–98.
Cervantes F, Vannucchi AM, Kiladjian JJ, et al. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013;122(25):4047–53.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Medinger, M., Passweg, J. Angiogenesis in myeloproliferative neoplasms, new markers and future directions. memo 7, 206–210 (2014). https://doi.org/10.1007/s12254-014-0142-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12254-014-0142-z