
Abstract
As an energy-intensive industry, the chlor-alkali process has caused numerous environmental issues due to heavy electricity consumption and pollution. Chlor-alkali industry has been upgraded from mercury, diaphragm electrolytic cell, to ion exchange membrane (IEM) electrolytic cells. However, several challenges, such as the selectivity of the anodic reaction, sluggish kinetics of alkaline hydrogen evolution, degradation of membranes, the reasonable design of electrolytic cell structure, remain to be addressed. For these reasons, this paper mainly reviews the research progress of the chlor-alkali industry from materials to devices, including hydrogen evolution anode, chlorine evolution cathode, IEM, and electrolytic cell system. Finally, the research directions and prospects in the chlor-alkali industry are proposed for its further improvement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The chlor-alkali process is one of the most basic chemical industries, mainly producing chlorine (Cl2) and sodium hydroxide (NaOH). The global annual production of chlorine exceeds 75 million tons. Each ton of chlorine consumes about 2200–2600 kW∙h of electricity, and the global chlor-alkali industry needs to consume over 150 TW∙h of electricity every year, accounting for about 10% of global electricity [1]. Chlorine has been used in a variety of applications, including the production of building materials such as polyvinyl chloride, organic synthesis, metallurgy, water treatment [2], and the manufacture of titanium dioxide [3]. NaOH is also a common chemical raw material that is widely used in the production of detergents, herbicides, pesticides, medicines, plastics, and soaps. However, the current chlor-alkali process is one of the industries with high energy consumption, releasing large amounts of pollutants and causing serious environmental problems. Therefore, energy saving and emission reduction in the chlor-alkali production process are important development directions.
The chlor-alkali industry has experienced a long development process from mercury, diaphragm electrolytic cells to ion exchange membrane (IEM) electrolytic cells. At present, membrane electrolytic cell technology accounts for approximately 81% of the global chlor-alkali capacity [4]. In mercury electrolysis cells, mercury as the liquid cathode reacts with sodium ions to form sodium amalgam (Na+ + Hg + e− → ∙NaHg), whereas Cl2 is formed on the anode. Then, NaHg is transferred to another electrolytic cell to release Na+ (NaHg → Na+ + Hg + e−), forming NaOH in the electrolyte [5, 6]. A diaphragm is introduced to separate the anode and cathode reactions to prevent the cathode product (NaOH) and the anode product (Cl2) from crossing over to generate sodium hypochlorite. The negatively charged membrane can inhibit the back diffusion of OH− [7]. Therefore, in diaphragm electrolyzers, asbestos felt is often used to separate the products (Cl2 and H2) [8, 9], although the chemical instability of asbestos itself causes the severe swelling of pure asbestos diaphragm under a high current load [7]. Therefore, these two technologies above rely on highly toxic mercury and asbestos respectively, resulting in serious environmental pollution [1, 10].
Membrane electrolysis cells use highly conductive IEMs instead of traditional asbestos felt to separate Cl2 and H2, which greatly reduces the operating voltage and pollutant emissions. Compared with diaphragm electrolytic cells, the membrane counterpart offers high-purity sodium hydroxide. Thus, the replacement of the diaphragm method with ion membrane technology is an inevitable trend. The perfluorinated membrane method is currently recognized as the most energy-efficient chlor-alkali process in the world. A cation-selective permeation membrane is placed between the anode and cathode compartments, and a saturated sodium chloride solution is injected into the anode compartment, which usually produces 32%–35% caustic soda (Fig. 1). This membrane rejects the passage of chloride ions (negatively charged) but allows sodium ions (positively charged) to pass through. The first-generation membrane can only work at low corrosive concentrations, whereas the state-of-the-art membrane is made of a perfluorosulfonic acid (PFSA) polymer layer, where a polytetrafluoroethylene (PTFE)-reinforced fabric and a perfluorocarboxylic acid (PFCA) polymer are bonded together. This membrane has low electrical resistance and high ion-selective permeability and can work in a highly corrosive electrolyte environment.

Schematic of the working principle for a renewable electricity-powered chlor-alkali electrolyzer
Hydrogen, which is usually treated as exhaust gas in the chlor-alkali industry, may become the major fuel used on a large scale in the future as the most promising clean energy. The chlor-alkali electrolyzer can be used as one of the most promising low-cost hydrogen sources in the near term, better than water electrolyzer, considering that the value of chlorine gas and chemical NaOH produced in the former technology is notably higher than that of the oxygen gas produced in the latter. Further coupled with cheap and clean electricity generated from renewable energy, chlor-alkali electrolyzers will become a promising green hydrogen production technique.
In general, all chlor-alkali reactions include three reactions: chlorine evolution reaction (CER) at the anode, hydrogen evolution reaction (HER) at the cathode, and NaOH generation in the electrolyte. Figure 1 shows a chlor-alkali electrolysis cell. The chlor-alkali electrolyzer is mainly composed of three parts: anode, cathode, and IEM. Although the energy consumption of state-of-the-art chlor-alkali electrolyzers has been greatly reduced with technological progress, all of its structural parts still need to be further optimized.
An in-depth understanding of the selectivity of the anode will help to further develop inexpensive, stable, and efficient chlorine evolution catalysts. The slow hydrogen evolution kinetics caused by the alkaline condition of the cathode is one of the main reasons for its high energy consumption. Therefore, the research on the alkaline HER cathode of chlor-alkali electrolysis cells is important to improve the efficiency of electric energy utilization. In addition, the use of oxygen depolarized cathodes (ODCs) to reduce the energy consumption of chlor-alkali electrolysis cells has achieved certain results, but their potential remains to be evaluated. An in-depth understanding of IEMs can not only guide specific industrial operating conditions and optimize the overall structural design of electrolytic cells to extend the service life of the membrane but also provide a basis for the design of high-performance membrane materials. In addition to the various structural units of the chlor-alkali electrolysis cell, the entire electrolysis system can still be optimized.
Cathode
In conventional chlor-alkali electrolyzers, HER occurs at the cathode, which can be employed as one of the most promising low-cost, green hydrogen-making technology. HER is complicated in the strong alkaline solution in chlor-alkali electrolyzers compared with the reaction in acidic solutions. First, the cathode materials need to withstand the highly corrosive alkaline conditions. Moreover, the slow kinetics of HER under alkaline conditions is the main reason for the high energy consumption. Thus, the design of highly efficient electrocatalysts under strongly basic conditions is the key to achieving low-energy chlor-alkali electrolyzers for green hydrogen production.
Hydrogen Energy
Hydrogen is a clean fuel and is one of the most promising energy carriers to replace traditional fossil fuels, possessing high heat value (120 MJ/kg) and no exhaust emissions (only produces water as by-products) at the point of use. Currently, hydrogen gas is mainly produced through petrifaction, such as the gasification of coal [11], natural gas steam reforming [12], and methanol cracking [13], with about 4% of global hydrogen produced from water electrolysis [14].
In a typical gasification process, coal is first gasified and then undergoes carbon monoxide vapor conversion and pressure swing adsorption purification to produce hydrogen. This process is less preferred due to the high temperature required, the pollution created, and complex operation. The most common method used in hydrogen production is the methane steam reforming process, which takes up 50% of the hydrogen produced worldwide [15]. This process is similar to the gasification of coal, and thus, the drawbacks are the same. Despite the low cost of hydrogen production from fossil fuels, the heavy carbon footprint and large investment in hardware equipment make them unattractive for future use. Three ways are used to produce hydrogen from methanol: methanol cracking, methanol-steam reforming, and partial oxidation of methanol [13]. Among these methods, methanol cracking is the most competitive due to its wider application range and single feedstock. Its advantage includes lower investment cost and less energy consumption than coal and natural gas. However, the cost of methanol feedstock is high, which results in a high unit cost of hydrogen production.
As for water electrolysis, the hydrogen production cost is the highest among different methods mainly due to the sluggish oxygen evolution reaction (OER) [16], which is estimated to be between 2.5 and 3.5 ¥/Nm3 hydrogen at the current price of electricity. Water electrolyzers can become a common method to produce hydrogen if the electricity price is decreased considerably by maturing wind and hydroelectric power supplies [17]. The chlor-alkali electrolyzer is potentially one of the most promising low-cost hydrogen-making technology, better than water electrolyzer, considering that the value of chlorine gas and chemical NaOH produced in the former technology is substantially higher than the oxygen gas produced by the latter [18, 19].
In modern chlor-alkali plants, unlike the products, namely, chlorine and sodium hydroxide, hydrogen is a by-product. In the USA, approximately 40%–50% of hydrogen from the chlor-alkali process is combusted for process heat, with 30%–40% sold to the merchant hydrogen market and the remainder (10%–30%) vented [20]. In China, nearly 1.8 billion m3 hydrogen is wasted in industrial chlor-alkali process [21]. Currently, the main applications for hydrogen produced in chlor-alkali electrolyzers include: (1) feedstock gas for the production of ammonia [22]; (2) hydrocracking and other petroleum processing industries [23]; (3) chemical and organic fine chemical synthesis; (4) production of high-purity compressed hydrogen for direct sale (for electronics, metallurgy, glass, and other industries); (5) fuel for steam production; (6) hydrogen fuel cell technology for the construction of hydrogen power plants [24]. Several companies have bottled and marketed hydrogen gas produced in chlor-alkali plants, which is then supplied to fuel cells [25]. Hydrogen fuel cells can be well coupled with chlor-alkali plants, which can recover 20% of the electrical energy and 10% of the thermal energy consumed in chlor-alkali electrolysis [26].
HER Fundamentals
Historically, HER is the most studied, particularly in acidic solutions. By contrast, limited research has been conducted on HER in alkaline solutions, that is, the operation condition of chlor-alkali electrolyzers, mainly due to the complicated reaction mechanism. The possible pathway of HER in highly alkaline media includes the following steps: electro-reduction of water molecules and hydrogen adsorption (Volmer reaction, Step 1); electrochemical desorption of hydrogen (Heyrovsky reaction, Step 2); chemical desorption (Tafel reaction, Step 3).
HER kinetics in alkaline solution is extremely sluggish and is usually 2–3 orders of magnitude lower than that of acidic media. The pH-dependent hydrogen binding energy (HBE) derived by cyclic voltammetry is considered a reasonable descriptor. HBE is generally considered to be an inherent property of every metal and is independent of pH. However, the electrode surface is covered with water. Thus, the adsorption/desorption of hydrogen is possibly accompanied by the desorption/resorption of water. Cheng et al. [27] attributed pH dependence to the changes in water adsorption energy, given that the hydrophobicity of Pt electrodes increases as the potential becomes negative. Therefore, water adsorption or surface hydrophilicity must be tuned by alloying or introducing hydrophilic surface groups (such as OH). In addition, several uncertain properties of the interface, such as the Schottky barrier [28] or mismatched lattice space [29], can hinder electron transfer and the transfer of reaction intermediates between the two phases, causing additional kinetic obstacles.
HER Catalysts
So far, platinum offers the most superior HER performance, but its limited abundance hinders its industrial applications. In practical chlor-alkali electrolyzers, low-carbon steel is employed as cathode material instead of platinum. Two main issues need to be addressed with the use of low-carbon steel. First, during the electrolysis process, the cathode surface becomes gradually covered by chromium oxide (Cr2O3), resulting in “blind current” loss. This condition will result in a large increase in overpotential and ultimately the precipitation of Ca and Mg products. Further, the steel cathode has a high corrosion rate due to the thermodynamic instability of iron in the alkaline solution. Nickel is also the most common catalyst used as cathode material in alkaline media, such as Ni foam. The complete replacement of platinum with low-cost active catalytic materials is one of the most important goals of modern electrocatalysis.
Meanwhile, numerous new materials have also been investigated for HER: (i) noble metals, such as Pd, Ir, Ru, Ag, etc.; (ii) inexpensive transition metal catalysts, including Fe, Co, Ni, Mn, Cu, Mo, and W, and their heterogamous nanostructures; (iii) non-metal catalysts, including B, C, N, P, and S [30]. These results have been summarized in the previous references [31, 32]. Nitrogen-doped transition metal materials exhibit a tunable local electronic structure and fast electron transfer capability. Lei et al. [33] developed an atomically dispersed Ni-anchored nitrogen-doped porous carbon (Ni–NC) matrix, which gave a minimum overpotential of ~ 150 mV at a current density of 10 mA/cm2. Experimental observations confirmed that the strong chemical coupling between Ni nanoparticles and Ni–NC regulates the electronic structure, promotes electron transfer on the constructed interface, and optimizes the process of hydrolysis and OH− adsorption. Nitrogen doping can effectively improve the alkaline HER activity. This atomic-level modulation strategy opens up a new way for the development of practical electrocatalysts using three-dimensional metal oxides in alkaline environments. Sun et al. [34] used N atoms to regulate ReS2. As shown in Fig. 2, the injection of N atoms can effectively promote electron transfer from Re to S atoms, leading to the redistribution of electrons, rendering Re atoms in an electron-deficient state, and increasing the hydrophilicity of O. The preferential sites of adsorption activation optimize the H adsorption/desorption process, increase the electronic state near the Fermi level of the material, and improve the charge transfer.

Schematic of the HER mechanism of a ReS2 and b N-ReS2 under alkaline conditions (reprinted from [34] with permission from RSC)
ODC
One of the important developments in the chlor-alkali industry is the introduction of ODC, which introduces oxygen into the electrolysis chamber through a porous cathode. Figure 3 shows the ODC electrolysis cell. If the oxygen reduction reaction (ORR) [35] at the ODC cathode is used to replace the HER in the traditional process, the electrical energy demand can theoretically be reduced by up to 30% [36], and the operating voltage can be reduced by approximately 1.0 V, as shown in Eqs. (4) and (5). The ODC process has also been widely used in the HCl electrolysis scenario, which consumes downstream HCl by-products to achieve chlorine recovery [37].

Comparison of chlor-alkali electrolysis with different active cathodes. a Hydrogen evolving cathode. b ODC (reprinted from [38] with permission from ACS)
ODC Electrode Structure and Working Principle
ODC is a gas diffusion electrode (GDE) that can significantly increase the reaction current densities [39]. The industrial ODC consists of a porous structure of silver particles and a PTFE supporting layer. The typical operating conditions include 10 M NaOH, temperature range from 80 °C to 90 °C [40], and current density of 400–600 mA/cm2 [41].
The ODC performance is related to the diffusion rate of oxygen and water activity [42]. Under industrial operating conditions, ORR only occurs near the gas–liquid interface [35, 43]. Given the water consumption and hydroxide ion accumulation, the phase balance changes significantly with the current density applied and oxygen solubility in the liquid electrolyte. This effect is enhanced at high reaction rates, which leads to complete depletion of oxygen and achievement of a limiting current. Röhe et al. [44] established a dynamic three-phase model of advanced chlor-alkali electrolysis porous ODC, which proved that water transport, especially that of hydroxide ions in the liquid electrolyte, is the overall limiting factor. The oxygen concentration is rapidly consumed near the gas–liquid interface [45]. Considering the non-ideal behavior of highly concentrated water, the water activity coefficient largely depends on ion concentration [46]. As sodium hydroxide concentration increases, water activity will decrease due to the formation of a solvated shell that combines water molecules with ions. For example, the highly concentrated electrolyte binds considerable water to the solvation shell of solute molecules and thus, decreases the abundance of free water molecules [47]. The decrease in water activity and oxygen solubility is also accompanied by the decrease in oxygen mass transport rate [48].
Research has focused on the correlation between the structural parameters of the porous gas diffusion layers (GDLs) and ORR performances. GDLs with large pores may promote gas supply and further facilitate the mass transport rate of oxygen to achieve an excellent electrode performance [49, 50]. Pore volume is another factor that determines the electrode performance [51]. Nara et al. [52] employed electrochemical impedance spectroscopy to further determine the correlation between the ODC performance and the pore structures, including the primary and secondary pores within the catalyst layer. Park and Popov [53] studied the influence of PTFE content of ODC on electrode activity and observed the volcanic dependence of PTFE content in the ODC performance.
ODC Electrode Materials
-
1.
Carbon-supported transition metal catalysts
Pt/C (usually 20 wt%) is a commonly used catalyst in fuel cells and has good activity and stability in acidic media [54,55,56]. However, metallic Pt is easily corroded by Cl ions; thus, a large amount of noble Pt will dissolve in the highly corrosive saturated Cl2 electrolyte [57]. The carbon-based rhodium-based chalcogenide RhxSy/C developed by E-TEK is the only ODC catalyst that maintains activity under concentrated Cl2 electrolyte, but this catalyst has low intrinsic activity [58]. Most of the existing carbon materials are dual-functional catalysts for ODC and chlorine evolution in hydrochloric acid electrolysis, such as CNx [59], Co-NSCx [60], Fe–N–C [58], etc. The main goal is to improve the tolerance to Cl ions. However, overall activity still needs to be improved.
-
2.
Silver catalysts
Under the harsh operating conditions of chlor-alkali electrolysis, carbon materials are easily corroded and suffer from low stability. Alternatively, silver exhibits an oxygen reduction activity similar to that of platinum under strongly alkaline conditions [61]; thus, silver can be used as a suitable substitute for noble platinum catalysts.
Optimizing silver-based GDEs will reduce energy consumption and enhance the long-term stability of chlor-alkali electrolyzers. GDEs are composed of silver particles, a complex pore system around them, and PTFE at the boundaries of the silver particles [62]. Figure 4 shows the three-phase interface and GDE electrode. When the hydrophilic silver particles support the penetration of the electrolyte, the pore channels containing hydrophobic PTFE keep the gas phase unobstructed. The existing GDE electrode is improved mainly through the increase in the three-phase interface (catalyst material, alkaline electrolyte, and O2-rich gas phase), thereby increasing the utilization rate of the catalyst. However, adjusting the three-phase boundary can be a complex task because the distribution of the electrolyte in the porous structure is still unknown. To address this issue, Paulisch et al. [48] used X-ray computed tomography to study the electrolyte infiltration and distribution during electrolysis. The results showed a potential-dependent electrolyte transport in the ODC and droplet formation on the gas diffusion side. Given the importance of determining the electrolyte distribution inside the porous structure, further research on in situ imaging technology is needed to accurately link the structural and electrochemical performances.

Schematic of three-phase interface and GDE structure (reprinted from [62] with permission from RSC)
Anode
Anode Reaction
In chlor-alkali electrolyzers, CER is the desirable reaction at the anode. The current efficiency of chlorine is the main measure of anode selectivity, whereas the OER is a side reaction that competes with CER. OER occurs directly at the anode, where water is oxidized to generate oxygen [16, 63]. The selectivity of chlorine and oxygen evolution is related to the inherent selectivity of anode materials, process conditions, and electrolyte compositions.
The anode reaction is mainly studied by conventional electrochemical techniques (such as voltammetry and polarization techniques), but theoretical methods are also used to study the intrinsic selectivity [13,14,15]. The composition of the evolved gas is usually determined by gas chromatography or O2 absorption method, such as that which uses pyrogallol. The concentrations of hypochlorite, chloride, and chlorate can be determined by titration of the electrolyte. In addition, differential electrochemical mass spectrometry can be used to measure the reaction products formed on the anode with high sensitivity [64,65,66].
CER Fundamentals
Equations (6) and (7), respectively, list two competing reactions on the anode: CER and OER. The four-electron transfer in OER is thermodynamically more favorable and can compete with CER, although the two-electron transfer CER exhibits faster kinetics [67]. The excess oxygen and hydrogen products can possibly be combined in the cell to form an explosive gas mixture, which renders oxygen a safety hazard [68]. Therefore, chlorine selectivity must be improved.
Numerous factors, including electrolytes, anode materials, and operating conditions, such as current densities and temperature, affect the selectivity of chlorine [68, 70]. Acidic conditions are favorable for CER, whereas alkaline conditions are favorable for OER [68]. The pH correlation between OER and CER can be represented by the phase diagram of the catalyst surface intermediates in equilibrium with Cl− (Fig. 5). Unlike the OER, which is a pH-dependent reaction, CER is pH independent. Furthermore, increasing the current density can increase chlorine selectivity, which is related to the suppression of OER in low local pH.

Surface phase diagram for IrO2(110) in equilibrium with Cl−, H+, and H2O at 298.15 K and aCl− = 1. The adsorbate phases are shown in the insets. Ir atoms are cyan, O atoms are red, H atoms are white, and Cl atoms are green (reprinted from [69] with permission from RSC)
Anode Electrode Material
Electrode materials with high activity for chlorine are usually also highly active for oxygen. Although these competing reactions are governed by the same factors, chlorine selectivity can still be optimized. The two reactions occur at a similar active site or form a common surface intermediate species, whereas CER and OER produce two competing intermediate products, namely, OCl and OOH, respectively [71].
Commercial Dimensionally Stable Anode (DSA) Electrodes
DSAs, which are mainly composed of noble metal Ir or Ru oxides, are currently the most advanced CER catalysts. The innovation of DSAs is called “one of the greatest technological breakthroughs in the field of electrochemistry in the past 50 years” [72]. Commercial DSA electrodes usually contain one or more other doping materials, such as TiO2, SnO2, CoOx, and SbOx, to reduce the amount of precious Ir or Ru [73,74,75]. Modern DSAs can work at current industrial densities for more than 10 years [72]. The surface of commercial DSAs is coated with an electrocatalytic layer containing a mixture of titanium dioxide and ruthenium dioxide with an atomic ratio of about 70: 30 and a CER selectivity of about 95% [76].
To investigate the CER process at the DSA electrode, Hansen et al. [69] constructed the Pourbaix diagram of IrO2 and RuO2 based on density functional theory (DFT) calculations. The electrochemical and thermodynamic methods were used to construct the linear relationship of various adsorption intermediates, and a possible reaction mechanism was deduced. All material dependencies were included in one descriptor, that is, the oxygen-binding energy (Fig. 6). The unit surface has two bridge sites and two cus sites. The binding of adsorbates at a bridge site is stronger than that at a cus site. Thus, most bridge sites are occupied by oxygen. The phase diagram of IrO2 shows that the surface is covered by OH groups at a low potential, which increases the oxidation potential of OH− in the solution. The OH adsorbed on the bridge site is first converted to O and finally to the thermodynamically favorable OOH. The direct formation of Cl adsorbate on the cus site requires a pH less than 3, whereas the bridge site requires an even lower pH. For a high-performance chlorine evolution catalyst, the free energy of formation of Cl adsorbed species is approximately 0 at a potential close to 1.36 V. The direct binding of Cl adsorbate at Ir fails to meet these conditions, whereas the intermediate ClOc is thermodynamically favorable at a potential higher than 1.5 V in the pH range from 0 to 3. Thus, the CER proceeds in two steps. Oc combines with Cl− to form a ClOc intermediate, which combines with another Cl− to form Cl2. This mechanism is also known as the Volmer–Heyvrosky mechanism. The surface phase diagram of RuO2 is complicated. A possible mechanism of the CER is that O2cc combines with Cl− to form a Cl(Oc)2 intermediate, and Cl(Oc)2 combines with another Cl− to form Oc and Cl2, that is, the Khrishtalik mechanism.

Most stable surface at pH = 0 and aCl− = 1 as a function of potential, U, and the surface reactivity descriptor, ΔE(Oc) (reprinted from [69] with permission from RSC)
Exner et al. [77] used the ab initio thermodynamic method to construct the surface Pourbaix diagram of the CER on RuO2(110) based on the DFT calculations and considered the solvation effect. Under CER conditions, that is, U > 1.36 V, no ORR occurs on the surface of RuO2(110). All uncoordinated Ru surface atoms (Rucus) are covered by top oxygen (Oot), and the adjacent Ru2f atoms are bridged by uncoordinated surface oxygen (Obr). In the ball-and-stick model, the active surface structure in CER and OER can be expressed as RuO2(110)–Oot.
In the first-order reaction kinetics, the reaction mechanism of RuO2(110)–Oot active surface is considered to be the Volmer–Heyrovsky pathway. The mechanism is mainly divided into the Volmer step of Cl adsorption and desorption on the Rucus–Oot active site and the Heyrovsky step of the adsorption of Cl on the Rucus–OClot precursor to recombine with another Cl ion to form Cl2 [70]. This phenomenon is consistent with the recent experimental study of CER on RuO2(110) single crystal [78]. The rate-limiting step is the Heyrovsky step under a low overpotential or Volmer step under a high overpotential [79]. OER is a relatively complicated four-electron reaction. This process mainly involves the formation of OHot, Oot, and OOHot adsorbates on Rucus sites.
DFT calculations showed that the formation of OOHot adsorbates is a rate-determining step. Exner et al. [80] proved this finding through experiments. Under industrial conditions, the OClot and OOHot adsorbates control the activity of CER and OER, respectively [70]. Thus, in addition to thermodynamic properties, the kinetic-based free energy of formation of OClot and OOHot adsorbents should also be considered in the improved design of catalysts for CER. Loading a single layer of TiO2(110) on RuO2(110) can increase the selectivity of CER, and the CER activity is also improved. Consistent with the theory above, Ti replaces the top Ru atom, which weakens the free adsorption energy of Oot by about 1.5 eV.
Other Metal Oxide Electrodes
Ir, whether as a pure oxide or dopant, can maintain a long-term stability [81]. A series of iridium-based double perovskite (Ba2BIrO6, where B = Pr, Nd, La, Sn, Y, Tb, Ce) has been synthesized and studied for OER selectivity and stability [82]. The results showed that CER was carried out through the Volmer–Tafel mechanism, where the activity was strongly affected by the concentration of chloride ions, showing a stepwise negative reaction order.
Highly efficient non-noble metal catalysts have also been developed to replace expensive Ru and Ir catalysts. Among these catalysts, crystalline transition metal antimonates (TMAs) are active and stable electrocatalysts [83]. Moreno-Hernandez et al. [84] reported that crystalline TMAs, such as NiSb2Ox, CoSb2Ox, and MnSb2Ox, are intermediate active catalysts for the electrochemical oxidation of chloride to chlorine under industrial chlor-alkali process conditions. The Faraday efficiencies of chlorine evolution were as follows: NiSb2Ox (96.0% ± 3.7%), CoSb2Ox (97.4% ± 3.0%), and MnSb2Ox (89.9% ± 0.8%). X-ray photoelectron spectroscopy showed that the valence state of the transition metal remained unchanged, whereas the valence of Sb changed from 5 to a mixed state of 5 and 3. CoSb2Ox exhibited the best stability and selectivity, and NiSb2Ox showed the highest intrinsic activity. Ha et al. [85] proposed that Co3O4 nanoparticles have better chlorine evolution selectivity and stability than DSAs. The reaction proceeds by the Krishtalik mechanism, in which the rate-determining step is the second step, that is, the Cl atoms adsorbed on the surface of cobalt atoms are discharged to form adsorbed Cl+ species.
Membranes
The IEM that is widely used in the chlor-alkali industry is a double-layer composite material composed of a thick sulfonic acid-based layer (PFSA) and a thin carboxylic acid-based layer (PFCA), which can be incorporated into symmetrical, dense, or non-porous membranes. Dense films are relatively void-free structures, and their permeability is inversely proportional to crystallinity. The early-aged chlor-alkali electrolysis cell employed PFSA-based IEMs, which suffered from low ion selectivity and current efficiency. By contrast, the PFCA-based IEM is usually used as proton exchange membranes in fuel cells [86,87,88] and has excellent current efficiency, but is only suitable for producing alkali metal hydroxides with a maximum yield of 35% by weight due to its low ion flux and high operating voltage. PFSA and PFCA are usually combined into a double-layer IEM to improve performance. Operation conditions influencing membrane performances, such as current densities and water contents, need to be optimized to advance the performance of chlor-alkali cells.
Key Parameters
The more uniform the distributions of temperature and brine concentration in the area near the IEM, the longer the service life of the IEM. A high current density can increase production capacity and reduce production costs. However, this condition speeds up the gas generation rate and causes the accumulation of the generated gas at the top of the cell, resulting in an increase in temperature. Given the increase in local temperature of electrolyzers, the internal channels of the membrane will be damaged, a phenomenon called the “membrane pinhole effect”. In addition, a pH gradient is built on the anode side surface of the membrane, whereas the salt concentration in the tank decreases. This condition will cause IEMs to blister, delaminate, and be permanently destroyed. A high operation current density will also increase the temperature of the ion membrane surface and the interior of the electrolytic cell. When the cell temperature is higher than 90 °C, the water in the electrolytic cell will be severely vaporized, and anolyte vaporization or boiling will deteriorate the performance of the ion membrane. Therefore, information about the distribution of cell temperature, membrane anode surface concentration gradient, and brine concentration is important to the operation, design, and development of highly efficient electrolyzers.
The water content in the membrane is determined by the nature and concentration of ion exchange groups, counterions, and the degree of cross-linking of the polymer. Two types of water absorption exist [89]: one is absorption by the film particles; the other is absorption by the combination of physical bonds and ions. The first type of water requires a very low heat; thus, it occurs at low temperatures, around 55–65 °C. The second type needs a slightly higher temperature. Structural properties, such as the equivalent weight of the polymer, free volume, the degree of aggregation of ionic clusters, or cluster size, will affect water absorption. The increase in water absorption will increase the distance between ion groups in the cluster due to the expansion phenomenon, which weakens the membrane mechanical strength.
Mg2+ and Ca2+ impurities in the brine easily contaminate the ionic membrane, which reduces efficiency, increases energy consumption, and shortens the service life of the membrane [90]. Ca ions can combine with carbonate to form triangular CaCO3 [9, 93]. This slow process will cause the efficiency of the electrolytic cell to gradually decrease. The state-of-the-art technology requires the use of an electrolyte containing calcium and magnesium below 20 ppb to enable the membrane function for over 4 years. Moreover, to prevent the formation of bubbles, industry operation often uses various techniques to cover a layer of mineral on the top of the membrane. However, the performance of the membrane in chlor-alkali electrolytic cells will degrade when the mineral layer detaches from the surface.
Advanced Membrane Design
Figure 7 shows the ion transport in a bilayer membrane. Under the long-term operation, particularly under abnormal conditions (such as failure of timely water supply in the cathode chamber, power interruption, reduction or interruption of brine feed, etc.), the composite membrane is easily delaminated, and blisters are formed between the two layers, resulting in increase in the operating voltage. The delamination between the sulfonic acid and carboxyl layers of the composite membrane is mainly due to the difference in their water permeability. The use of a blend of PFSA and PFCA as an intermediate layer between the sulfonic acid and carboxyl layers was proposed to minimize the difference in water transport between the two layers.

Multicomponent ion transport in a bilayer cation-exchange membrane (reprinted from [91] with permission from Springer Nature)
Wang et al. [92] studied the effect of blend layers with different ratios of PFSA to PFCA on the performance of composite membranes. The distribution morphology of the blend membrane has a great influence on its ion transport performance and water absorption capacity. The maximum amount of water absorption of PFCA is smaller than that of PFSA. However, the diffusion path of water in PFCA is straighter than that in PFSA. Thus, a large diffusion coefficient can reach equilibrium faster in the former. In addition, water absorption of the membrane in 32% sodium hydroxide solution is considerably less than that in deionized water, indicating that the highly concentrated sodium hydroxide solution significantly reduces the water absorption of the IEM in the electrolytic cell. As the content of PFCA in the blend film increases, the proton conductivity drops rapidly because the proton conductivity of sulfonate is higher than that of carbonate. The proton conductivity of PFSA is directly related to water content, and the increase in PFCA content will reduce the water content. The introduction of the blend layer can not only enhance the intermolecular binding force between PFSA and PFCA layers but also balance the water transfer. Given the different water permeabilities of the two materials, an interlayer stress will be generated due to the electro-osmotic water during the electrolysis process. The blend film can effectively reduce this interlayer stress, which increases its resistance to peeling damage and achieves long-term stability.
Electrolyzers
Membrane-Free Method
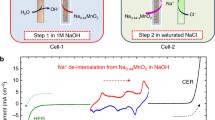
Given the above disadvantages of IEMs, membrane-less electrolysis was proposed as an alternative technology. The mercury electrolytic cell technology used in the early chlor-alkali industry was a typical membrane-less process, but the high toxicity of mercury prevented its further development. However, the use of the previous mercury electrolysis cell implies that the reversible sodium-ion storage reaction can decouple the chlor-alkali process. Hou et al. [93] proposed the Na0.44MnO2 electrode for Na+ insertion and extraction as a redox mediator (Fig. 8). The chlor-alkali process was decoupled into two separate steps: H2/NaOH formation and Cl2 generation. The first step is the cathodic HER (H2O → H2) and the anode Na+ deintercalation reaction (Na0.44MnO2 → Na0.44−xMnO2), which produces NaOH in the electrolyte, as shown in Eqs. (6) and (8), respectively. The second step is the cathode Na+ intercalation reaction (Na0.44−xMnO2 → Na0.44MnO2) and the production of Cl2 (Cl → Cl2) (Eqs. (2) and (9), respectively). Technically, the two-step reaction can be carried out in the same electrolytic cell. However, as the NaOH concentration increases, the CER process at the electrode will shift to the OER side reaction. Compared with membrane electrolysis, the Faraday efficiency of hydrogen production is higher (nearly 100%), whereas the efficiency of chlorine production is about 90.2%, which is lower than that of membrane electrolysis (97.4%).

Schematic of the operation mechanism of a membrane-free electrolysis cell (reprinted from [93] with permission from Nat Commun)
Step 1:
Step 2:
Coupling with Other Systems
The ODC electrode can be employed as the cathode to optimize the electrolysis system and improve energy efficiency. A strategy is to use bifunctional electrodes to switch between HER and ODC electrodes at any time [94]. However, a certain downtime will occur when electrode cleaning is necessary. Alternatively, chlor-alkali electrolysis cells can possibly be combined with water electrolyzers or hydrogen fuel cells [94]. The O2 produced in water electrolyzers can be used to directly supply ODC, thereby saving part of the cost. Coupled with the H2 fuel cell, the generated electrical energy can be used to compensate for part of the electricity consumption in chlor-alkali electrolysis cells.
The combination of the chlor-alkali process with CO2 reduction reaction (CO2RR) into value-added products while achieving negative carbon emissions and mitigating the greenhouse effect has also been proposed. In a single CO2 reduction process, CO2RR occurs at the cathode [95], and the anode undergoes the OER process. CO2RR accepts electrons to break the C=O bonds and generates a series of basic anions (OH−, HCO3−, and HCOO−) [96], as shown in Eqs. (10–12), whereas the anode generates acid to neutralize the cathode products [95, 97,98,99,100,101]. Utilizing the CO2RR process to replace the cathode HER in the chlor-alkali electrolytic cells, CO2 reduction products, Cl2, and NaOH can be obtained. At present, most electrolytic cells adopt a zero-gap configuration to minimize the ohmic loss of the electrolyte. The integration of CO2RR and chlor-alkali can be achieved in an electrolytic cell similar to the H-cell, but this process is limited by the slow diffusion of CO2 in saturated solutions [62], and its current density is far below that used in practical applications.
The GDE structure and flow cell have been developed to overcome the slow CO2 transport issue. In the GDE structure [62], CO2 is supplied in a gaseous state, and the path of CO2 diffusion to the catalyst is extremely short. Thus, its concentration is maintained in the catalytic layer of the cathode, thereby achieving a high current density. Liu et al. [102] carried out a CO2RR experiment in 3.5 wt% (approximately 0.6 M) NaCl electrolyte with a GDE to simulate the seawater medium used in chlor-alkali electrolyzers (Fig. 9). The high formate faradaic efficiency of over 80% and a high formate production rate of ~0.5 A/cm2 have been achieved within more than 0.5 V potential range. Moreover, the highest formate selectivity of 95% was achieved in saltwater because the HER side reaction was deeply suppressed along with the increased local pH.

Schematic model of a the chlor-alkali electrolyzer coupled with CO2RR and b the performance with tin oxide catalysts converting CO2 into formate (reprinted from [102] with permission from J Mater Chem A)
Conclusion and Outlook
The above discussion summarizes the research progress, including those of the anode, cathode, IEM, and electrolysis system, of chlor-alkali electrolyzers. Although the chlor-alkali industry has a history of over 100 years, it remains a high-energy-consuming industry today, consuming nearly 10% of global electricity every year. This industry can be improved from the following aspects.
For the anode reaction, the mechanism of CER/OER must be further understood. For example, combining modern experimental and theoretical methods to study anode activity and selectivity can provide further insights into the structure-performance relationship. The combination of theoretical modeling and modern characterization methods (such as X-ray spectroscopy) can help explore the details of the electrode reaction process and improve the selectivity and activity of the anode. For the cathode, currently, a huge amount of hydrogen as by-products is being vented to the atmosphere directly due to the insufficient market size to consume the hydrogen produced by the chlor-alkali industry. Moreover hydrogen generated above is usually mixed with impurities (water vapor, nitrogen, oxygen, and chlorine), which limits its usage. Further purification processes, such as pressure swing adsorption, can produce high-purity hydrogen, which has a large market in electronic industry, petrochemical industry, metal smelting, scientific research, and other fields. Particularly, large amount of pure hydrogen is needed to feed fuel cells to generate clean electricity to drive future hydrogen society.
For IEMs, one can design a self-cleaning membrane to avoid the accumulation of precipitation on the surface. The service life of the membrane can be extended by optimizing the operation conditions, such as the quality of brine. Moreover, the addition of circulating electrolytes can result in the uniform distribution of the temperature and concentration of the brine in the chlor-alkali pool. This condition can be realized by a reasonable electrolytic cell design, such as the introduction of an external forced circulation or installation of an inner weir, which cannot eliminate gas accumulation at the top of the anode chamber. The passage of gas and liquid into the separation chamber must be improved to enhance liquid exhaustion. Chlor-alkali electrolyzers can be coupled with other systems (such as fuel cells and CO2 electrolyzers). These combined technologies need to be further evaluated based on the relationship between the values of different products from CO2 reduction and energy consumption. Moreover, hydrogen, which is usually treated as exhaust gas in the chlor-alkali industry, may become the major fuel used on a large scale in the future as a promising clean energy.
References
Crook J, Mousavi A (2016) The chlor-alkali process: a review of history and pollution. Environ Forensics 17(3):211–217
Fauvarque J (1996) The chlorine industry. Pure Appl Chem 68(9):1713–1720
Lakshmanan S, Murugesan T (2014) The chlor-alkali process: work in progress. Clean Technol Environ Policy 16(2):225–234
Chen YQ, Manzhos S (2016) Voltage and capacity control of polyaniline based organic cathodes: an ab initio study. J Power Sources 336:126–131
Cao YL, Yang HX, Ai XP et al (2003) The mechanism of oxygen reduction on MnO2-catalyzed air cathode in alkaline solution. J Electroanal Chem 557:127–134
Cao YL, Xiao LF, Wang W et al (2011) Reversible sodium ion insertion in single crystalline manganese oxide nanowires with long cycle life. Adv Mater 23(28):3155–3160
Tan C (2005) Development and application of modified diaphragm. Chlor-Alkali Ind 11:21–24 (in Chinese)
Bazinet L, Araya-Farias M (2005) Effect of calcium and carbonate concentrations on cationic membrane fouling during electrodialysis. J Colloid Interface Sci 281(1):188–196
Casademont C, Pourcelly G, Bazinet L (2007) Effect of magnesium/calcium ratio in solutions subjected to electrodialysis: characterization of cation-exchange membrane fouling. J Colloid Interface Sci 315(2):544–554
Dötzel O, Schneider L (2002) Non-asbestos diaphragms in chlor-alkali electrolysis. Chem Eng Technol 25(2):167
Stiegel GJ, Ramezan M (2006) Hydrogen from coal gasification: an economical pathway to a sustainable energy future. Int J Coal Geol 65(3–4):173–190
Xu JG, Froment GF (1989) Methane steam reforming, methanation and water-gas shift: I. Intrinsic kinetics. Aiche J 35(1):88–96
de Wild PJ, Verhaak MJFM (2000) Catalytic production of hydrogen from methanol. Catal Today 60(1–2):3–10
Rossmeisl J, Logadottir A, Nørskov JK (2005) Electrolysis of water on (oxidized) metal surfaces. Chem Phys 319(1–3):178–184
Kayfeci M, Keçebaş A, Bayat M (2019) Chapter 3-hydrogen production. Solar hydrogen production: processes, systems and technologies (1st Edition):45–83
Kong FD, Zhang S, Yin GP et al (2013) IrO2-graphene hybrid as an active oxygen evolution catalyst for water electrolysis. Int J Hydrog Energy 38(22):9217–9222
Levene JI, Mann MK, Margolis RM et al (2007) An analysis of hydrogen production from renewable electricity sources. Sol Energy 81(6):773–780
Lee DY, Elgowainy AA, Dai Q (2017) Life cycle greenhouse gas emissions of by-product hydrogen from chlor-alkali plants. Office of Scientific and Technical Information (OSTI), USA
Elgowainy A (2017) Resourcing byproduct hydrogen from industrial operations. H2@Scale Workshop, Houston
Lee DY, Elgowainy A, Dai Q (2018) Life cycle greenhouse gas emissions of hydrogen fuel production from chlor-alkali processes in the United States. Appl Energy 217:467–479
Yu H (2018) Hydrogen energy production from chlor-alkali electrolyzers. Chlor-Alkali Ind (in Chinese)
Ozturk M, Dincer I (2021) An integrated system for ammonia production from renewable hydrogen: a case study. Int J Hydrog Energy 46(8):5918–5925
Félix G, Quitian A, Rodríguez E et al (2017) Methods to calculate hydrogen consumption during hydrocracking experiments in batch reactors. Energy Fuels 31(11):11690–11697
Sharaf OZ, Orhan MF (2014) An overview of fuel cell technology: fundamentals and applications. Renew Sustain Energy Rev 32:810–853
Brinkmann T, Giner-Santonja G, Schorcht F et al (2014) Best available techniques (BAT) reference document for the production of chlor-alkali. Publications Office of the European Union, Denmark
Guandalini G, Foresti S, Campanari S et al (2017) Simulation of a 2 MW PEM fuel cell plant for hydrogen recovery from chlor-alkali industry. Energy Procedia 105:1839–1846
Cheng T, Wang L, Merinov BV et al (2018) Explanation of dramatic pH-dependence of hydrogen binding on noble metal electrode: greatly weakened water adsorption at high pH. J Am Chem Soc 140(25):7787–7790
Wang YH, Chen L, Yu XM et al (2017) Superb alkaline hydrogen evolution and simultaneous electricity generation by Pt-decorated Ni3N nanosheets. Adv Energy Mater 7(2):1601390
Wang PT, Jiang KZ, Wang GM et al (2016) Phase and interface engineering of platinum-nickel nanowires for efficient electrochemical hydrogen evolution. Angewandte Chemie Int Ed 55(41):12859–12863
Mahmood N, Yao Y, Zhang JW et al (2018) Electrocatalysts for hydrogen evolution in alkaline electrolytes: mechanisms, challenges, and prospective solutions. Adv Sci 5(2):1700464
Zheng Y, Jiao Y, Vasileff A et al (2018) The hydrogen evolution reaction in alkaline solution: from theory, single crystal models, to practical electrocatalysts. Angew Chem Int Ed Engl 57(26):7568–7579
Safizadeh F, Ghali E, Houlachi G (2015) Electrocatalysis developments for hydrogen evolution reaction in alkaline solutions-a review. Int J Hydrog Energy 40(1):256–274
Lei CJ, Wang Y, Hou Y et al (2019) Efficient alkaline hydrogen evolution on atomically dispersed Ni–Nx species anchored porous carbon with embedded Ni nanoparticles by accelerating water dissociation kinetics. Energy Environ Sci 12(1):149–156
Sun QZ, Zhang B, Diao LC et al (2020) Engineering the electronic structure of 1T’-ReS2 through nitrogen implantation for enhanced alkaline hydrogen evolution. J Mater Chem A 8(23):11607–11616
Zhao DJ, Zhang S, Yin GP et al (2012) Effect of Se in Co-based selenides towards oxygen reduction electrocatalytic activity. J Power Sources 206:103–107
Kintrup J, Millaruelo M, Trieu V et al (2017) Gas diffusion electrodes for efficient manufacturing of chlorine and other chemicals. Electrochem Soc Interface 26(2):73–76
Ding JS, Hua WQ, Hu BB et al (2011) Closed loop recycling of chlorine for sustainable development of polyurethane industry. Trans Tianjin Univ 17(4):298–304
Liu JJ, Yang C, Liu CG et al (2014) Design of pore structure in gas diffusion layers for oxygen depolarized cathode and their effect on activity for oxygen reduction reaction. Ind Eng Chem Res 53(14):5866–5872
Erikson H, Sarapuu A, Tammeveski K (2019) Oxygen reduction reaction on silver catalysts in alkaline media: a minireview. ChemElectroChem 6(1):73–86
Lipp L, Gottesfeld S, Chlistunoff J (2005) Peroxide formation in a zero-gap chlor-alkali cell with an oxygen-depolarized cathode. J Appl Electrochem 35(10):1015–1024
Kuwertz R, Gonzalez Martinez I, Vidaković-Koch T et al (2013) Energy-efficient chlorine production by gas-phase HCl electrolysis with oxygen depolarized cathode. Electrochem Commun 34:320–322
Zhang CZ, Fan FRF, Bard AJ (2009) Electrochemistry of oxygen in concentrated NaOH solutions: solubility, diffusion coefficients, and superoxide formation. J Am Chem Soc 131(1):177–181
Zhao DJ, Zhang S, Yin GP et al (2013) Tungsten doped Co-Se nanocomposites as an efficient non precious metal catalyst for oxygen reduction. Electrochim Acta 91:179–184
Röhe M, Kubannek F, Krewer U (2019) Processes and their limitations in oxygen depolarized cathodes: a dynamic model-based analysis. Chemsuschem 12(11):2373–2384
Röhe M, Botz A, Franzen D et al (2019) The key role of water activity for the operating behavior and dynamics of oxygen depolarized cathodes. ChemElectroChem 6(22):5671–5681
Chavan N, Pinnow S, Polcyn GD et al (2015) Non-isothermal model for an industrial chlor-alkali cell with oxygen-depolarized cathode. J Appl Electrochem 45(8):899–912
Clausmeyer J, Botz A, Öhl D et al (2016) The oxygen reduction reaction at the three-phase boundary: nanoelectrodes modified with Ag nanoclusters. Faraday Discuss 193:241–250
Franzen D, Ellendorff B, Paulisch MC et al (2019) Influence of binder content in silver-based gas diffusion electrodes on pore system and electrochemical performance. J Appl Electrochem 49(7):705–713
Morimoto T, Suzuki K, Matsubara T et al (2000) Oxygen reduction electrode in brine electrolysis. Electrochim Acta 45(25–26):4257–4262
Tseng CJ, Lo SK (2010) Effects of microstructure characteristics of gas diffusion layer and microporous layer on the performance of PEMFC. Energy Convers Manag 51(4):677–684
Passalacqua E, Squadrito G, Lufrano F et al (2001) Effects of the diffusion layer characteristics on the performance of polymer electrolyte fuel cell electrodes. J Appl Electrochem 31(4):449–454
Nara H, Momma T, Osaka T (2013) Impedance analysis of the effect of flooding in the cathode catalyst layer of the polymer electrolyte fuel cell. Electrochim Acta 113:720–729
Park S, Popov BN (2009) Effect of cathode GDL characteristics on mass transport in PEM fuel cells. Fuel 88(11):2068–2073
Kiros Y, Pirjamali M, Bursell M (2006) Oxygen reduction electrodes for electrolysis in chlor-alkali cells. Electrochim Acta 51(16):3346–3350
Marković NM, Schmidt TJ, Stamenković V et al (2001) Oxygen reduction reaction on Pt and Pt bimetallic surfaces: a selective review. Fuel Cells 1(2):105–116
Shao YY, Zhang S, Engelhard MH et al (2010) Nitrogen-doped graphene and its electrochemical applications. J Mater Chem 20(35):7491
Masa J, Xia W, Muhler M et al (2015) On the role of metals in nitrogen-doped carbon electrocatalysts for oxygen reduction. Angewandte Chemie Int Ed 54(35):10102–10120
Li JK, Jia QY, Ghoshal S et al (2017) Highly active and stable Fe–N–C catalyst for oxygen depolarized cathode applications. Langmuir 33(37):9246–9253
Mamtani K, Jain D, Co AC et al (2017) Investigation of chloride poisoning resistance for nitrogen-doped carbon nanostructures as oxygen depolarized cathode catalysts in acidic media. Catal Lett 147(12):2903–2909
Singh V, Nagaiah TC (2019) In situ incorporation of cobalt nanoclusters and nitrogen into the carbon matrix: a bifunctional catalyst for the oxygen depolarized cathode and chlorine evolution in HCl electrolysis. J Mater Chem A 7(16):10019–10029
Moussallem I, Pinnow S, Wagner N et al (2012) Development of high-performance silver-based gas-diffusion electrodes for chlor-alkali electrolysis with oxygen depolarized cathodes. Chem Eng Process Process Intensif 52:125–131
Burdyny T, Smith WA (2019) CO2 reduction on gas-diffusion electrodes and why catalytic performance must be assessed at commercially-relevant conditions. Energy Environ Sci 12(5):1442–1453
Zhang LH, Fan Q, Li K et al (2020) First-row transition metal oxide oxygen evolution electrocatalysts: regulation strategies and mechanistic understandings. Sustain Energ Fuels 4(11):5417–5432
Petrykin V, Macounová K, Okube M et al (2013) Local structure of Co doped RuO2 nanocrystalline electrocatalytic materials for chlorine and oxygen evolution. Catal Today 202:63–69
Halck NB, Petrykin V, Krtil P et al (2014) Beyond the volcano limitations in electrocatalysis—oxygen evolution reaction. Phys Chem Chem Phys 16(27):13682–13688
Macounová KM, Simic N, Ahlberg E et al (2015) Electrochemical water-splitting based on hypochlorite oxidation. J Am Chem Soc 137(23):7262–7265
Koper MTM (2011) Thermodynamic theory of multi-electron transfer reactions: implications for electrocatalysis. J Electroanal Chem 660(2):254–260
Karlsson RK, Cornell A (2016) Selectivity between oxygen and chlorine evolution in the chlor-alkali and chlorate processes. Chem Rev 116(5):2982–3028
Hansen HA, Man IC, Studt F et al (2010) Electrochemical chlorine evolution at rutile oxide (110) surfaces. Phys Chem Chem Phys 12(1):283–290
Exner KS (2019) Controlling stability and selectivity in the competing chlorine and oxygen evolution reaction over transition metal oxide electrodes. ChemElectroChem 6(13):3401–3409
Over H (2012) Surface chemistry of ruthenium dioxide in heterogeneous catalysis and electrocatalysis: from fundamental to applied research. Chem Rev 112(6):3356–3426
Trasatti S (2000) Electrocatalysis: understanding the success of DSA (R). Electrochim Acta 45(15–16):2377–2385
Nanni LC, Polizzi S, Benedetti A et al (1999) Morphology, microstructure, and electrocatalytic properties of RuO2-SnO2 thin films. J Electrochem Soc 146(1):220–225
Xiong K, Deng ZH, Li L et al (2013) Sn and Sb co-doped RuTi oxides supported on TiO2 nanotubes anode for selectivity toward electrocatalytic chlorine evolution. J Appl Electrochem 43(8):847–854
Trasatti S (1984) Electrocatalysis in the anodic evolution of oxygen and chlorine. Electrochim Acta 29(11):1503–1512
Exner KS (2020) Beyond dimensionally stable anodes: single-atom catalysts with superior chlorine selectivity. ChemElectroChem 7(7):1528–1530
Exner KS, Anton J, Jacob T et al (2014) Chlorine evolution reaction on RuO2(110): ab initio atomistic thermodynamics study—Pourbaix diagrams. Electrochim Acta 120:460–466
Kuo DY, Paik H, Nelson JN et al (2019) Chlorine evolution reaction electrocatalysis on RuO2(110) and IrO2(110) grown using molecular-beam epitaxy. J Chem Phys 150(4):041726
Exner KS, Anton J, Jacob T et al (2016) Full kinetics from first principles of the chlorine evolution reaction over a RuO2(110) model electrode. Angew Chem Int Ed 55(26):7501–7504
Exner KS, Sohrabnejad-Eskan I, Anton J et al (2017) Full free energy diagram of an electrocatalytic reaction over a single-crystalline model electrode. ChemElectroChem 4(11):2902–2908
Spöri C, Kwan JTH, Bonakdarpour A et al (2017) The stability challenges of oxygen evolving catalysts: towards a common fundamental understanding and mitigation of catalyst degradation. Angewandte Chemie Int Ed 56(22):5994–6021
Vos J, Liu ZC, Speck FD et al (2019) Selectivity trends between oxygen evolution and chlorine evolution on iridium-based double perovskites in acidic media. ACS Catal 9(9):8561–8574
Moreno-Hernandez IA, MacFarland CA, Read CG et al (2017) Crystalline nickel manganese antimonate as a stable water-oxidation catalyst in aqueous 1.0 M H2SO4. Energy Environ Sci 10(10):2103–2108
Moreno-Hernandez IA, Brunschwig BS, Lewis NS (2019) Crystalline nickel, cobalt, and manganese antimonates as electrocatalysts for the chlorine evolution reaction. Energy Environ Sci 12(4):1241–1248
Ha H, Jin K, Park S et al (2019) Highly selective active chlorine generation electrocatalyzed by Co3O4 nanoparticles: mechanistic investigation through in situ electrokinetic and spectroscopic analyses. J Phys Chem Lett 10(6):1226–1233
Mogg L, Zhang S, Hao GP et al (2019) Perfect proton selectivity in ion transport through two-dimensional crystals. Nat Commun 10:4243
Mogg L, Hao GP, Zhang S et al (2019) Atomically thin micas as proton-conducting membranes. Nat Nanotechnol 14(10):962–966
Liang X, Wu L, Xu TW (2018) Role of ionomer in membrane electrode assembly for proton exchange membrane fuel cells. Sci Sin-Chim 48(9):1040–1057
Zeynali ME, Mohammadi F, Rabiee A (2015) Structural analysis and defect evaluation of ion exchange composite membranes used in electrolysis of sodium chloride in chlor-alkali process. Iran Polym J 24(2):85–93
Le Faucheur S, Vasiliu D, Catianis I et al (2016) Environmental quality assessment of reservoirs impacted by Hg from chlor-alkali technologies: case study of a recovery. Environ Sci Pollut Res 23(22):22542–22553
Moshtarikhah S, Oppers NAW, Groot MT et al (2017) Multicomponent ion transport in a mono- and bilayer cation-exchange membrane at high current density. J Appl Electrochem 47(2):213–221
Wang J, Wang XJ, Dou P et al (2015) Morphology and properties of perfluorosulfonic acid polymer/perfluorocarboxylic acid polymer blend membranes. Polym Eng Sci 55(1):180–189
Hou M, Chen L, Guo Z et al (2018) A clean and membrane-free chlor-alkali process with decoupled Cl2 and H2/NaOH production. Nat Commun 9(1):438
Brée LC, Bulan A, Herding R et al (2020) Techno-economic comparison of flexibility options in chlorine production. Ind Eng Chem Res 59(26):12186–12196
Kuang SY, Li ML, Xia R et al (2020) Stable surface-anchored Cu nanocubes for CO2 electroreduction to ethylene. ACS Appl Nano Mater 3(8):8328–8334
Guo JH, Sun WY (2020) Integrating nickel-nitrogen doped carbon catalyzed CO2 electroreduction with chlor-alkali process for CO, Cl2 and KHCO3 production with enhanced techno-economics. Appl Catal B Environ 275:119154
Zhang S, Fan Q, Xia R et al (2020) CO2 reduction: from homogeneous to heterogeneous electrocatalysis. Acc Chem Res 53(1):255–264
Gao DF, Zhang Y, Zhou ZW et al (2017) Enhancing CO2 electroreduction with the metal–oxide interface. J Am Chem Soc 139(16):5652–5655
Zhang S, Kang P, Meyer TJ (2014) Nanostructured tin catalysts for selective electrochemical reduction of carbon dioxide to formate. J Am Chem Soc 136(5):1734–1737
Xia R, Zhang S, Ma XB et al (2020) Surface-functionalized palladium catalysts for electrochemical CO2 reduction. J Mater Chem A 8(31):15884–15890
Liu S, Yang HB, Hung SF et al (2020) Elucidating the electrocatalytic CO2 reduction reaction over a model single-atom nickel catalyst. Angew Chem Int Ed Engl 59(2):798–803
Liu H, Su YQ, Kuang SY et al (2021) High efficient CO2 electrolysis within a wide operation window using octahedral tin oxide single crystals. J Mater Chem A 9:7848–7856
Acknowledgements
The work was supported by the National Nature Science Foundation of China (Nos. 2193800 and 22078232) and the Science and Technology Major Project of Tianjin (Nos. 18ZXJMTG00180 and 19ZXNCGX00030).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, K., Fan, Q., Chuai, H. et al. Revisiting Chlor-Alkali Electrolyzers: from Materials to Devices. Trans. Tianjin Univ. 27, 202–216 (2021). https://doi.org/10.1007/s12209-021-00285-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12209-021-00285-9