
Abstract
Patulin is a mycotoxin that exhibits a number of toxic effects in animals. The main sources of patulin intake in human diet that was shown for EU consumers are apple juice and nectar and for this reason, apple-based food is most often monitored for this mycotoxin. However, the presence of patulin in other fruits, including stone fruits, and soft fruits has been reported as well. Most of them are seasonal, suitable for consumption for short time, and are usually processed in order to be commercially available throughout the year. Patulin can also be generated during food storage and remains stable over food processing procedures. Therefore, constant monitoring of different fruit-based products ought to be carried out to provide proper estimation of human exposure to this toxin. The preferred approach used for mycotoxin determination in a variety of foods is liquid chromatography (LC) allowing quantitative analysis of patulin content. This paper presents recently proposed LC methods with ultraviolet and mass spectrometric detection for patulin quantification in fruits and derived products. We focus on developments in sample preparation and the applied analytical conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
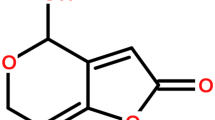
Patulin (4˗hydroxy˗4H-furo[3,2˗c]pyran-2(6H)-one) (Fig. 1) is a heterocyclic lactone with molecular weight of 154.12 g mol−1 and low volatility. It is resistant to heat (Harrison 1988) and quite stable in aqueous media at 105–125 °C in the pH range from 3.5 to 5.5 (Lovett and Peeler 1973). However, it is much less stable in higher pH. Collin’s group has reported that 50% of patulin degrades within 1 h at 100 °C in aqueous medium at pH 6 (Collin et al. 2008). They have also noted that sulfites promote patulin destruction at higher pH with complete patulin degradation achieved at pH 7 within 3-h incubation at 25 °C in the presence of 50 ppm SO2. The significant reduction of patulin in apple-based juices by exposure to UV light (Zhu et al. 2014), microwaves (Zhang et al. 2006), high hydrostatic pressure (Hao et al. 2016), or in the presence of ascorbic acid (Kokkinidou et al. 2014) has also been reported.

Chemical structure of patulin and 5-hydroxymethylfurfural (5-HMF)
Patulin is produced by several species of fungus belonging mainly to the genera of Aspergillus, Penicillium, and Byssochlamys; however, others also synthesize this mycotoxin (Anderson et al. 1979; Steiman et al. 1989; Okeke et al. 1993; Samson et al. 2009). In case of deterioration of fruits and their derived products, Penicillium and Byssochlamys are the most studied fungi with Penicillium expansum being the most common contaminant. It is found in healthy as well as in rotten apple fruit (Hasan 2000; Hammami et al. 2017) and other fruits like infected pears, peaches, or apricots (Neri et al. 2010; Reddy et al. 2010). This fungus was also isolated from grapes (Tančinová et al. 2015) and the generated patulin accumulates also in plums, strawberries, and kiwifruits (Neri et al. 2010; Reddy et al. 2010; Wei et al. 2017). Considering Byssochlamys strains (B. fulva and B. nivea) showing high thermal resistance, they survive heat treatment during food processing and cause deterioration of fruit juices (Rice et al. 1977; Rice 1980; Roland and Beuchat 1984; Sant’Ana et al. 2010).
Several studies have shown toxic, mutagenic, carcinogenic, and teratogenic properties of patulin (Schumacher et al. 2005). In animal studies, patulin administration causes damage to different organs including brain, kidney, liver (de Melo et al. 2012; Song et al. 2014; Boussabbeh et al. 2015), thymus (Arzu Koçkaya et al. 2009), and skin (Saxena et al. 2009). Despite several negative effects of patulin observed in animal tissues, the molecular mechanism of toxicity is still not fully known. Different triggers are being considered; however, most reports point at the high reactivity of this mycotoxin with nucleophilic molecules, especially sulfhydryl groups on proteins and glutathione (GSH) (Fliege and Metzler 1999, 2000) and subsequent DNA damage (Glaser and Stopper 2012), proteotoxicity (Guerra-Moreno and Hanna 2017), or dysregulation of protein expression (Assunção et al. 2016) leading to inflammation (Luft et al. 2008). It is also worth noting that co-occurrence of different mycotoxins enhances toxic properties associated with individual ones. As shown on cells (Zouaoui et al. 2015), even low level of patulin might be deleterious when accompanied by other toxins, namely sterigmatocystin or beauvericin.
Negative impact of patulin on living cells and organs brings an interest and need for determination of this mycotoxin in food. Numerous analytical protocols utilizing among others near-infrared fluorescence assay (Pennacchio et al. 2015), thin-layer chromatography (Welke et al. 2009), gas chromatography-mass spectrometry (Kharandi et al. 2013), and liquid chromatography with various detection modalities (UV absorbance, mass spectrometry) have been developed for patulin quantification, especially in apple-based products. However, the works published within the last 10 years clearly indicate that a steady monitoring of patulin contamination should not be limited to apples and apple-based products but must be carried out in all fruit commodities to provide proper assessment of health risk. Patulin determination in complex food matrices (like highly pigmented fruits, jams, marmalades, dried rings) sets new challenges for quantification analysis due to low sensitivity resulting from complex matrices. To overcome these analytical problems, additional improvements in current protocols and development of new methodologies for sample preparation, as well as advanced analytical equipment (i.e., liquid chromatograph coupled with mass spectrometry) are necessary. Future approaches should meet both the food safety and green chemistry requirements. Toward this goal, the modern methods such as ultra-high-performance liquid chromatography (UHPLC) coupled with mass spectrometric detectors offer sufficient selectivity for rapid multi-analyte quantification in complicated food matrices. Despite the indisputable advantages of liquid chromatography-mass spectrometry (LC-MS) protocols, a low efficiency of patulin ionization in a mass spectrometer source could cause poor sensitivity of the method. Thus, multi-target LC-MS assays may fail in case of patulin quantification at low concentration levels. Meanwhile, the applicability of high-performance liquid chromatography (HPLC) with UV absorbance detection has been intensively investigated to meet the challenges of patulin analysis in a variety of foods. This article reviews the methods reported within last decade, showing patulin determination in fruits and fruit-based products using liquid chromatography coupled with UV absorbance and mass spectrometric detection. The developments in an initial sample preparation for chromatographic analysis and the applied analytical conditions are considered in this review.
Patulin Occurrence in Fruit Commodities
The risk of consumer exposure to patulin is associated with consumption of the molded as well as visually clean products infected with fungus (Sant’Ana et al. 2008), due to accumulation of mycotoxin in both the lesion and other parts of the spoiled fruit (Hasan 2000; Wei et al. 2017). Also, consumer risk increases with the possible production of this mycotoxin during fruits storage (Reddy et al. 2010) and food processing due to patulin resistance to applied conditions. For this reason, different fruits and derived products (juices, purees, ciders, jams, marmalades, vinegars, dried rings) should be monitored for patulin contamination. The regulations regarding accepted patulin level in food products depend on the country and are summarized in Table 1. EU recommendations are more detailed and demanding compared to other countries, thus we discuss human exposure to patulin following the EU limits.
The analysis of reports show frequent occurrence of patulin, although at different level, in apples and various apple-based food commodities from over the World, i.e., Qatar (Hammami et al. 2017), Portugal (Barreira et al. 2010), Spain (Murillo-Arbizu et al. 2009; Marsol-Vall et al. 2014), Italy (Ritieni 2003; Piemontese et al. 2005), Czech Republic (Vaclavikova et al. 2015), Romania (Catana et al. 2011; Oroian et al. 2014), Tunisia (Zaied et al. 2013; Zouaoui et al. 2015), Brazil (Sargenti and Almeida 2010), Argentina (Funes and Resnik 2009), United States (Harris et al. 2009), Iran (Jalali et al. 2010; Forouzan and Madadlou 2014), Pakistan (Iqbal et al. 2018), South Korea (Cho et al. 2010), and Northeast China (Yuan et al. 2010). According to published data, patulin concentration in the infected apples could reach 8.8–120.4 mg kg−1 (Piemontese et al. 2005; Wu et al. 2008; Gaspar and Lucena 2009) (Celli et al. 2009). The apple puree tested by Ritieni contained 15.9–74.2 μg kg−1 of patulin in three out of six tested samples (Ritieni 2003) with only one sample meeting the EU standards and not exceeding 25 μg kg−1 of the mycotoxin (European Commission 2006a). Funes and Resnik have found that four out of eight apple puree samples contained 22–221 μg kg−1 of patulin (Funes and Resnik 2009). Alarmingly high contamination of apple jams with patulin has been reported in Tunisia (about 33% with average concentration about 302 μg L−1) (Zouaoui et al. 2015). The major concern associated with patulin contamination regards apple juices, since loss of patulin through processing (pressing, milling, pasteurization processes, enzymatic and micro-filtration treatments) is relatively small (Yuan et al. 2010). Table 2 illustrates patulin content in apple and other fruit juices from different regions in the world.
Due to the high consumption of fruit products during the first years of life, children are especially vulnerable population to patulin exposure. Therefore, recommendations are more stringent regarding fruit products dedicated for infants and young children (see Table 1). As shown in Table 3, baby food often displays patulin level above the EU limits (set at 10 μg L−1 or 10 μg kg−1). To highlight some, 28% of tested products from Tunisia contained sevenfold more of patulin than the maximum permitted limit (average contamination of 68 μg L−1) (Zaied et al. 2013). Similarly, in China, patulin was detected in 19 out of 30 analyzed baby food products with a highest level reaching 67.3 μg L−1 (Yuan et al. 2010). Several other products dedicated for babies made out of apples that were tested in Italy contained considerable amount of patulin (see Table 3). This indicates necessity to develop prophylactic actions and programs for food surveillance in order to better protect the youngest from exposure to toxin.
The fungi producing patulin, in addition to apple, contaminate also other fruit commodities that constitute another source of this mycotoxin. High amount of patulin (above the EU recommended level) was found in pears (some at 620 μg kg−1) in Pakistan (Iqbal et al. 2018) or pear juices in Tunisia and in Italy (Bonerba et al. 2010a; Zouaoui et al. 2015). Based on the reports, contamination of pear jams (Zouaoui et al. 2015), mixed juices (Spadaro et al. 2008; Bonerba et al. 2010a; Zouaoui et al. 2015), and dried fruit products need to be considered as possibly containing patulin level above the EU safety limit (Ji et al. 2017).
Lately, a growing number of work focused on patulin determination in pigmented fruits such as hawthorns, red grapes, plums, sour cherries, and berries (strawberries, raspberries, blueberries, blackberries) (Zhou et al. 2012; Abu-Bakar et al. 2014; Vaclavikova et al. 2015; Ji et al. 2017; Iqbal et al. 2018; Sadok et al. 2018). The limited number of validated analytical methods for patulin quantification in this difficult fruit material entails the necessity to introduce new strategies of sample preparation for efficient removing of interfering compounds. Comprehensive studies conducted in different countries and employing significant amount of collected samples are still awaiting in order to determine the contamination of soft fruits by patulin and to assess the risk for consumers.
Preparation of Fruits and Their Related Products for Patulin Quantification
The new trends and improvements regarding patulin determination by LC in fruit commodities appear in recent publications. They address numerous issues regarding sample preparation and chromatographic separation aimed to increase specificity and limits of detection/quantification of patulin in the complex fruit matrices. The important problem in fruit analysis is the great content of sugars (sucrose, glucose, fructose, xylose). They are also rich in organic acids (citric, fumaric, tartaric) that determine taste and flavor and typically contain low amounts of fat and proteins (Ruby et al. 2000; Sturm et al. 2003). Some fruits (mainly berries) are rich sources of flavonoids and other phenolic compounds possessing antioxidant activity (Määttä et al. 2003; Pavlović et al. 2013), but fruits might be also a source of toxic elements, mycotoxins, or pesticides (Pavlović et al. 2013; Stachniuk et al. 2017).
High complexity of some fruit matrices impacts patulin analysis, i.e., it disturbs LC separation, decreases recovery, and ionization efficiency of the target compounds during LC-MS analysis. To overcome these problems, different sample pre-treatment methods have been implemented for the extraction of patulin from fruit matrix. A liquid–liquid extraction (LLE) with ethyl acetate was traditionally used for apple juice pre-treatment. However, LLE is a time-consuming approach and requires a large amount of the organic solvent that is not preferred in modern analytical chemistry. Furthermore, LLE procedure involves several extraction steps of the sample with fresh volumes of an organic solvent, which favors the co-elution of impurities (Valle-Algarra et al. 2009) - a common problem observed during analysis of the highly pigmented fruits. The dark berries, currants, or grapes owe their color to anthocyanins that appear red in acidic conditions and turn blue when the pH increases (Khoo et al. 2017). The orange-colored fruits such as apricot, grapefruit, and mango are rich in β-carotene (Khoo et al. 2011), while apples are poorer in carotenoids (Caller and Mackinney 1965) but their peel might contain up to 100–2160 mg of anthocyanins per kilogram (Horbowicz et al. 2008). In the apple’s peel, however, total pigment content is always higher than in the flesh independently from the fruit cultivars (green, yellow, and red) (Delgado-Pelayo et al. 2014). Pigments are co-extracted with patulin and can interfere in chromatographic analysis (peak overlapping, high matrix effects), contaminate LC system as well as a mass spectrometer. Figure 2 presents extracts obtained from different fruits after extraction with ethyl acetate (commonly used in LLE) and acetonitrile (commonly used in QuEChERS). As can be seen, the amount of extracted from the fruit pigments depends on pigment type, pH of a fruit pulp, and solvent used for extraction. The carotenoid pigments are more easily extracted using ethyl acetate, whereas extraction of anthocyanins is favored in acetonitrile. It should be also noted that lower pH facilitates pigments co-extraction (see Fig. 2). Thus, for patulin analysis, the final extract should be further purified to avoid overwhelming matrix effects and other unwanted impacts. Since traditional methods used for patulin extraction were not satisfactory, recently the new methodologies have been proposed. The summary of investigations regarding the important aspects of sample preparation for patulin analysis in fruits is presented below.

Comparison of extracts obtained from different fruits after extraction with ethyl acetate (vials on a left side) or acetonitrile (vials on a right side). The homogenized fruit pulps (10 g) were extracted with 20 mL of proper organic solvent, shaken for 15 min using a see-saw rocker, and centrifuged (8228×g, 10 min). The pH value was estimated from three independent measurements for a grained fruit pulp before extraction
Pectin Removal
Pectin collectively relates to a group of heterogeneous polysaccharides present in primary cell walls and in the non-woody parts of many terrestrial plants. This soluble fiber is widely used in food industry as the gelling, stabilizing, and thickening agent for jams, jellies, confectionery, and fruit juices production (Srivastava and Malviya 2011). Citrus fruits (grapefruits, lemons, oranges) and apples contain much more pectin than cherries or grapes (Baker 1997). The enzymatic hydrolysis of pectin improves both patulin recoveries and a clarity of fruit juices or puree (MacDonald et al. 2000; Funes and Resnik 2009). Depectinization is usually performed before chromatographic analyses of cloudy juices and solid apple products. The enzymatic hydrolysis of pectin is carried out using pectinase with or without addition of amylase. The samples are incubated overnight at room temperature or for 2 h at 40 °C, centrifuged, and subjected to further preparation steps like extraction, purification. This approach allowed to obtain recovery results for patulin analysis from cloudy apple juices that complied with EU criteria (50–120% for < 20 μg kg−1 of patulin spiked; 70–105% for 20–50 μg kg−1 of patulin spiked; 75–105% for > 50 μg kg−1 of patulin spiked) (European Commission 2006b; Cho et al. 2010). Good 85.2–88.9% recovery was also obtained for fruit juice (for 8.0–50.0 μg kg−1 of patulin spiked) (Spadaro et al. 2008). Finally, employing depectinization followed by a solid-phase extraction (SPE) clean-up allowed for almost 100% recovery of patulin from apple marmalade, jam, jelly, and pear marmalade (Funes and Resnik 2009). It is important to note that depectinization step may in some cases cause a significant patulin loss, probably due to binding to proteins present in the solid residues of cloudy apple juice (Baert et al. 2007). Other authors suggested that the pectin hydrolysate may interfere with SPE clean-up resulting in low patulin recoveries (from 53.1 to 63.9%) obtained for dried apple rings (Katerere et al. 2008). Funes and Resnik have observed the mycotoxin loss for apple puree at 10 and 50 μg kg−1 of patulin spiked after depectinization followed by SPE clean-up. Nevertheless, the obtained recoveries (73–75%) still met EU recommendations (Funes and Resnik 2009).
Liquid-Liquid Extraction
The first step in analysis of food is the extraction procedure. It affects concentration and purity of the target compound in the extracted sample. One of the commonly used extraction method is a liquid-liquid extraction (LLE). In LLE, one or more species are separated between two immiscible or partially miscible with each other solvents. This approach has found numerous applications in analysis of aqueous samples, including studies focused on mycotoxins determination in a variety of foods.
The LLE using ethyl acetate followed by the sodium carbonate clean-up is a part of the Association of Official Analytical Chemists (AOAC) official method 2000.02 recommended for apple juice and apple puree preparation for HPLC-UV analysis (MacDonald et al. 2000). Recently, this method with or without some modification was applied for the mycotoxin extraction from apple and other fruit juices (Li et al. 2007b; Spadaro et al. 2008; Barreira et al. 2010; Cho et al. 2010; Yuan et al. 2010; Forouzan and Madadlou 2014; Zouaoui et al. 2015; Hammami et al. 2017) as well as for solid samples (apples, pears, grapes, pineapples, baby food) (Bonerba et al. 2010b; Karakose et al. 2015; Iqbal et al. 2018). The extraction step is frequently followed by clean-up of the collected organic phase using sodium carbonate (1.5–2.0% w/v) (Li et al. 2007b; Bonerba et al. 2010b; Cho et al. 2010; Yuan et al. 2010; Zaied et al. 2013; Forouzan and Madadlou 2014; Karakose et al. 2015; Zouaoui et al. 2015; Iqbal et al. 2018) to remove the interfering polyphenols (Marsol-Vall et al. 2014). Unfortunately, sodium carbonate increases sample pH and causes patulin degradation, as patulin is more stable in acidic medium. To avoid the problem, the use of other salts such as sodium sulfate, sodium hydrogen carbonate (Spadaro et al. 2008; Barreira et al. 2010), and alternative SPE clean-up steps (De Clercq et al. 2016) have also been tasted and incorporated into the extraction procedure.
The disadvantage of LLE is high consumption of organic solvents and relatively long time of sample preparation for analysis. The example procedure was performed to extract patulin from 10 g of different apple-based products (apple juices, apple juice concentrate, mixed apple juice, baby foods) with 25 mL of ethyl acetate. After 3 min of vigorous shaking and 5 min centrifugation, the aqueous phase was re-extracted twice with 20 mL of ethyl acetate. The organic layers were combined and further purified by several additions of sodium carbonate (3 × 2 mL) and one 5 mL portion of ethyl acetate, followed by shaking. After pH adjustment, evaporation to dryness, reconstitution, and filtration, the samples were subjected to chromatographic analysis (Yuan et al. 2010). This approach resulted in excellent analyte recoveries for apple juice (92.0 and 98.0% for 10 and 100 μg kg−1 of patulin spiked, respectively). However, this methodology is quite laborious, involves a large ethyl acetate volume (70 mL per one sample), and generates large quantities of hazardous wastes. Bonerba and others have utilized LLE for baby foods pre-treatment (Bonerba et al. 2010b). The sample (10 g) was diluted in 10 mL of water and then extracted thrice with 50, 25, and 15 mL of ethyl acetate. Each time, the sample was shaken for 10 min and centrifuged for 5 min to separate phases. The organic phase was collected and shaken with 9 mL of sodium carbonate solution. After phase separation, bottom layer (sodium carbonate solution) was again extracted with 10 mL of ethyl acetate. The combined organic layer was subjected for dehydration, evaporation, reconstitution in a solvent, SPE purification, and filtration. This multi-step protocol has provided good patulin recoveries for baby food matrix (95% mean recovery for mycotoxin spiked at 5–20 μg L−1). The downside of this procedure is a large volume of ethyl acetate per one sample (100 mL) and a long time required for sample delivery to chromatographic analysis. Other examples of LLE application in patulin determination in fruit matrices are presented in Table 4.
Recently, some interesting developments in LLE have been applied for patulin determination. The good alternative for traditional LLE during the pre-treatment of juices can be an ultrasonic technique (Sargenti and Almeida 2010). In this method, the mixture of two non-miscible solvents (water from fruit matrix and an organic solvent) is sonicated (see Fig. 3a). The technique allows for rapid extraction of several samples simultaneously and reduces the amount of material and organic solvent (Sargenti and Almeida 2010). The technique has been successfully applied for patulin extraction from apple juice using ethyl acetate (Sargenti and Almeida 2010) and whole apple using ammonium acetate-acetic acid solution in methanol-water (95:5, v/v) (Christensen et al. 2009).

Scheme of a ultrasonic-assisted liquid-liquid extraction (ULLE), b dispersive liquid-liquid microextraction (DLLME), and c vortex-assisted liquid-liquid extraction (VALLME) methodology
Another promising and environmentally friendly method is the dispersive liquid-liquid microextraction (DLLME). It is based on injection of the extraction and disperser solvent mixture into the aqueous sample containing target compounds. As a result, the generated fine droplets display greater surface area between the extraction solvent and the aqueous sample. This effect enhances extraction efficiency. Following centrifugation of a cloudy solution, the sedimented phase is collected for analyte determination. The advantage of this method is its simplicity, reduction to microliters in amount of organic solvents, and short preparation time. The proposed methodology provides high extraction efficiency and good reproducibility. The main steps of DLLME are presented in Fig. 3b. The method has been used for patulin quantification in apple juices and concentrates (Farhadi and Maleki 2011). In this work, acetonitrile as a dispersive solvent (1 mL) and chloroform (0.5 mL) as an extraction solvent were applied.
The vortex-assisted liquid–liquid microextraction (VALLME) is an alternative LLE-based approach. Dispersion of microvolumes of an extraction solvent into the aqueous sample is achieved by vortex agitation. The generation of fine droplets speeds up extraction process of the target compounds due to shorter diffusion distance and greater extraction surface (Yiantzi et al. 2010). After phase separation, the organic layer can be collected and directly applied into the chromatographic system. The principles of this technique are schematically presented in Fig. 3c. It was successfully applied for patulin isolation from apples, mango, and grapes juices (Abu-Bakar et al. 2014).
Solid-Phase Extraction
The solid-phase extraction (SPE) is another example of an environment-friendly approach in sample preparation for chromatographic analysis. The growing interest in SPE techniques is related to multiple advantages over the traditional LLE, i.e., reduction in sample amount and organic solvents required for analysis, high recovery, and rapid procedure (Li et al. 2007b). SPE is based on dissolving or suspending of sample in a solvent (such as acetonitrile) and passing the mixture through a solid phase. It separates analytes based on difference in compounds affinity to the sorbent. This approach allows for isolation, concentration, as well as purification of target molecule. SPE has been appreciated by researchers studying patulin contamination in variety of fruit products (juices, purees, jams) and was used separately (Li et al. 2007b; Funes and Resnik 2009; Catana et al. 2011; Zhou et al. 2012) or in combination with liquid-liquid extraction (Spadaro et al. 2008; Barreira et al. 2010; Zaied et al. 2013; Karakose et al. 2015; Seo et al. 2015; De Clercq et al. 2016) (Tamura et al. 2012). Nevertheless, like any other technique, SPE has also some limitations. It was not ideal for clarified juice, dried fruits, or other complex fruit-based products (Ji et al. 2017).
There are some commercially available SPE columns dedicated for patulin analysis in food, i.e., MycoSep 228 SPE column was evaluated for patulin determination in domestic apple and hawthorn beverages (Li et al. 2007a). In order to enhance the efficiency of patulin separation from other fruit matrix components, novel sorbents have been developed as an alternative solution to the commercially available SPE columns. Different polymers as solid-phase sorbents have been investigated to clean-up and pre-concentrate patulin from fruit products. Zhou and co-workers developed the home-made polyvinylpolypyrrolidone-florisil (PVPP-F) SPE columns for apple jam, apple, and hawthorn juices pre-treatment (Zhou et al. 2012).
Molecularly imprinted polymers (MIPs) have been also proposed and used by Khorrami and Taherkhani (2011). The oxindole molecule served as a dummy template to synthesize MIPs capable of selective binding of the mycotoxin from apple juice. The polymer was prepared in the non-covalent approach based on a free radical polymerization of methacrylic acid (functional monomer) and ethylene glycol dimethacrylate (cross-linker). Unfortunately, the proposed method for MIPs preparation is quite complicated and no significant improvement of recoveries was observed (from 84.31 to 88.89%). Zho and co-workers have proposed an alternative approach for MIPs preparation by a radical so called “grafting from” polymerization method (Zhao et al. 2011). A silica surface was first pre-grafted with amino groups and in further steps the synthesis of MIP was carried out in the presence of 6-hydroxynicotinic acid (a template substitute), acrylamide (functional monomer), and ethylene glycol dimethacrylate (cross-linker). The final material was successfully applied as the patulin-selective SPE sorbent. The obtained recoveries of analyte during patulin analysis in apple juice were from 90.08 to 96.59% (Zhao et al. 2011).
QuEChERS
QuEChERS (acronym of Quick, Easy, Cheap, Effective, Rugged, and Safe) is gaining significant popularity as the sample pre-treatment method for chromatographic analysis due to advantages included in its name. Traditionally, the QuEChERS methodology involves an initial extraction of 10 g of a homogenized sample using 10 mL of acetonitrile followed by the separation step after addition of a salt mixture (4 g of magnesium sulfate anhydrous and 1 g of sodium chloride). The acetonitrile extract (1 mL) is further subjected to purification by dispersive solid-phase extraction (dSPE) employing 150 mg of anhydrous magnesium sulfate and 25 mg of primary secondary amine (PSA) sorbent (Anastassiades et al. 2003). Dispersive SPE shows many advantages comported to traditional SPE such as elimination of conditioning and elution steps, decreased sorbent consumption, and obligatory use of additional equipment, i.e., vacuum/pressure or flow control devices. In dSPE clean-up, the sorbents are chosen to selectively retain co-extracted, interfering compounds from the sample matrix and to allow analyte retention in the liquid phase. Several dSPE sorbents have been tested to obtain satisfactory recoveries and accurate results, i.e., PSA sorbent effectively removes many polar matrix components, such as organic acids, polar pigments, and sugars from the food extracts. The carbon-based sorbents provide good removal of carotenoids, chlorophyll, and sterols, whereas addition of octadecyl silica (C18) is recommended for samples rich in fat and waxes (Rejczak and Tuzimski 2015).
The original QuECHERS procedure has been modified since then for better adaptation into the unique applications. In case of patulin quantification in fruit matrices, the extraction solvents and composition of the salt mixture used for separation and purification steps have been optimized. Instead of acetonitrile (Marsol-Vall et al. 2014), methanol (Vaclavikova et al. 2015), mixture of water, and acetonitrile (Desmarchelier et al. 2011) or acetonitrile acidified with acetic acid (Sadok et al. 2018) can be used for patulin extraction. The improvements in matrix compounds separation could be achieved by introduction of buffering salts (sodium citrate, sodium hydrogen citrate sesquihydrate) into traditional mixture of magnesium sulfate anhydrous and sodium chloride for better pH control and improved recovery rate (Marsol-Vall et al. 2014; Sadok et al. 2018). The purification step is performed using dSPE (Desmarchelier et al. 2011; Marsol-Vall et al. 2014; Sadok et al. 2018) and rarely by traditional SPE columns (Tamura et al. 2012).
Despite many advantages, QuEChERS protocol consumes relatively large amount of the extraction solvents compared to mentioned above DLLME and VALLME approaches. Furthermore, the efficiency of extraction strongly depends on type and amount of salts used in salting-out step. The procedure requires optimization step to determine the optimal conditions that will prevent co-extraction of some undesirable compounds from the sample matrix (e.g., sugars, pigments) (Rejczak and Tuzimski 2015). Furthermore, QuEChERS methodology requires subsequent clean-up of the obtained extract that extends preparation time and cost of analysis. The multi-step nature of QuEChERS favors target compound loss. It is often controlled by addition of an internal standard at an early stage of sample preparations step. Unfortunately, it introduces a new factor that needs to be investigated during method development.
QuEChERS methodology has been recently applied for patulin determination in different fruits: apples, pears, peaches, apricots, bananas, strawberries, grapes, plums, raspberries, blackberries, blueberries, sour cherries (Anastassiades et al. 2003; Desmarchelier et al. 2011; Marsol-Vall et al. 2014; Sadok et al. 2018; Vaclavikova et al. 2015; Tamura et al. 2012).
Matrix Solid-Phase Dispersion
In the case of solid and semi-solid sample preparation for chromatographic analysis, another technique, namely matrix solid-phase dispersion (MSPD), seems to be worth considering. In this approach, sample is mechanically dispersed in a solid support (mainly a C18 or C8-bonded silica), creating a unique stationary phase that is then packed into an empty cartridge. The applied sample is eluted with a proper solvent and the obtained eluent is subjected for further actions (evaporation, reconstruction, analysis). The great advantage of the technique is the simultaneous extraction and clean-up of the target substances. However, this approach is fairly laborious regarding sample/dispersant mixing and column packing with obtained material. Furthermore, it requires relatively large volumes of organic solvents for analyte elution (10–15 mL per sample). In some cases, like during SPE, a column should be washed before sample elution. This extra step increases total time of sample preparation and amount of the organic solvents (Wu et al. 2008). MSPD methodology has been tested for patulin extraction from rot apple and apple juice concentrate (Wu et al. 2008), as well as for other mycotoxin analysis in baby food (Rubert et al. 2012).
Importance of Sample pH Control
Patulin is more stable in the slightly acidic medium (Valle-Algarra et al. 2009) and optimal pH ensures satisfactory target molecule recoveries. There are three critical points within a whole preparation step at which pH should be taken into account (presented in Fig. 4). The initial pH adjustment might be ensured at an extraction step. As shown recently, the extraction using acetonitrile acidified with acetic acid (1%, v/v) has provided better patulin recoveries in strawberry matrix compared to a pure acetonitrile (Sadok et al. 2018). Improvement of patulin recovery in compotes and apple purees has been also observed after addition of NaH2PO4 to ethyl acetate-hexane mixture (94:6, v/v) (Valle-Algarra et al. 2009). It makes the sample pH slightly acidic and protects patulin molecule from degradation. As suggested, acidified solution helps in extraction of the mycotoxin by breaking interactions between toxins and other constituents (e.g., proteins and sugars) (Rahmani et al. 2009). On the other hand, the samples with lower pH show tendency to produce extracts with higher amount of the co-extracting compounds (Rejczak and Tuzimski 2015). This negative effect has been also shown in Fig. 2. At the later steps, after extract clean-up, pH of the sample should be controlled to avoid analyte loss, since some clean-up steps might increase sample pH and endanger the stability of base-sensitive compounds like patulin. This is commonly observed during QuEChERS protocol after PSA clean-up in dSPE procedure. In this case, pH of the purified extract could reach values in the range from 8 to 9 (Rejczak and Tuzimski 2015). The similar situation occurs after extract purification using Na2CO3 in a typical LLE protocol. For this reason, sample adjustment with glacial acetic acid to pH 4 is also practiced after different extract purification procedures (Wu et al. 2008; Yuan et al. 2010; Zouaoui et al. 2015; Iqbal et al. 2018). The last step when pH is kept low is the final reconstitution of a sample before analysis. The mobile phase used for sample reconstitution is frequently acidified with acetic acid (Spadaro et al. 2008; Funes and Resnik 2009; Barreira et al. 2010; Cho et al. 2010; Forouzan and Madadlou 2014).

Summary of the fruit sample preparation for patulin analysis with indication of pH check point
Patulin Quantification Using Liquid Chromatography
Several chromatographic techniques are used for patulin analysis in food. Among the applied methods of mycotoxin determination, the thin-layer chromatography (TLC), high-performance liquid chromatography (HPLC) coupled with spectroscopic detectors, ultra-high-performance liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS), and gas chromatography (GC) are most often utilized. HPLC with UV absorbance detectors and LC-MS/MS systems are frequently used for patulin quantification in fruit commodities, therefore we have summarized below the developments in this field within recent decade.
HPLC Methods with UV Absorbance Detection
The analysis using the HPLC coupled with ultraviolet (UV) or diode array (DAD) detectors present the most popular chromatographic techniques used for patulin determination in different fruits and their products (see Table 5). The majority of published articles in recent 10 years employing HPLC show applicability of this method for mycotoxin quantification at concentration of 10 μg kg−1 or 10 μg L−1 that meets the EU recommendations for baby food (see Table 3). The HPLC separation is frequently achieved using a mobile phase composed of water and small amount of acetonitrile (5–10%, v/v). Since patulin is more stable in acidic conditions, acidification of the mobile phase with perchloric acid (Spadaro et al. 2008; Gaspar and Lucena 2009; Barreira et al. 2010) and acetic acid (Sargenti and Almeida 2010; Karakose et al. 2015; De Clercq et al. 2016; Sadok et al. 2018) is also used. Due to the molecule maximal absorbance at 276 nm, this wavelength is used for patulin identification and quantification purpose (Li et al. 2007b; Spadaro et al. 2008; Barreira et al. 2010; Bonerba et al. 2010b; Sargenti and Almeida 2010; Cho et al. 2010; Khorrami and Taherkhani 2011; Catana et al. 2011; Farhadi and Maleki 2011; Zaied et al. 2013; Marsol-Vall et al. 2014; Forouzan and Madadlou 2014; Zouaoui et al. 2015; Karakose et al. 2015; De Clercq et al. 2016; Ji et al. 2017; Sadok et al. 2018; Iqbal et al. 2018). Unfortunately, detection based on this wavelength suffers from low selectivity due to obstructing phenolic compounds, especially 5-hydroxymethylfurfural (5-HMF) (structure shown in Fig. 1) present in the analyzed sample. This aldehyde is formed during thermal treatment of food as an intermediate product of acid-catalyzed degradation of hexose and decomposition of 3-deoxyosone in the Maillard reaction (Fallico et al. 2008). In literature, there are reports showing negative and positive influence of 5-HMF on human and animal health, such as mutagenic (Monien et al. 2012), carcinogenic (Zhang et al. 1993), antioxidative (Li et al. 2009), and anti-allergic (Alizadeh et al. 2017) effects. In fresh fruits, 5-HMF is nearly undetectable, in contrast to processed food, where it is found at significant levels (Kus et al. 2005). Thus, it might be a good marker to evaluate thermal damages or aging in food products. 5-HMF is also the most common interfering contaminant observed during mycotoxin determination in apple-derivative samples (Cho et al. 2010; Lee et al. 2014). Patulin and 5-HMF exhibit strong UV absorption, similar retention time under chromatographic conditions, and consequently tendency to peak overlapping. The structures of both compounds were compared in Fig. 1. The improvements in separation of patulin and 5-HMF peaks presented the analytical challenge undertaken in many HPLC-UV studies. Finally, the optimal conditions for apple and apple-based products analysis have been established (Gaspar and Lucena 2009; Abu-Bakar et al. 2014; Lee et al. 2014; Karakose et al. 2015).
LC-MS Methods
The liquid chromatography–mass spectrometry (LC-MS) is overall used in food analysis because of its versatility, specificity, and selectivity. The majority of studies focusing on patulin monitoring in fruits utilize the LC coupled with triple quadrupole electrospray tandem mass spectrometer (LC-QQQ) operated in negative-ion mode. The ion of m/z 153 (corresponding to a patulin molecule after proton lost, [M–H]−) is frequently chosen as the precursor ion characteristic for patulin. The mobile phase composed of water and methanol or water and acetonitrile with addition of acetic acid (Tamura et al. 2012; Hammami et al. 2017) or ammonium acetate (Christensen et al. 2009; Malachová et al. 2014; Seo et al. 2015; Vaclavikova et al. 2015) is used for this mycotoxin analysis. Unfortunately, due to high polarity of target molecule, the LC-MS analysis suffers from low sensitivity. It is a result of patulin poor ionization under both electrospray (ESI) and atmospheric pressure chemical ionization (APCI) conditions (Vaclavikova et al. 2015). Matrix effects, observed as a suppression or enhancement of signal from a target molecule, must be also considered when performing LC-MS analysis of complex matrices, i.e., fruit samples. The application of an isotopically labeled standard of patulin and a matrix-matched methodology minimize these effects and are generally employed when developing analytical methods for patulin quantification.
The LC-MS analytical protocols are most often developed for multi-target analyses. Such approach compromises sensitivity of the method and might be insufficient for patulin quantification at legislative limits set by EU for baby food. For example, LC-QQQ has been applied for simultaneous determination of 33 pesticides or degradation products together with patulin in apples (Christensen et al. 2009). The achieved limit of quantification (LOQ) for patulin was 40 μg kg−1 with recoveries in the range from 86.0 to 101.0%. In another work, the LC-MS/MS approach has been used for determination of 295 fungal and bacterial metabolites along with patulin quantification (Malachová et al. 2014). The method has been validated on four different food matrices: apple purees, hazelnuts, maize, and green pepper. The obtained LOQ for patulin in apple matrix was 119.7 μg kg−1 with recoveries of 77%. Tamura and co-workers have applied LC-QQQ protocol for patulin and 13 other mycotoxins quantification in white and red wines after SPE clean-up. LOQ for patulin was 5 μg L−1 with recoveries of 78% (Tamura et al. 2012). To overcome sensitivity drawback, several single-target LC-MS methods that employed more sophisticated sample pre-treatment have been proposed for patulin quantification. Application of the in-tube solid-phase microextraction (SPME) coupled with LC-MS has resulted in an excellent detection limit of 23.5 ng L−1 in water (Kataoka et al. 2009). The method was used for patulin determination in different fruit juices (apple, grape, orange, blueberry, lemon, pear, mango, coconut) and dried fruits (apple, apricot, kiwi, prune, pineapple, papaya, mango, fig). Recoveries estimated for apple juice matrix was above 92%. In addition, involvement of QuEChERS technique for apple and pear-based commodities preparation before LC-MS/MS analysis resulted in better recoveries reaching 94–104% and LOQs up to 10 μg L−1 for all tested matrices (Desmarchelier et al. 2011). The more advanced LC method with triple quadrupole analyzer has also been applied for individual patulin determination in four different apple matrices (juice, fruit, puree, and compote) (Beltrán et al. 2014). The obtained LOQs for all matrices were in the range from 2 to 15 μg kg−1 with average recoveries from 71 to 108%. The LC-MS/MS has been successfully used for patulin monitoring in a variety of fresh fruits including apples, pears, apricots, peaches, bananas, grapes, plums, strawberries, raspberries, blueberries, blackberries, sour cherries, and their products (juices, pulps) (Vaclavikova et al. 2015). The following transitions were recorded in case of the mycotoxin analysis: 213.0 > 152.9 (quantifier), 213.0 > 109.0 (qualifier). Employing a combination of the QuEChERS procedure and the SPE clean-up enabled an effective removal of matrix components and pre-concentration of patulin that resulted in limits of quantification ranging from 1 to 2.5 μg kg−1and good recoveries. In some work, the authors have utilized LLE followed by SPE clean-up and LC-MS/MS quantification of patulin in apple juice, ground fruit, and jam using (Seo et al. 2015). The method provides satisfactory LOQs in the range from 0.8 to 2.4 μg kg−1. Most recently, LC-MS/MS has been also applied for single-target monitoring of patulin content in apples and apple-based products (Hammami et al. 2017).
New Directions in Patulin Analysis: Summary Remarks
The apple juices are frequently monitored for patulin presence due to high probability of contamination by this mycotoxin. The LLE approach set by AOAC was sufficient for patulin isolation from juice matrix (MacDonald et al. 2000). However, the method employs several extraction steps using large volumes of ethyl acetate and sodium carbonate that promotes patulin degradation. Therefore, other extraction methods like ULLE, DLLME, and VLLME have been adopted for the mycotoxin isolation from fruit matrix. These methodologies provide good extraction efficiency simultaneously with reduction of organic solvent consumption. Furthermore, they are faster and easier to perform compared to the traditional LLE that importantly meets green chemistry recommendations. However, LLE-derived techniques may fail in term of chromatographic analysis of patulin in more complex matrices like dried or pigmented fruits (berries, sour cherries) or their products. It brings the necessity to adjust existing or to develop new analytical protocols, i.e., more sophisticated sample preparation. Different sample components that strongly affect chromatographic peak resolution and ionization efficiency are a reason for unsatisfactory recovery and in consequence make patulin quantification difficult at the legislative levels. Recent advances in this field mainly focused on adjustment of SPE and QuEChERS. It allowed for development of effective protocols for removal of interfering compounds (like phenolic molecules) from the complex fruit matrices. Application of molecularly imprinted polymers (MIPs) as SPE sorbents and introduction of different modifications into the original QuEChERS protocol have been proposed in order to improve sample clean-up and patulin pre-concentration.
Although HPLC-UV method is still preferably used for this mycotoxin analysis in fruit commodities, it suffers from extended time of analysis (vital for good peak resolution) and consequently quite large mobile phase consumption. Therefore, the UHPLC systems followed by mass spectrometry detection are favored over HPLC-UV method that is being slowly displaced in organic compounds analysis.
Importantly, due to patulin poor ionization in a mass spectrometer source, this toxin is frequently omitted from a list of multi-target mycotoxin analysis. Instead, it is individually determined employing separate protocol. It seems to be the one of the main obstacles that inhibits development of multi-mycotoxin approaches and great care must be paid to resolve the patulin sensitivity problem on the future path of LC-MS-based methods.
Conclusions
Increased public awareness of health and food quality is reflected in studies currently undertaken by many researchers. Mycotoxin contamination is one of the popular topic and it includes patulin quantification by LC in different fruit commodities. Consumption of the mycotoxin-contaminated fruits presents health risk and mycotoxin level should be strictly controlled, especially in baby food. The recent developments allowed for highlighting frequent occurrence of patulin in a variety of fruits including mostly studied apples, other stone fruits, as well as pigmented fruits and derived products.
The need for monitoring of complex fruit matrix to guarantee an effective control of food toxin level resulted in improvements of chemical tools. Developments in analytical approaches focus on new strategies for sample pre-treatment and chromatographic analysis to achieve detection levels meeting the regulatory recommendations. The trend in using multi-compound analysis presents the very attractive approach in terms of time and cost. Additional work needs to be done to identify optimal analytical conditions allowing for testing of the complex fruit matrices with low amount of patulin contamination. Until then, the single compound analysis for patulin quantification might be necessary to meet the analytical standards for baby food testing according to the EU recommendations.
References
Abu-Bakar N-B, Makahleh A, Saad B (2014) Vortex-assisted liquid-liquid microextraction coupled with high performance liquid chromatography for the determination of furfurals and patulin in fruit juices. Talanta 120:47–54. https://doi.org/10.1016/j.talanta.2013.11.081
Alizadeh M, Khodaei H, Mesgari Abbasi M, Saleh-Ghadimi S (2017) Assessing the effect of 5-hydroxymethylfurfural on selected components of immune responses in mice immunised with ovalbumin. J Sci Food Agric 97:3979–3984. https://doi.org/10.1002/jsfa.8261
Anastassiades M, Lehotay SJ, Štajnbaher D, Schenck FJ (2003) Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J AOAC Int 86:412–431
Anderson MS, Dutton MF, Harding K (1979) Production and degradation of patulin by paecilomyces species, a common contaminant of silage. J Sci Food Agric 30:229–232. https://doi.org/10.1002/jsfa.2740300303
Arzu Koçkaya E, Selmanoǧlu G, Özsoy N, Gül N (2009) Evaluation of patulin toxicity in the thymus of growing male rats. Arh Hig Rada Toksikol 60:411–418. https://doi.org/10.2478/10004-1254-60-2009-1973
Assunção R, Alvito P, Kleiveland CR, Lea TE (2016) Characterization of in vitro effects of patulin on intestinal epithelial and immune cells. Toxicol Lett 250–251:47–56. https://doi.org/10.1016/j.toxlet.2016.04.007
Baert K, De Meulenaer B, Kasase C et al (2007) Free and bound patulin in cloudy apple juice. Food Chem 100:1278–1282. https://doi.org/10.1016/j.foodchem.2005.10.012
Baker RA (1997) Reassessment of some fruit and vegetable pectin levels. J Food Sci 62:225–229
Barreira MJ, Alvito PC, Almeida CMM (2010) Occurrence of patulin in apple-based-foods in Portugal. Food Chem 121:653–658. https://doi.org/10.1016/j.foodchem.2009.12.085
Beltrán E, Ibáñez M, Sancho JV, Hernández F (2014) Determination of patulin in apple and derived products by UHPLC-MS/MS. study of matrix effects with atmospheric pressure ionisation sources. Food Chem 142:400–407. https://doi.org/10.1016/j.foodchem.2013.07.069
Bonerba E, Ceci E, Conte R, Tantillo G (2010a) Survey of the presence of patulin in fruit juices. Food Addit Contam Part B Surveill 3:114–119. https://doi.org/10.1080/19393210.2010.490882
Bonerba E, Conte R, Ceci E, Tantillo G (2010b) Assessment of dietary intake of patulin from baby foods. J Food Sci 75:T123–T125. https://doi.org/10.1111/j.1750-3841.2010.01743.x
Boussabbeh M, Ben Salem I, Prola A et al (2015) Patulin induces apoptosis through ROS-mediated endoplasmic reticulum stress pathway. Toxicol Sci 144:328–337. https://doi.org/10.1093/toxsci/kfu319
Caller M, Mackinney G (1965) The carotenoids of certain fruits (apple, pear, cherry, strawberry). J Food Sci 30:393–395. https://doi.org/10.1111/j.1365-2621.1965.tb01774.x
Catana M, Catana L, Lilios G et al (2011) Determination of patulin in apple juice. Rom J Food Sci 1:65–69
Celli M, Coelho A, Wosiacki G et al (2009) Patulin determination in apples with rotten areas. World Mycotoxin J. https://doi.org/10.3920/WMJ2008.1038
Cho MS, Kim K, Seo E et al (2010) Occurrence of patulin in various fruit juices from South Korea: an exposure assessment. Food Sci Biotechnol 19:1–5. https://doi.org/10.1007/s10068-010-0001-6
Christensen HB, Poulsen ME, Rasmussen PH, Christen D (2009) Development of an LC-MS/MS method for the determination of pesticides and patulin in apples. Food Addit Contam Part A 26:1013–1023
Collin S, Bodart E, Badot C et al (2008) Identification of the main degradation products of patulin generated through heat detoxication treatments. J Inst Brew 114:167–171. https://doi.org/10.1002/j.2050-0416.2008.tb00322.x
De Clercq N, Van Pamel E, Van Coillie E et al (2016) Optimization and validation of a method without alkaline clean-up for patulin analysis on apple puree agar medium (APAM) and apple products. Food Anal Methods 9:370–377. https://doi.org/10.1007/s12161-015-0190-y
de Melo FT, de Oliveira IM, Greggio S et al (2012) DNA damage in organs of mice treated acutely with patulin, a known mycotoxin. Food Chem Toxicol 50:3548–3555. https://doi.org/10.1016/j.fct.2011.12.022
Delgado-Pelayo R, Gallardo-Guerrero L, Hornero-Méndez D (2014) Chlorophyll and carotenoid pigments in the peel and flesh of commercial apple fruit varieties. Food Res Int 65:272–281. https://doi.org/10.1016/j.foodres.2014.03.025
Desmarchelier A, Mujahid C, Racault L et al (2011) Analysis of patulin in pear- and apple-based foodstuffs by liquid chromatography electrospray ionization tandem mass spectrometry. J Agric Food Chem 59:7659–7665. https://doi.org/10.1021/jf201461r
European Commission (2006a) Commission Regulation 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. 1–35
European Commission (2006b) Commission regulation 401/2006 of 23 February 2006 laying down the methods of sampling and analysis for the official control of the levels of mycotoxins in foodstuffs. Off J Eur Union 12–34
Fallico B, Arena E, Zappala M (2008) Degradation of 5-hydroxymethylfurfural in honey. J Food Sci 73. https://doi.org/10.1111/j.1750-3841.2008.00946.x
FAO (2003) Worldwide regulations for mycotoxins in food and feed in 2003. FAO Food Nutr Pap Rome 1–165. https://doi.org/10.1017/CBO9781107415324.004
Farhadi K, Maleki R (2011) Dispersive liquid-liquid microextraction followed by HPLC-DAD as an efficient and sensitive technique for the determination of patulin from apple juice and concentrate samples. J Chin Chem Soc 58:340–345. https://doi.org/10.1002/jccs.201190035
Fliege R, Metzler M (1999) The mycotoxin patulin induces intra- and intermolecular protein crosslinks in vitro involving cysteine, lysine, and histidine side chains, and α-amino groups. Chem Biol Interact 123:85–103. https://doi.org/10.1016/S0009-2797(99)00123-4
Fliege R, Metzler M (2000) Electrophilic properties of patulin. N-acetylcysteine and glutathione adducts. Chem Res Toxicol 13:373–381. https://doi.org/10.1021/tx9901480
Forouzan S, Madadlou A (2014) Incidence of patulin in apple juices produced in west Azerbayjan Province, Iran. J Agric Sci Technol 16:1613–1622
Funes GJ, Resnik SL (2009) Determination of patulin in solid and semisolid apple and pear products marketed in Argentina. Food Control 20:277–280. https://doi.org/10.1016/j.foodcont.2008.05.010
Gaspar EMSM, Lucena AFF (2009) Improved HPLC methodology for food control—furfurals and patulin as markers of quality. Food Chem 114:1576–1582. https://doi.org/10.1016/j.foodchem.2008.11.097
Glaser N, Stopper H (2012) Patulin: mechanism of genotoxicity. Food Chem Toxicol 50:1796–1801. https://doi.org/10.1016/j.fct.2012.02.096
Government of Singapore (2018) Food (Amendment) Regulations 2018 (G.N. No. S 146/2018)
Guerra-Moreno A, Hanna J (2017) Induction of proteotoxic stress by the mycotoxin patulin. Toxicol Lett 276:85–91. https://doi.org/10.1016/j.toxlet.2017.05.015
Hammami W, Al Thani R, Fiori S et al (2017) Patulin and patulin producing Penicillium spp. Occurrence in apples and apple-based products including baby food. J Infect Dev Ctries 11:343–349. https://doi.org/10.3855/jidc.9043
Hao H, Zhou T, Koutchma T et al (2016) High hydrostatic pressure assisted degradation of patulin in fruit and vegetable juice blends. Food Control 62:237–242. https://doi.org/10.1016/j.foodcont.2015.10.042
Harris KL, Bobe G, Bourquin LD (2009) Patulin surveillance in apple cider and juice marketed in Michigan. J Food Prot 72:1255–1261
Harrison MA (1988) Presence and stability of patulin in apple products: a review. J Food Saf 9:147–153. https://doi.org/10.1111/j.1745-4565.1988.tb00515.x
Hasan HAH (2000) Patulin and aflatoxin in brown rot lesion of apple fruits and their regulation. World J Microbiol Biotechnol 16:607–612. https://doi.org/10.1023/A:1008982511653
Horbowicz M, Grzesiuk A, Dębski H, Kosson R (2008) Anthocyanins of fruits and vegetables—their occurrence, analysis and role in human. Veg Crop Res Bull 68:5–22. https://doi.org/10.2478/v10032-008-0001-8
Iqbal SZ, Malik S, Asi MR et al (2018) Natural occurrence of patulin in different fruits, juices and smoothies and evaluation of dietary intake in Punjab, Pakistan. Food Control 84:370–374. https://doi.org/10.1016/j.foodcont.2017.08.024
Jalali A, Khorasgani ZN, Goudarzi M, Khoshlesan N (2010) HPLC determination of patulin in apple juice: a single center study of southwest area of Iran. J Pharmacol Toxicol 5. https://doi.org/10.3923/jpt.2010.208.214
Ji X, Li R, Yang H et al (2017) Occurrence of patulin in various fruit products and dietary exposure assessment for consumers in China. Food Control 78:100–107. https://doi.org/10.1016/j.foodcont.2017.02.044
Karakose A, Sanli S, Sanli N, Bulduk I (2015) Evaluation of patulin in commercial baby foods by solid phase extraction and liquid chromatography PDA detection. Czech J Food Sci 33:52–57. https://doi.org/10.17221/198/2014-CJFS
Kataoka H, Itano M, Ishizaki A, Saito K (2009) Determination of patulin in fruit juice and dried fruit samples by in-tube solid-phase microextraction coupled with liquid chromatography-mass spectrometry. J Chromatogr A 1216:3746–3750. https://doi.org/10.1016/j.chroma.2009.03.017
Katerere DR, Stockenström S, Shephard GS (2008) HPLC-DAD method for the determination of patulin in dried apple rings. Food Control 19:389–392. https://doi.org/10.1016/j.foodcont.2007.04.015
Kharandi N, Babri M, Azad J (2013) A novel method for determination of patulin in apple juices by GC-MS. Food Chem 141:1619–1623. https://doi.org/10.1016/j.foodchem.2013.05.080
Khoo HE, Prasad KN, Kong KW et al (2011) Carotenoids and their isomers: color pigments in fruits and vegetables. Molecules 16:1710–1738
Khoo HE, Azlan A, Tang ST, Lim SM (2017) Anthocyanidins and anthocyanins: colored pigments as food, pharmaceutical ingredients, and the potential health benefits. Food Nutr Res 61:1361779. https://doi.org/10.1080/16546628.2017.1361779
Khorrami AR, Taherkhani M (2011) Synthesis and evaluation of a molecularly imprinted polymer for pre-concentration of Patulin from apple juice. Chromatographia 73(Suppl):S151–S156. https://doi.org/10.1007/s10337-010-1892-3
Kokkinidou S, Floros JD, Laborde LF (2014) Kinetics of the thermal degradation of patulin in the presence of ascorbic acid. J Food Sci 79. https://doi.org/10.1111/1750-3841.12316
Kus S, Gogus F, Eren S (2005) Hydroxymethyl furfural content of concentrated food products. Int J Food Prop 8:367–375
Lee TP, Sakai R, Manaf NA et al (2014) High performance liquid chromatography method for the determination of patulin and 5-hydroxymethylfurfural in fruit juices marketed in Malaysia. Food Control 38:142–149. https://doi.org/10.1016/j.foodcont.2013.10.018
Li F, Zhao S, Chin L et al (2007a) Determination of patulin in apple and hawthorn beverages by solid-phase filtration column and liquid chromatography. Journal of AOAC International, In, pp 167–172
Li J, Wu R, Hu Q, Wang J (2007b) Solid-phase extraction and HPLC determination of patulin in apple juice concentrate. Food Control 18:530–534. https://doi.org/10.1016/j.foodcont.2005.12.014
Li YX, Li Y, Qian ZJ et al (2009) In vitro antioxidant activity of 5-HMF isolated from marine red alga Laurencia undulata in free radical mediated oxidative systems. J Microbiol Biotechnol 19:1319–1327. https://doi.org/10.4014/jmb.0901.0004
Lovett J, Peeler JT (1973) Effect of pH on the thermal destruction kinetics of patulin in aqueous solution. J Food Sci 38:1094–1095. https://doi.org/10.1111/j.1365-2621.1973.tb02163.x
Luft P, Oostingh GJ, Gruijthuijsen Y et al (2008) Patulin influences the expression of Th1/Th2 cytokines by activated peripheral blood mononuclear cells and T cells through depletion of intracellular glutathione. Environ Toxicol 23:84–95. https://doi.org/10.1002/tox.20309
Määttä KR, Kamal-Eldin A, Riitta Törrönen A (2003) High-performance liquid chromatography (HPLC) analysis of phenolic compounds in berries with diode array and electrospray ionization mass spectrometric (MS) detection: Ribes species. J Agric Food Chem 51:6736–6744. https://doi.org/10.1021/jf0347517
MacDonald S, Long M, Gilbert J et al (2000) Liquid chromatographic method for determination of patulin in clear and cloudy apple juices and apple puree: collaborative study. J AOAC Int 83:1387–1394
Malachová A, Sulyok M, Beltrán E et al (2014) Optimization and validation of a quantitative liquid chromatography-tandem mass spectrometric method covering 295 bacterial and fungal metabolites including all regulated mycotoxins in four model food matrices. J Chromatogr A 1362:145–156. https://doi.org/10.1016/j.chroma.2014.08.037
Marsol-Vall A, Delpino-Rius A, Eras J et al (2014) A fast and reliable UHPLC-PDA method for determination of patulin in apple food products using QuEChERS extraction. Food Anal Methods 7:465–471. https://doi.org/10.1007/s12161-013-9648-y
Ministry of Health of the Russian Federation (2001) Hygienic requirements for Safety and Nutrition value of Food Products. Sanitary and Epidemiological Rulea and Regulations (SanPin 2.3.2.1078–01)
Monien BH, Engst W, Barknowitz G et al (2012) Mutagenicity of 5-Hydroxymethylfurfural in V79 cells expressing human SULT1A1: identification and mass spectrometric quantification of DNA adducts formed. Chem Res Toxicol 25:1484–1492. https://doi.org/10.1021/tx300150n
Murillo-Arbizu M, Amézqueta S, González-Peñas E, de Cerain AL (2009) Occurrence of patulin and its dietary intake through apple juice consumption by the Spanish population. Food Chem 113:420–423. https://doi.org/10.1016/j.foodchem.2008.07.054
National Health and Family Planning Commission of the People’s Republic of China and China Food and Drug Administration (2017) GB 2761–2017, National Food Safety Standard Maximum Levels of Mycotoxins in Foods
Neri F, Donati I, Veronesi F et al (2010) Evaluation of Penicillium expansum isolates for aggressiveness, growth and patulin accumulation in usual and less common fruit hosts. Int J Food Microbiol 143:109–117. https://doi.org/10.1016/j.ijfoodmicro.2010.08.002
Okeke B, Seigle-Murandi F, Steiman R et al (1993) Identification of mycotoxin-producing fungal strains: a step in the isolation of compounds active against Rice fungal diseases. J Agric Food Chem 41:1731–1735. https://doi.org/10.1021/jf00034a040
Oroian M, Amariei S, Gutt G (2014) Patulin in apple juices from the Romanian market. Food Addit Contam Part B Surveill 7:147–150. https://doi.org/10.1080/19393210.2013.861518
Pavlović AV, Dabić DČ, Momirović NM et al (2013) Chemical composition of two different extracts of berries harvested in Serbia. J Agric Food Chem 61:4188–4194. https://doi.org/10.1021/jf400607f
Pennacchio A, Varriale A, Esposito MG et al (2015) A near-infrared fluorescence assay method to detect patulin in food. Anal Biochem 481:55–59. https://doi.org/10.1016/j.ab.2015.04.027
Piemontese L, Solfrizzo M, Visconti A (2005) Occurrence of patulin in conventional and organic fruit products in Italy and subsequent exposure assessment. Food Addit Contam 22:437–442. https://doi.org/10.1080/02652030500073550
Rahmani A, Jinap S, Soleimany F (2009) Qualitative and quantitative analysis of mycotoxins. Compr Rev Food Sci Food Saf 8:202–251. https://doi.org/10.1111/j.1541-4337.2009.00079.x
Reddy KRN, Spadaro D, Lore A et al (2010) Potential of patulin production by Penicillium expansum strains on various fruits. Mycotoxin Res 26:257–265. https://doi.org/10.1007/s12550-010-0064-5
Rejczak T, Tuzimski T (2015) A review of recent developments and trends in the QuEChERS sample preparation approach. Open Chem 13:980–1010
Rice SL (1980) Patulin production by Byssochlamys spp. in canned grape juice. J Food Sci 45:485–488. https://doi.org/10.1111/j.1365-2621.1980.tb04081.x
Rice S, Beuchat L, Worthington R (1977) Patulin production by Byssochlamys spp. in fruit juices. Appl Environ Microbiol 34:791–796
Ritieni A (2003) Patulin in Italian commercial apple products. J Agric Food Chem 51:6086–6090. https://doi.org/10.1021/jf034523c
Roland JO, Beuchat LR (1984) Biomass and patulin production by Byssochlamys nivea in apple juice as affected by sorbate, benzoate, SO2 and temperature. J Food Sci 49:402–406. https://doi.org/10.1111/j.1365-2621.1984.tb12432.x
Rubert J, Soler C, Mañes J (2012) Application of an HPLC-MS/MS method for mycotoxin analysis in commercial baby foods. Food Chem 133:176–183. https://doi.org/10.1016/j.foodchem.2011.12.035
Ruby J, Nathan PT, Balasingh J, Kunz TH (2000) Chemical composition of fruits and leaves eaten by short-nosed fruit bat, Cynopterus sphinx. J Chem Ecol 26:2825–2841. https://doi.org/10.1023/A:1026446011693
Sadok I, Szmagara A, Staniszewska MM (2018) The validated and sensitive HPLC-DAD method for determination of patulin in strawberries. Food Chem 245:364–370. https://doi.org/10.1016/j.foodchem.2017.10.093
Samson RA, Houbraken J, Varga J, Frisvad JC (2009) Polyphasic taxonomy of the heat resistant ascomycete genus Byssochlamys and its Paecilomyces anamorphs. Persoonia Mol Phylogeny Evol Fungi 22:14–27. https://doi.org/10.3767/003158509X418925
Sant’Ana A d S, Rosenthal A, de Massaguer PR (2008) The fate of patulin in apple juice processing: a review. Food Res Int 41:441–453
Sant’Ana AS, Simas RC, Almeida CAA et al (2010) Influence of package, type of apple juice and temperature on the production of patulin by Byssochlamys nivea and Byssochlamysfulva. Int J Food Microbiol 142:156–163. https://doi.org/10.1016/j.ijfoodmicro.2010.06.017
Sargenti SR, Almeida C (2010) Determination of patulin in apple juice by HPLC using a simple and fast sample preparation method. Eclética Quím 35:14–21. https://doi.org/10.1590/S0100-46702010000200002
Saxena N, Ansari KM, Kumar R et al (2009) Patulin causes DNA damage leading to cell cycle arrest and apoptosis through modulation of Bax, p53 and p21/WAF1 proteins in skin of mice. Toxicol Appl Pharmacol 234:192–201. https://doi.org/10.1016/j.taap.2008.09.033
Schumacher DM, Metzler M, Lehmann L (2005) Mutagenicity of the mycotoxin patulin in cultured Chinese hamster V79 cells, and its modulation by intracellular glutathione. Arch Toxicol 79:110–121. https://doi.org/10.1007/s00204-004-0612-x
Seo M, Kim B, Baek SY (2015) An optimized method for the accurate determination of patulin in apple products by isotope dilution-liquid chromatography/mass spectrometry. Anal Bioanal Chem 407:5433–5442. https://doi.org/10.1007/s00216-015-8705-3
Song E, Su C, Fu J et al (2014) Selenium supplementation shows protective effects against patulin-induced brain damage in mice via increases in GSH-related enzyme activity and expression. Life Sci 109:37–43. https://doi.org/10.1016/j.lfs.2014.05.022
Spadaro D, Garibaldi A, Gullino ML (2008) Occurrence of patulin and its dietary intake through pear, peach, and apricot juices in Italy. Food Addit Contam Part B Surveill 1:134–139. https://doi.org/10.1080/02652030802363790
Srivastava P, Malviya R (2011) Sources of pectin, extraction and its applications in pharmaceutical industry - an overview. Indian J Nat Prod Resour 2:10–18
Stachniuk A, Szmagara A, Czeczko R, Fornal E (2017) LC-MS/MS determination of pesticide residues in fruits and vegetables. J Environ Sci Health B 52:446–457
Steiman R, Seigle-Murandi F, Sage L, Krivobok S (1989) Production of patulin by Micromycetes. Mycopathologia 105:129–133. https://doi.org/10.1007/BF00437244
Sturm K, Koron D, Stampar F (2003) The composition of fruit of different strawberry varieties depending on maturity stage. Food Chem 83:417–422. https://doi.org/10.1016/S0308-8146(03)00124-9
Tamura M, Takahashi A, Uyama A, Mochizuki N (2012) A method for multiple mycotoxin analysis in wines by solid phase extraction and multifunctional cartridge purification, and ultra-high-performance liquid chromatography coupled to tandem mass spectrometry. Toxins (Basel) 4:476–486. https://doi.org/10.3390/toxins4060476
Tančinová D, Felšöciová S, Rybárik Ľ et al (2015) Colonization of grapes berries and cider by potential producers of patulin. Potravinarstvo 9:138–142. https://doi.org/10.5219/460
Vaclavikova M, Dzuman Z, Lacina O et al (2015) Monitoring survey of patulin in a variety of fruit-based products using a sensitive UHPLC–MS/MS analytical procedure. Food Control 47:577–584. https://doi.org/10.1016/j.foodcont.2014.07.064
Valle-Algarra FM, Mateo EM, Gimeno-Adelantado JV et al (2009) Optimization of clean-up procedure for patulin determination in apple juice and apple purees by liquid chromatography. Talanta 80:636–642. https://doi.org/10.1016/j.talanta.2009.07.040
Wei DM, Xu J, Shou DF et al (2017) Penicillium and patulin distribution in pears contaminated with Penicillium expansum. Determination of patulin in pears by UHPLC-MS/MS. J Integr Agric 16:1645–1651. https://doi.org/10.1016/S2095-3119(16)61543-5
Welke JE, Hoeltz M, Dottori HA, Noll IB (2009) Quantitative analysis of patulin in apple juice by thin-layer chromatography using a charge coupled device detector. Food Addit Contam Part A Chem Anal Control Expo Risk Assess 26:754–758. https://doi.org/10.1080/02652030802662746
Wu RN, Dang YL, Niu L, Hu H (2008) Application of matrix solid-phase dispersion-HPLC method to determine patulin in apple and apple juice concentrate. J Food Compos Anal 21:582–586. https://doi.org/10.1016/j.jfca.2008.05.010
Yiantzi E, Psillakis E, Tyrovola K, Kalogerakis N (2010) Vortex-assisted liquid-liquid microextraction of octylphenol, nonylphenol and bisphenol-A. Talanta 80:2057–2062. https://doi.org/10.1016/j.talanta.2009.11.005
Yuan Y, Zhuang H, Zhang T, Liu J (2010) Patulin content in apple products marketed in Northeast China. Food Control 21:1488–1491. https://doi.org/10.1016/j.foodcont.2010.04.019
Zaied C, Abid S, Hlel W, Bacha H (2013) Occurrence of patulin in apple-based-foods largely consumed in Tunisia. Food Control 31:263–267. https://doi.org/10.1016/j.foodcont.2012.10.005
Zhang XM, Chan CC, Stamp D et al (1993) Initiation and promotion of colonic aberrant crypt foci in rats by 5-hydroxymethy1-2-furaldehyde in thermolyzed sucrose. Carcinogenesis 14:773–775. https://doi.org/10.1093/carcin/14.4.773
Zhang X, Li Y, Shi J et al (2006) Patulin destabilization in acid solution using microwave. Trans Chinese Soc Agric Mach 37:64–67
Zhao D, Jia J, Yu X, Sun X (2011) Preparation and characterization of a molecularly imprinted polymer by grafting on silica supports: a selective sorbent for patulin toxin. Anal Bioanal Chem 401:2259–2273. https://doi.org/10.1007/s00216-011-5282-y
Zhou Y, Kong W, Li Y et al (2012) A new solid-phase extraction and HPLC method for determination of patulin in apple products and hawthorn juice in China. J Sep Sci 35:641–649. https://doi.org/10.1002/jssc.201100919
Zhu Y, Koutchma T, Warriner K, Zhou T (2014) Reduction of patulin in apple juice products by UV light of different wavelengths in the UVC range. J Food Prot 77:963–971. https://doi.org/10.4315/0362-028X.JFP-13-429
Zouaoui N, Sbaii N, Bacha H, Abid-Essefi S (2015) Occurrence of patulin in various fruit juice marketed in Tunisia. Food Control 51:356–360. https://doi.org/10.1016/j.foodcont.2014.09.048
Acknowledgments
This work was supported by the European Union from European Regional Development Fund under the Operational Programme Development of Eastern Poland 2007-2013 (agreement POPW.01.03.00-06-003/09-00) and the Polish National Science Centre grant for AS (DEC-2017/01/X/ST4/00722).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Ilona Sadok declares that she has no conflict of interest. Anna Stachniuk declares that she has no conflict of interest. Magdalena Staniszewska declares that she has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Informed consent is not applicable in this study.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sadok, I., Stachniuk, A. & Staniszewska, M. Developments in the Monitoring of Patulin in Fruits Using Liquid Chromatography: an Overview. Food Anal. Methods 12, 76–93 (2019). https://doi.org/10.1007/s12161-018-1340-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-018-1340-9