
Abstract
Metabolic dysfunction-associated fatty liver disease (MAFLD) is the most common liver disease worldwide. The risk of developing MAFLD varies among individuals, due to a combination of environmental inherited and acquired genetic factors. Genome-wide association and next-generation sequencing studies are leading to the discovery of the common and rare genetic determinants of MAFLD. Thanks to the great advances in genomic technologies and bioinformatics analysis, genetic and epigenetic factors involved in the disease can be used to develop genetic risk scores specific for liver-related complications, which can improve risk stratification. Genetic and epigenetic factors lead to the identification of specific sub-phenotypes of MAFLD, and predict the individual response to a pharmacological therapy. Moreover, the variant transcripts and protein themselves represent new therapeutic targets. This review will discuss the current status of research into genetic as well as epigenetic modifiers of MAFLD development and progression.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Metabolic dysfunction-associated fatty liver disease (MAFLD) defines fatty liver disease related to systemic metabolic dysregulation due to insulin resistance [1]. MAFLD is a multifactorial disease that encompasses a spectrum of pathological conditions, ranging from simple steatosis (MAFL), MASH, and fibrosis/cirrhosis which can lead ultimately to hepatocellular carcinoma (HCC) [2]. This condition was previously known as non-alcoholic fatty liver disease (NAFLD), and is now also referred to as metabolic dysfunction-associated steatotic liver disease (MASLD) [3]. Between 1999 and 2022, MAFLD-related mortality rate increased from 0.2 to 1.7 per 100,000 individuals. Hence, it is estimated that the number of MAFLD death will rise along with the risk of type 2 diabetes (T2D) and cardiovascular disease [4]. People with MAFLD have metabolic dysfunction triggered by excess adiposity and insulin resistance, and features of the metabolic syndrome including dyslipidemia, hypertension, hyperglycemia, and pro-inflammatory state [5]. Indeed, a positive energy balance due to excess in food intake and sedentary lifestyle leads to excess adiposity and potentially ectopic lipid accumulation in the liver and insulin resistance [6]. Adipose tissue insulin resistance causes increased lipolysis with excess circulating free fatty acids (FFA) flux to the liver, while at the same time hyperinsulinemia promotes de novo lipogenesis in hepatocytes. When the ability of hepatocytes to get rid of excess lipids by secretion of lipoproteins and oxidation is overwhelmed, hepatic fat accumulation drives lipotoxicity, lipid peroxidation, and elevated reactive oxygen species (ROS) production [7]. In addition, intestinal dysbiosis and enhanced intestinal permeability with bacterial translocation into the liver contribute to inflammation [7]. In patients with MAFLD, T2D affects liver disease progression and advanced fibrosis leading to a major risk of adverse outcomes, and it is also associated with higher cardiovascular mortality [5].
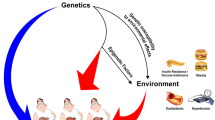
The risk of developing MAFLD varies even among individuals with insulin resistance, due to a combination of environmental and inherited genetic factors. Environmental factors, such as smoking, air pollution, and the exposure to toxins, are implicated in the development of liver disease [7] (Fig. 1). In the last decades, major advances in genomics technologies and bioinformatics have led to a better comprehension of the causes behind liver disease [8]. Genome-wide association studies (GWAS) identified the main common genetic variants modulating hepatocellular lipid metabolism. However, all in all these loci explained less than 25% of disease heritability [9]. The molecular understanding of liver disease continues to be redefined since the introduction of “next-generation sequencing” (NGS) in clinical practice, which also permits to identify rare variants with a strong impact on protein function and to diagnose a considerable number of cases previously classified as “cryptogenic” [10, 11]. This review aims to provide an update of the current knowledge on MAFLD genetics and epigenetics. Taking into consideration that the majority of the literature focuses on NAFLD, we will extrapolate data from literature related to different analysis such as GWAS and whole-exome sequencing (WES) in NAFLD and discuss the recent discoveries and limitations of these approaches, including biological understanding, risk prediction, and drug development (Fig. 2).

Heterogeneous factors lead to MAFLD, including ethnicity, sex, dietary habits, genetic predisposition, age, gut microbiota, and metabolic status. MAFLD is present if hepatic steatosis occurs with either obesity or overweight (BMI > 25 kg/m2 in white and >23 kg/m2 in Asian individuals), type 2 diabetes mellitus or evidence of metabolic dysregulation. At least two metabolic risk factors should be present for definition of metabolic dysregulation: waist circumference ≥102/88 cm in white male and female or ≥90/80 cm in Asian male and female; prediabetes; inflammation with elevated high-sensitive serum C-reactive protein level; elevated blood pressure or specific drug treatment; decreased HDL-cholesterol levels; increased plasma triglycerides levels. Heterogeneous factors lead to MAFLD, including ethnicity, sex, dietary habits, genetic predisposition, age, gut microbiota, and metabolic status

Genomic approaches to investigate MAFLD genetic determinants: GWAS to discovery the main common genetic variants; biological and pathways analysis to provide details regarding GWAS-prioritized tissues, and genes; PheWAS of MAFLD-risk-increasing alleles to identify distinct biological subgroupings; Mendelian randomization (MR) to estimate variant-MAFLD casual effect; PRS to stratify MAFLD risk in individuals with metabolic disorders; NGS techniques to identify rare variants involved in disease progression
MAFLD heritability
Epidemiological studies in multi-ethnic cohorts, and familial and twin studies demonstrated that inherited factors play an important role in determining MAFLD predisposition [12, 13]. The risk of advanced fibrosis is more than 12-fold higher in first-degree relatives of patients with MAFLD cirrhosis than in those without MAFLD irrespective of metabolic triggers [12, 14]. Long et al. [15] demonstrated that a greater proportion of individuals with a parental history of MAFLD had hepatic steatosis as compared to those without MAFLD in parents. In keeping, twin studies led to the estimation that about 38 and 100% of hepatic fat content and MAFLD variability depend on inherited factors [14]. Advances in nuclear magnetic resonance techniques and in the measurement of hepatic fat and fibrosis by transient elastometry showed again that these traits are inherited for about 50% [13, 16]. Studies on multi-ethnic cohorts have demonstrated that there is a major inter-ethnic variability in MAFLD susceptibility which is high in people of Hispanic and East Asian ancestry, intermediate in Europeans, and lower in individuals with African ancestry [17, 18]. Interestingly, the rs738409 variant encoding for PNPLA3 p.I148M accounts for a large fraction of the inter-ethnic variability in MAFLD predisposition (see below). However, a recent study suggests that the protection from chronic liver disease in African ancestry may be mostly accounted for by still unknown rare genetic variants [19, 20].
Gene loci associated with MAFLD development by GWAS
In the last 15 years, GWAS allowed to identify the “low hanging fruit”, that is the main common genetic determinants of MAFLD [9, 21]. The first hits were highlighted in the patatin-like phospholipase domain-containing 3 (PNPLA3) [22], transmembrane 6 superfamily member 2 (TM6SF2) [23], glucokinase regulator (GCKR) [24], and membrane-bound O-acyl-transferase 7 (MBOAT7) genes [25]. Variants at these loci modulate lipid handling by hepatocytes, e.g., substrate delivery for de novo lipogenesis, formation of lipid droplets, utilization of lipid for mitochondrial energy, compartmentalization of fatty acid, catabolism, assembly of very low-density lipoprotein, and their secretion [26]. The PNPLA3 p.I148M variant (rs738409 C>G) accounts for the largest fraction of genetic predisposition to MAFLD and it increases the susceptibility to the entire spectrum of hepatic damage associated with MAFLD, including MASH, fibrosis, and HCC [22, 27]. The presence of the p.I148M variant facilitates the accumulation of hepatic fat without affecting adiposity and insulin resistance [22] and it increases the risk of liver-related mortality in MAFLD patients and in the general population [28]. The carriage of rs58542926 C>T encoding for the p.E167K variant of TM6SF2 also causes hepatic triglyceride accumulation in intracellular lipid droplets. TM6SF2 is a transmembrane protein tightly bound to the endoplasmic reticulum, which stabilizes apolipoprotein B (ApoB) through two intraluminal loops [29]. The variant destabilizes the protein [29] hampering ApoB stability, lipidation, and secretion [30, 31]. The TM6SF2 variant also predisposes to the full spectrum of liver damage, that is MASH, severe fibrosis, and HCC [23, 32], but at the same time protects against cardiovascular disease through the reduction in lipoprotein secretion [33]. The rs641738 C>T variant situated 500 bases-downstream of the MBOAT7 gene is linked to fat deposition in the liver and the development of MAFLD, inflammation, fibrosis, and HCC [34]. The rs641738 variant is associated with lower hepatic mRNA and protein expression of MBOAT7, which is involved in the remodeling of phospholipids. The resulting increase in intracellular triglycerides is due to the induction a non-canonical pathway for triglyceride synthesis mediated by a futile cycle in phosphatidyl inositol metabolism [25]. The p.P446L GCKR variant (rs1260326) gene [24] curtails the ability to inhibit glucokinase in response to fructose-6-phosphate, thereby causing a constitutive activation of hepatic glucose uptake. This process reduces circulating glucose, but enhances the production of malonyl-CoA, which is a substrate for lipogenesis, blocking the oxidation of fatty acid and promoting fat accumulation in the liver [24].
In the last 2 years, recruitment of larger biobank cohorts led to pinpoint novel genetic determinants of MAFLD. Chen et al. [35] conducted a GWAS meta-analysis of MAFLD identified by imaging and diagnostic codes across diverse ancestries 17 new loci; the study highlighted new MAFLD-associated variants in or near fat mass and obesity associated (FTO), torsin family 1-member B (TOR1B), cordon-bleu WH2 repeat protein like 1 (COBLL1)/growth factor receptor-bound protein 14 (GRB14), insulin receptor (INSR), and sterol regulatory element-binding transcription factor 1 (SREBF1). The alteration in the expression of these genes affects insulin resistance, triglyceride, and cholesterol accumulation. By a phenome-wide association study (PheWAS), authors identified seven distinct clusters among the MAFLD variants and their associations with related phenotypes and cellular localization of the resulting protein product. In particular, alterations in PNPLA2, INSR, SREBF1, and COBLL1 were grouped in “insulin resistance MAFLD subtype”; mutations in GCKR and TRIB1 in “alteration of glucose level subtype”, variants in GPAM, MARC1, TOR1B, ADH1B, and MBOAT7 in “triglyceride diversion/reduction subgroup”, APOE variants in “high/normal lipoprotein”, FTO variants in “low lipid burn subtype”, alterations in MTTP in “intestinal absorption” subgroup, and mutations in PNPLA3, TM6SF2, and PTPRD in “Low lipoprotein output” in subtype [35].
Furthermore, variants associated with cirrhosis trough alcohol consumption were found in alcohol dehydrogenase 1B (ADH1B), genes involved in de novo lipogenesis and retinol metabolism [35]. Moreover, Sveinbjornsson et al. [36] using magnetic resonance imaging, a meta-analysis of determinants of clinical MAFLD and integrating the results multiomics data pinpointed several genetic determinants as in a gene involved in lipogenesis, Apolipoprotein H (APOH) and cholesterol synthesis and Glucuronidase Beta (GUSB) which is related to glycosaminoglycan metabolism. Recently, a variant in the pleckstrin and Sec7 domain-containing 3 (PSD3) gene (rs71519934) was reported to reduce MAFLD susceptibility and it was associated with protection against MAFLD in individuals at risk. PSD3 expression level is increased in MAFLD patients and its downregulation leads to a reduction in hepatocellular lipid content in mice and in several hepatocyte cell lines including human primary hepatocytes [37]. Gene variants associated with MAFLD predisposition are reported in Table 1.
Polygenic risk score for MAFLD risk prediction
Polygenic risk scores (PRS) were developed to summarize the effect variants for MAFLD, such as those in PNPLA3–TM6SF2–GCKR–MBOAT7, e.g., the hepatic fat PRS, or PRS-HFC, and then adjusted for a protected variant in and 17-β hydroxysteroid dehydrogenase 13 (HSD17B13) (PRS-5) [38,39,40], to improve MAFLD risk stratification. These PRS allowed to demonstrate that genetic predisposition derived by rare combinations of common variants contribute to severe or early onset liver disease phenotypes [38]. The integration of genetics with clinical fibrosis scores refines individual risk and prediction for liver disease, mainly in subjects at risk for MAFLD. PRS provide evidence that common genetic variants capture additional prognostic insights not showed by validated clinical/biochemical parameters [40]. Moreover, PRS improve the accuracy of HCC detection and may help stratify HCC risk in individuals with dysmetabolism, including those without severe liver fibrosis [38].
Rare variants predisposing to MAFLD
In the last 5 years, the advent of NGS studies has led to the identification of genes whose rare loss-of-function (LoF) variants most frequently contribute to MAFLD. Rare variants in Apolipoprotein B (APOB) predispose to MAFLD and they are responsible for the development of severe MAFLD and hypobetalipoproteinemia. Familial hypobetalipoproteinemia (FHBL) is a codominant disorder of lipoprotein metabolism derived from mutations in the APOB gene encoding for ApoB and characterized by low levels of LDL-cholesterol. FHBL can be caused by truncations of full-length ApoB-100 as short as 5% (apoB-5) and as long as 89% (ApoB-89) [33, 41, 42]. Rare variants in MTTP gene have been linked with susceptibility to MAFLD and rare and loss-of-function mutations in MTTP result in abetalipoproteinaemia. The gene encodes for microsomal triglyceride transfer protein (MTP) which forms a heterodimer with protein disulfide isomerase (PDI), and catalyzes the lipidation and assembly of ApoB [43]. These data suggest that lipoprotein retention in hepatocytes plays a key role in MAFLD. In addition, rare LoF mutations in autophagy related 7 (ATG7) gene modulating lipophagy and mitophagy in hepatocytes have been implicated in disease progression [44].
Gene loci associated with MAFLD progression
Through the evaluation as outcomes of severe liver disease phenotypes, GWAS have also led to the discovery of genetic variants implicated in MAFLD progression to steatohepatitis, fibrosis, and cirrhosis, besides in hepatic fat accumulation per se. The mechanisms are related to interferences with oxidative stress, cell senescence, fibrogenesis, insulin signaling, glucose metabolism, inflammation, and lipotoxicity [45]. Oxidative stress and deranged mitochondrial respiratory complex activity and oxidation, namely mitochondrial dysfunction, are considered a main contributor to liver injury and MAFLD progression [46]. Indeed, among the determinants of liver damage and cirrhosis risk in Europe are Homeostatic Iron Regulator (HFE) p.C282Y and p.H63D variants (rs1800562, rs1799945) associated with hemochromatosis, type 1 [47]. Excess tissue iron predisposes to the development and progression of MAFLD by catalyzing oxidative stress [48]. Other genes whose mutations are implicated in iron disorders and in MAFLD are ceruloplasmin (CP), serpin family A member 1 (SERPINA1), and proprotein convertase subtilisin/kexin type 7 (PCSK7), which is involved in hepatic inflammation by modulating multiple pathways, such as lipid, iron metabolism, and fibrogenesis [44, 49, 50]. Other pathways are also involved. For example, variants in Fibronectin Type III Domain Containing 5 (FNDC5) have been linked with fibrosis progression: the gene encodes for a myokine named Irisin involved in hepatic stellate cell activation and fibrogenesis [51, 52]. Moreover, concerning hepatic inflammation, two variants in interferon IFN-λ3/IFN-λ4 region in linkage disequilibrium among them were confirmed to be associated with more severe fibrosis in MAFLD by modulating the activation of innate immunity and inflammation [53, 54]. Alterations in insulin signaling lead to a more severe fibrosis: examples are variants in Ectonucleotide Pyrophosphatase/Phosphodiesterase 1 (ENPP1), Insulin Receptor Substrate 1 (IRS1), and Tribbles homolog 1 (TRIB1) [55, 56]. The main modulator of steatohepatitis affects HSD17B13, a lipid droplet (LD)-associated protein that is mainly expressed in hepatocytes. LoF variants (mainly rs72613567: TA) of this gene mitigate the progression of MAFLD, reducing the risk of steatohepatitis, cirrhosis, and HCC. Genetic variants in HSD17B13 result in a loss of expression and/or of enzymatic activity, toward retinol, steroid hormones, and other pro-inflammatory lipid mediators, and increase retinol–retinol binding protein (RBP4)–transthyretin (TTR) transport from hepatocytes [57]. However, despite this evidence, the function of HSD17B13 and how its absence protects against MASH remains obscure. Finally, concerning the role of rare variants, those in ATG7 were identified as modifiers of MAFLD progression in Europeans by enhancing specifically the risk of hepatocellular ballooning (e.g., rs143545741 C>T and rs36117895 T>C) [58], as well as LoF mutations in Telomerase Reverse Transcriptase (TERT) were associated with liver senescence and development of HCC [59]. Variants/genes modulating MAFLD progression are presented in Table 2.
Sex-genotype epistasis in MAFLD
Excess adiposity is a main trigger of MAFLD genetic susceptibility [58, 60]. Sex hormones have also a major role in modulating liver fat content [61]. Estrogens protect against MAFLD, accounting for a lower prevalence of this condition in female before menopause. In older male, lower testosterone levels are associated with frailty and increased risk of MAFLD, while, in both sexes, lower sex hormone-binding globulin (SHBG) circulating levels are associated with MAFLD [62]. High androgen levels in female with polycystic ovary syndrome (PCOS) lead to a markedly increased risk of MAFLD, as well as insulin resistance and obesity [63]. Recently, Cherubini et al. [64] demonstrated that there is an interaction between sex and the common genetic variant PNPLA3 p.I148M in determining the development and severity of MAFLD. This relation, documented both at the epidemiological and a molecular level, contributes explaining why a subset of Female develop rapidly progressive MAFLD at menopause. Indeed, estrogens protect premenopausal female against MAFLD operating on lipid metabolism at a systemic level and in hepatocytes through estrogen receptor alpha (ERα) [65]. Instead, following menopause, lower estrogens cannot inhibit de novo lipogenesis, favoring import and accumulation of lipid in the liver [64]. Postmenopausal female carrying of PNPLA3 p.I148M variant has a persistent induction and accumulation of the mutant PNPLA3 protein in hepatocytes and consequently enhanced hepatic fat accumulation and fibrogenesis [64] (Fig. 3).

Genetic determinants of MAFLD, classified according to the biological processes by which the encoded proteins are thought to contribute to the pathogenesis. Red arrows indicate pathological processes/lipid fluxes, whereas green arrows indicate beneficial pathways. Pathophysiological processes are indicated in red, while genes, and cellular and liver compartments in black
Acquired somatic variants in MAFLD
Somatic mutations are non-heritable gene variants that occur in aged or chronically injured somatic cells, and this phenomenon is also observed in livers from individuals with MAFLD. The acquisition of somatic variants and clonal expansion leads to the progression of chronic liver diseases into HCC [66, 67]. Somatic alterations occurring in genes involved in lipid metabolism are also implicated in MAFLD progression to cirrhosis. Indeed, in individuals with the most advanced liver disease, there are evidences of selection of somatic variants in Forkhead Box O1 (FOXO1), Cell Death Inducing DFFA Like Effector B (CIDEB) and Glycerol-3-Phosphate Acyltransferase, Mitochondrial (GPAM), involved in the regulation of lipid synthesis and the antioxidant response, leading to a reduction of hepatic fat accumulation possibly accounting for “burnt-out steatohepatitis”, as an adaptive response against chronic lipotoxicity [68, 69]. On the other hand, somatic variants accumulation occurring in genes involved in chromatin remodeling [i.e., AT-Rich Interaction Domain 1A (ARID1A), AT-Rich Interaction Domain 2 (ARID2), and Lysine Methyltransferase 2C (KMT2C)] and in cell differentiation and migration (i.e., RAS, MAPK, AKT, mTOR, and MET pathways) contribute to hepatic inflammation and carcinogenesis [67, 70]. Importantly, while germline common and rare variants affect primarily hepatocyte triglyceride homeostasis and inflammation that are the initial disease stage, the somatic ones are mostly present in the late stage.
In regards to HCC, it is not surprising that somatic mutations in TERT promoter account for 60% of HCC cases [71]. Alterations in TERT promoter cause the reactivation of telomerase reverse transcriptase causing telomere re-elongation and immortalization of the neoplastic clone; these variants occur not only in cancer tissue but also in early cirrhotic tissue (6–19%) highlighting their importance in the disease progression and tumorigenesis [72]. Similarly, another frequent gene affected by somatic variants is Tumor Protein P53 (TP53) (45% of HCC cases), a tumor suppressor protein involved in the maintenance of genome integrity inducing cell cycle arrest, apoptosis, and senescence in response to cellular stress [73]. Aberrant reactivation of Wnt-β-catenin pathway due to somatic alterations in Catenin Beta 1 (CTNNB1) is present in 30% of the cases, while for 10% of the cases mutations involved Axin 1 (AXIN1) gene [74, 75]. Furthermore, recent studies have shown an excess of somatic variants predisposing to clonal haematopoiesis of indeterminate potential (CHIP), a precursor of hematologic cancer, in individuals with MAFLD. Somatic mutations in Tet Methylcytosine Dioxygenase 2 (TET2), ASXL Transcriptional Regulator 1 (ASXL1), and Janus Kinase 2 (JAK2) genes are implicated in CHIP [76]. In general CHIP may induce chronic liver disease progression via an aberrant inflammatory response [77, 78]. Also, CHIP is relatively common in patients with solid tumor malignancies and it is associated with adverse outcomes of hematologic malignancies. However, it is fair to say that the risk associated with CHIP for progressive MAFLD is negligible as compared to blood cancer. Several examples of genes in which somatic variants occur are reported in Table 3.
Epigenetic alterations in MAFLD
Epigenetic factors are stable modifications of chromosomes/DNA that modify gene expression and cause phenotypic variation without direct alteration of DNA base sequence. Epigenetic alterations encompass DNA methylation, histone modifications, and modulation of gene expression by microRNAs (miRNA) and other non-coding RNAs [79]. DNA methylation occurs when methyl groups are covalently bound to cytosine to produce 5-methylcytosine near to guanine (CpG dinucleotides), which is most frequently located at the promoter region of genes. This reaction is catalyzed by DNA methyltransferases (DNMTs). Hypermethylation of CpG islands is associated with gene silencing, while hypomethylation leads to gene activation [79]. Some studies have reported a role of global hypomethylation and differential methylation in the progression of MAFLD, highlighting also specific methylation shifts at transcriptional start of genes involved in lipid metabolism and energy homeostasis. There is evidence of changes in methylation of genes regulating lipid and cholesterol transport (APO family members and STARD) and the metabolic hormone fibroblast growth factor 21 (FGF21), which is high expressed in the liver and modulates systemic energy values acting in macronutrient metabolism [80]. Pirola et al. [81] showed that silencing of mitochondrial gene NADH dehydrogenase 6 (MT-ND6) by promoter hypermethylation correlated with MAFLD severity. Furthermore, hypermethylation of the hepatic promoter of the peroxisome proliferative activated receptor (PPAR)-gamma coactivator one alpha (PGC1-α) gene, a transcriptional regulator of mitochondrial fatty acid oxidation was associated with peripheral insulin resistance and fasting insulin levels of MAFLD patients [82]. Two studies highlighted a general hypomethylation status of hepatic DNA in patients with MAFLD compared to individuals with healthy liver, and a more marked demethylation in patients with advanced compared to milder MAFLD [83, 84]. Conversely, PNPLA3 was reportedly hypermethylated in patients with MAFLD [85], but evidence is controversial and specific hypomethylation may be linked to higher gene expression in people carrying at risk genotypes and in female [64, 86]. GWAS analyses evidenced that hypomethylated loci are near to genes involved in cancer and immunoresponse, whereas hypermethylated regions occur close to genes associated with lipid metabolism [87, 88]. Another epigenetics modification consists of the addition of methyl groups, acetyl groups or phosphoryl groups to histone proteins leading to an alteration of the physical structure of chromatin and changing the ability of recruitment of other proteins. Some studies reported a correlation with aberrant histone methylation and acetylation profiles and metabolic syndrome and alteration in the expression of specific histone lysine methyltransferases (KMT) and demethylases (KDMs) during MAFLD [89, 90]. Histone acetyltransferases (HATs) promote gene transcription, by increasing the accessibility to DNA, whereas histone deacetylase (HDACs) repress it. The HAT p300 activates the glucose-responsive lipogenic activators ChREBP promoting the lipogenesis and steatosis. Conversely, Histone Deacetylase 3 (HDAC3), Sirtuin 1 (SIRT1), and Sirtuin 6 (SIRT6) protect against MAFLD by deacetylating promoter histones of lipogenic genes [91, 92]. Alterations in miRNA expression have also been associated with MAFLD development and progression. miRNAs are a class of endogenous non-coding functional RNAs implicated in the regulation of gene expression by interacting with complementary non-coding regions of genes and other RNAs. Cell death and degeneration during MAFLD lead to the release of different miRNAs which can regulate an array of biological processes, such as lipid metabolism, glucose catabolism, inflammation, cell proliferation and apoptosis, adipocyte differentiation, and insulin resistance [93, 94]. Alterations in miRNAs involved in the regulation of hepatic cholesterol and lipid metabolism contribute to the development of metabolic disorder, atherosclerosis, and cardiovascular disease. In particular, miR-122 has been causally involved in MAFLD development: it represents the 70% of hepatic miRNAs and it plays an important role in the regulation of genes associated with liver regeneration, lipid, and cholesterol metabolism. Lower levels of miR-122 in hepatocytes are implicated in the activation of fibrotic pathways and in the reduction of lipid secretion [95]. Conversely, circulating levels of miR-122 are increased in patients with compared to those with simple steatosis and general population, due to the release of circulating miRNAs from hepatocytes [96]. Also, miR-192, and miR-375, miR-19a/b and miR-125b, which are overexpressed in subjects with MAFLD, participate in the development of MASH. Specifically, miR-192 is induced by TGFβ1 contributing to fibrosis development, miR-375 regulates glucose levels, and the requirement for adaptive β cell expansion, while miR-19a/b related to NF-κB signaling and miR-125b are associated with cardiovascular disease and inflammation [93]. Also, miR-10b, miR-144, miR-146b, and miR-155 participate in hepatic inflammation and liver damage by regulating PPAR-α, Toll-like receptors (TLR), and Tumor Necrosis Factorα (TNFα) [97]. Another risk factor for MAFLD development is the upregulation of miR-29 a/b/c which leads to insulin resistance through the block of Akt pathway and insulin signaling [98]. Yu et al. [99] showed that overexpression of miR-33a/b in hepatocytes determines triglycerides accumulation and promote steatosis, whereas overproduction of miR-34 a/b/c promotes lipid metabolism by targeting acyl-CoA synthetase long-chain family member 1 (ACSL1) [100]. Recently, a role of miR-21 in the transition to HCC was reported, mediated by silencing of HMG box-containing protein 1 (HBP1) and of the consequent activation of p53 [93]. Apart from these, other miRNAs reportedly dysregulated in MAFLD are shown in Table 4.
Conclusion
Genetic variation plays a key role in determining the susceptibility to the development and progression of MAFLD extending to liver-related disease and overall mortality. Genetic determinants have an effect size comparable and synergic to that of the main metabolic risk factors, such as obesity and type 2 diabetes. Thanks to genomic studies subsets of patients with different pathophysiology, risk of liver-related complications and response to treatment can now be profiled. In the coming years, the search of genetic mutations will continue increasing the number of individuals in GWAS and introducing novel strategies as Mendelian randomization methods and meta-analysis allowing to explore the reasons for heterogeneity of the genetic effects across datasets. The high gene x environment interactions observed in the genetic architecture of MAFLD, and the rise of prevalence of people at risk (i.e., obese, insulin resistant) and with severe disease may lead to the identification of new loci and precision medicine strategies. In conclusion, genomic studies are revolutionizing the comprehension of MAFLD leading the way to new tools for targeted screening of high-risk individuals, also improving patient stratification for clinical trials, for prognostication and clinical management (Fig. 4).

Genomic studies to develop new MAFLD therapeutics and precision medicine approach
Data availability
This is a review article utilizing data from published papers and other public database and, as such, data availability statement is not applicable.
References
Eslam M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol 2020;73(1):202–209. https://doi.org/10.1016/j.jhep.2020.03.039
Serfaty L, Lemoine M. Definition and natural history of metabolic steatosis: clinical aspects of NAFLD, NASH and cirrhosis. Diabetes Metab 2008;34(6 Part 2):634–637. https://doi.org/10.1016/S1262-3636(08)74597-X
Rinella ME, et al. A multi-society Delphi consensus statement on new fatty liver disease nomenclature. J Hepatol 2023. https://doi.org/10.1016/j.jhep.2023.06.003
Ilyas F, et al. Increasing nonalcoholic fatty liver disease–related mortality rates in the United States from 1999 to 2022. Hepatol Commun 2023;7(7):e00207. https://doi.org/10.1097/hc9.0000000000000207
Kaya E, Yilmaz Y. Metabolic-associated fatty liver disease (MAFLD): a multi-systemic disease beyond the liver. J Clin Transl Hepatol 2022;10(2):329–338. https://doi.org/10.14218/JCTH.2021.00178
Rocha R, Cotrim HP, Carvalho FM, Siqueira AC, Braga H, Freitas LA. Body mass index and waist circumference in non-alcoholic fatty liver disease. J Hum Nutr Diet 2005;18(5):365–370. https://doi.org/10.1111/j.1365-277X.2005.00634.x
Juanola O, Martínez-López S, Francés R, Gómez-Hurtado I. Non-alcoholic fatty liver disease: metabolic, genetic, epigenetic and environmental risk factors. Int J Environ Res Public Health 2021;18(10):5227. https://doi.org/10.3390/ijerph18105227
Valenti L, Ronzoni L. Genetics: a new clinical tool for the hepatologist. Liver Int 2022;42(4):724–726. https://doi.org/10.1111/liv.15205
Trépo E, Valenti L. Update on NAFLD genetics: from new variants to the clinic. J Hepatol 2020;72(6):1196–1209. https://doi.org/10.1016/j.jhep.2020.02.020
Pelusi S, et al. Clinical exome sequencing for diagnosing severe cryptogenic liver disease in adults: a case series. Liver Int 2022;42(4):864–870. https://doi.org/10.1111/liv.15185
Zheng M et al. Advancing diagnosis and management of liver disease in adults through exome sequencing. EBioMedicine 2023;95:104747. [Online]. Available: https://pan.ukbb.broadinstitute.org/
Caussy C, et al. Nonalcoholic fatty liver disease with cirrhosis increases familial risk for advanced fibrosis. J Clin Invest 2017;127(7):2697–2704. https://doi.org/10.1172/JCI93465
Loomba R, et al. Heritability of hepatic fibrosis and steatosis based on a prospective twin study. Gastroenterology 2015;149(7):1784–1793. https://doi.org/10.1053/j.gastro.2015.08.011
Schwimmer JB, et al. Heritability of nonalcoholic fatty liver disease. Gastroenterology 2009;136(5):1585–1592. https://doi.org/10.1053/j.gastro.2009.01.050
Long MT, et al. Parental non-alcoholic fatty liver disease increases risk of non-alcoholic fatty liver disease in offspring. Liver Int 2019;39(4):740–747. https://doi.org/10.1111/liv.13956
Dongiovanni P, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med 2018;283(4):356–370. https://doi.org/10.1111/joim.12719
Guerrero R, Vega GL, Grundy SM, Browning JD. Ethnic differences in hepatic steatosis: an insulin resistance paradox? Hepatology 2009;49(3):791–801. https://doi.org/10.1002/hep.22726
Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2015. https://doi.org/10.1002/hep.28431/suppinfo
Kubiliun MJ, Cohen JC, Hobbs HH, Kozlitina J. Contribution of a genetic risk score to ethnic differences in fatty liver disease. Liver Int 2022;42(10):2227–2236. https://doi.org/10.1111/liv.15322
Romeo S, Valenti L. African genetic ancestry and protection against fatty liver disease. Liver Int 2022;42:2122–2123
Eslam M, Valenti L, Romeo S. Genetics and epigenetics of NAFLD and NASH: clinical impact. J Hepatol 2018;68:268–279
Romeo S, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40(12):1461–1465. https://doi.org/10.1038/ng.257
Kozlitina J, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46(4):352–356. https://doi.org/10.1038/ng.2901
Speliotes EK, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 2011;7(3):e1001324. https://doi.org/10.1371/journal.pgen.1001324
Mancina RM, et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of european descent. Gastroenterology 2016;150(5):1219–1230.e6. https://doi.org/10.1053/j.gastro.2016.01.032
Sookoian S, Pirola CJ, Valenti L, Davidson NO. Genetic pathways in nonalcoholic fatty liver disease: insights from systems biology. Hepatology 2020;72(1):330–346. https://doi.org/10.1002/hep.31229
Dongiovanni P, et al. PNPLA3 I148M polymorphism and progressive liver disease. World J Gastroenterol 2013;19(41):6969–6978. https://doi.org/10.3748/wjg.v19.i41.6969
Grimaudo S, et al. Association between PNPLA3 rs738409 C>G variant and liver-related outcomes in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2020;18(4):935–944.e3. https://doi.org/10.1016/j.cgh.2019.08.011
Li BT, et al. Disruption of the ERLIN–TM6SF2–APOB complex destabilizes APOB and contributes to non-alcoholic fatty liver disease. PLoS Genet 2020;16(8):e1008955. https://doi.org/10.1371/JOURNAL.PGEN.1008955
Prill S, et al. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein B secretion in human hepatic 3D spheroids. Sci Rep 2019;9(1):11585. https://doi.org/10.1038/s41598-019-47737-w
Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH. Inactivation of Tm6sf2, a gene defective in fatty liver disease, impairs lipidation but not secretion of very low density lipoproteins. J Biol Chem 2016;291(20):10659–10676. https://doi.org/10.1074/jbc.M116.719955
Liu YL, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun 2014;5:4309. https://doi.org/10.1038/ncomms5309
Pelusi S, et al. Rare pathogenic variants predispose to hepatocellular carcinoma in nonalcoholic fatty liver disease. Sci Rep 2019;9(1):3682. https://doi.org/10.1038/s41598-019-39998-2
Buch S, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat Genet 2015;47(12):1443–1448. https://doi.org/10.1038/ng.3417
Chen Y, et al. Genome-wide association meta-analysis identifies 17 loci associated with nonalcoholic fatty liver disease. Nat Genet 2023. https://doi.org/10.1038/s41588-023-01497-6
Sveinbjornsson G, et al. Multiomics study of nonalcoholic fatty liver disease. Nat Genet 2022;54(11):652–1663
Mancina RM, et al. PSD3 downregulation confers protection against fatty liver disease. Nat Metab 2022;4(1):60–75. https://doi.org/10.1038/s42255-021-00518-0
Bianco C, et al. Non-invasive stratification of hepatocellular carcinoma risk in non-alcoholic fatty liver using polygenic risk scores. J Hepatol 2021;74(4):775–782. https://doi.org/10.1016/j.jhep.2020.11.024
Bianco C, Tavaglione F, Romeo S, Valenti L. Genetic risk scores and personalization of care in fatty liver disease. Curr Opin Pharmacol 2021;61:6–11. https://doi.org/10.1016/j.coph.2021.08.014
De Vincentis A, et al. A polygenic risk score to refine risk stratification and prediction for severe liver disease by clinical fibrosis scores. Clin Gastroenterol Hepatol 2022;20(3):658–673. https://doi.org/10.1016/j.cgh.2021.05.056
Haas ME, et al. Machine learning enables new insights into genetic contributions to liver fat accumulation. Cell Genomics 2021;1(3):100066. https://doi.org/10.1016/j.xgen.2021.100066
Burnett JR, Hooper AJ, Hegele RA. APOB-related familial hypobetalipoproteinemia. GeneReviews® [Internet] 2021;1993–2023
Grove JI, et al. Identification and characterisation of a rare MTTP variant underlying hereditary non-alcoholic fatty liver disease. JHEP Rep 2023;5(8):100764. https://doi.org/10.1016/j.jhepr.2023.100764
Dongiovanni P, et al. PCSK7 gene variation bridges atherogenic dyslipidemia with hepatic inflammation in NAFLD patients. J Lipid Res 2019;60(6):1144–1153. https://doi.org/10.1194/jlr.P090449
Wojcik GL, et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature 2019;570(7762):514–518. https://doi.org/10.1038/s41586-019-1310-4
Chen Z, Tian R, She Z, Cai J, Li H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic Biol Med 2020;152:116–141. https://doi.org/10.1016/j.freeradbiomed.2020.02.025
Valenti L, et al. HFE genotype, parenchymal iron accumulation, and liver fibrosis in patients with nonalcoholic fatty liver disease. Gastroenterology 2010;138(3):905–912. https://doi.org/10.1053/j.gastro.2009.11.013
Milic S, et al. The role of iron and iron overload in chronic liver disease. Med Sci Monit 2016;22:2144–2151. https://doi.org/10.12659/MSM.896494
Corradini E, et al. Ceruloplasmin gene variants are associated with hyperferritinemia and increased liver iron in patients with NAFLD. J Hepatol 2021;75(3):506–513. https://doi.org/10.1016/j.jhep.2021.03.014
Vujkovic M, et al. A multiancestry genome-wide association study of unexplained chronic ALT elevation as a proxy for nonalcoholic fatty liver disease with histological and radiological validation. Nat Genet 2022;54(6):761–771. https://doi.org/10.1038/s41588-022-01078-z
Petta S, et al. Fibronectin type III domain-containing protein 5 rs3480 A>G polymorphism, irisin, and liver fibrosis in patients with nonalcoholic fatty liver disease. J Clin Endocrinol Metab 2017;102(8):2660–2669. https://doi.org/10.1210/jc.2017-00056
Metwally M, et al. A polymorphism in the Irisin-encoding gene (FNDC5) associates with hepatic steatosis by differential miRNA binding to the 3′UTR. J Hepatol 2019;70(3):494–500. https://doi.org/10.1016/j.jhep.2018.10.021
Petta S, et al. Interferon lambda 4 rs368234815 TT>dG variant is associated with liver damage in patients with nonalcoholic fatty liver disease. Hepatology 2017;66:1885–1893. https://doi.org/10.1002/hep.29395/suppinfo
Eslam M, Ahlenstiel G, George J. Interferon lambda and liver fibrosis. J Interf Cytokine Res 2019;39(10):627–635
Kitamoto A, et al. Association of polymorphisms in GCKR and TRIB1 with nonalcoholic fatty liver disease and metabolic syndrome traits. Endocr J 2014;64(7):683–689
Dongiovanni P, et al. Genetic variants regulating insulin receptor signalling are associated with the severity of liver damage in patients with non-alcoholic fatty liver disease. Gut 2010;59(2):267–273
Abul-Husn NS, et al. A protein-truncating HSD17B13 variant and protection from chronic liver disease. N Engl J Med 2018;378(12):1096–1106. https://doi.org/10.1056/nejmoa1712191
Baselli GA, et al. Rare ATG7 genetic variants predispose patients to severe fatty liver disease. J Hepatol 2022;77(3):596–606. https://doi.org/10.1016/j.jhep.2022.03.031
Calado RT, et al. A spectrum of severe familial liver disorders associate with telomerase mutations. PLoS ONE 2009;4(11):e7926. https://doi.org/10.1371/journal.pone.0007926
Stender S, Kozlitina J, Nordestgaard BG, Tybjærg-Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet 2017;49(6):842–847. https://doi.org/10.1038/ng.3855
Tavaglione F, Jamialahmadi O, Valenti L, Romeo S. Fatty liver disease genetic risk variants and interference on sex hormones. Liver Int 2023;43(5):958–961. https://doi.org/10.1111/liv.15562
Jaruvongvanich V, Sanguankeo A, Riangwiwat T, Upala S. Testosterone, sex hormone-binding globulin and nonalcoholic fatty liver disease: a systematic review and meta-analysis. Ann Hepatol 2017;16(3):382–394. https://doi.org/10.5604/16652681.1235481
Falzarano C, et al. Nonalcoholic fatty liver disease in women and girls with polycystic ovary syndrome. J Clin Endocrinol Metab 2022;107(1):258–272. https://doi.org/10.1210/clinem/dgab658
Cherubini A, et al. Interaction between estrogen receptor-α and PNPLA3 p. I148M variant drives fatty liver disease susceptibility in women. Nat Med 2023;29:2643–2655. https://doi.org/10.1038/s41591-023-02553-8
Shen M, Shi H. Sex hormones and their receptors regulate liver energy homeostasis. Int J Endocrinol 2015;2015:294278. https://doi.org/10.1155/2015/294278
Brunner SF, et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature 2019;574:538–542. https://doi.org/10.17632/ktx7jp8sch.1
Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 2015;149(5):1226–1239.e4. https://doi.org/10.1053/j.gastro.2015.05.061
Valenti L, Romeo S, Pajvani U. A genetic hypothesis for burnt-out steatohepatitis. Liver Int 2021;41(12):2816–2818. https://doi.org/10.1111/liv.15103
Ng SWK, et al. Convergent somatic mutations in metabolism genes in chronic liver disease. Nature 2021;598(7881):473–478. https://doi.org/10.1038/s41586-021-03974-6
Garcia-Lezana T, Lopez-Canovas JL, Villanueva A. Signaling pathways in hepatocellular carcinoma. Adv Cancer Res 2021;149:63–101. https://doi.org/10.1016/bs.acr.2020.10.002
Nault JC, et al. Molecular classification of hepatocellular adenoma associates with risk factors, bleeding, and malignant transformation. Gastroenterology 2017;152(4):880–894.e6. https://doi.org/10.1053/j.gastro.2016.11.042
Donati B, Valenti L. Telomeres, NAFLD and chronic liver disease. Int J Mol Sci 2016;17(3):383. https://doi.org/10.3390/ijms17030383
Muller PAJ, Vousden KH. P53 mutations in cancer. Nat Cell Biol 2013;15(1):2–8. https://doi.org/10.1038/ncb2641
Rebouissou S, et al. Proliferation markers are associated with MET expression in hepatocellular carcinoma and predict tivantinib sensitivity in vitro. Clin Cancer Res 2017;23(15):4364–4375. https://doi.org/10.1158/1078-0432.CCR-16-3118
Satoh S, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. 2000. [Online]. Available: http://genetics.nature.com/supplementary_info/
Genovese G, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014;371(26):2477–2487. https://doi.org/10.1056/nejmoa1409405
Wong WJ, et al. Clonal hematopoiesis and risk of chronic liver disease. Nature 2023;616:747–754. https://doi.org/10.1101/2022.01.17.22269409
Kessler MD, et al. Common and rare variant associations with clonal haematopoiesis phenotypes. Nature 2022;612(7939):301–309. https://doi.org/10.1038/s41586-022-05448-9
Ferrari A, et al. Epigenome modifiers and metabolic rewiring: new frontiers in therapeutics. Pharmacol Ther 2019;193:178–193. https://doi.org/10.1016/j.pharmthera.2018.08.008
Mwinyi J, et al. NAFLD is associated with methylation shifts with relevance for the expression of genes involved in lipoprotein particle composition. Biochim Biophys Acta Mol Cell Biol Lipids 2017;1862(3):314–323. https://doi.org/10.1016/j.bbalip.2016.12.005
Pirola CJ, et al. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut 2013;62(9):1356–1363. https://doi.org/10.1136/gutjnl-2012-302962
Lee JH, Friso S, Choi SW. Epigenetic mechanisms underlying the link between non-alcoholic fatty liver diseases and nutrition. Nutrients 2014;6(8):3303–3325. https://doi.org/10.3390/nu6083303
Murphy SK, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology 2013;145(5):1076–1087. https://doi.org/10.1053/j.gastro.2013.07.047
de Mello VD, et al. Human liver epigenetic alterations in non-alcoholic steatohepatitis are related to insulin action. Epigenetics 2017;12(4):287–295. https://doi.org/10.1080/15592294.2017.1294305
Kitamoto T, et al. Targeted-bisulfite sequence analysis of the methylation of CpG islands in genes encoding PNPLA3, SAMM50, and PARVB of patients with non-alcoholic fatty liver disease. [Online]. Available: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Donati B, et al. The rs2294918 E434K variant modulates patatin-like phospholipase domain-containing 3 expression and liver damage. Hepatology 2015. https://doi.org/10.1002/hep.28370/suppinfo
Hotta K, et al. Identification of the genomic region under epigenetic regulation during non-alcoholic fatty liver disease progression. Hepatol Res 2018;48(3):E320–E334. https://doi.org/10.1111/hepr.12992
Gerhard GS, et al. Differentially methylated loci in NAFLD cirrhosis are associated with key signaling pathways. Clin Epigenet 2018;10(1):93. https://doi.org/10.1186/s13148-018-0525-9
Klose RJ, Kallin EM, Zhang Y. JmjC-domain-containing proteins and histone demethylation. Nat Rev Genet 2006;7(9):715–727. https://doi.org/10.1038/nrg1945
Ferrante AW. Improving metabolism by throwing out all the JNK. Science 2013;339(6116):147–148. https://doi.org/10.1126/science.1233223
Bricambert J, Miranda J, Benhamed F, Girard J, Postic C, Dentin R. Salt-inducible kinase 2 links transcriptional coactivator p300 phosphorylation to the prevention of ChREBP-dependent hepatic steatosis in mice. J Clin Invest 2010;120(12):4316–4331. https://doi.org/10.1172/JCI41624
Li Y, et al. Hepatic SIRT1 attenuates hepatic steatosis and controls energy balance in mice by inducing fibroblast growth factor 21. Gastroenterology 2014;146(2):539–549. https://doi.org/10.1053/j.gastro.2013.10.059
Dongiovanni P, Meroni M, Longo M, Fargion S, Fracanzani AL. MiRNA signature in NAFLD: a turning point for a non-invasive diagnosis. Int J Mol Sci 2018;19(12):3966. https://doi.org/10.3390/ijms19123966
Szabo G, Bala S. MicroRNAs in liver disease. Nat Rev Gastroenterol Hepatol 2013;10(9):542–552. https://doi.org/10.1038/nrgastro.2013.87
Horvath S, et al. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci U S A 2014;111(43):15538–15543. https://doi.org/10.1073/pnas.1412759111
Cermelli S, Ruggieri A, Marrero JA, Ioannou GN, Beretta L. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS ONE 2011;6(8):e23937. https://doi.org/10.1371/journal.pone.0023937
Pogribny IP, et al. Difference in expression of hepatic microRNAs miR-29c, miR-34a, miR-155, and miR-200b is associated with strain-specific susceptibility to dietary nonalcoholic steatohepatitis in mice. Lab Investig 2010;90(10):1437–1446. https://doi.org/10.1038/labinvest.2010.113
Jampoka K, Muangpaisarn P, Khongnomnan K, Treeprasertsuk S, Tangkijvanich P, Payungporn S. Serum miR-29a and miR-122 as potential biomarkers for non-alcoholic fatty liver disease (NAFLD). Microrna 2018;7(3):215–222
Yu J, Peng J, Luan Z, Zheng F, Su W. MicroRNAs as a novel tool in the diagnosis of liver lipid dysregulation and fatty liver disease. Molecules 2019;24(2):230. https://doi.org/10.3390/molecules24020230
He Y, Huang C, Zhang S, Sun X, Long X, Li J. The potential of microRNAs in liver fibrosis. Cell Signal 2012;24(12):2268–2272. https://doi.org/10.1016/j.cellsig.2012.07.023
Acknowledgements
Italian Ministry of Health (Ministero della Salute), Ricerca Finalizzata 2016, RF-2016-02364358 (“Impact of whole exome sequencing on the clinical management of patients with advanced non-alcoholic fatty liver and cryptogenic liver disease”), Italian Ministry of Health, Ricerca Finalizzata 2021 “Targeting the epigenetic regulators Suv420h1/2 in hepatocytes to treat non-alcoholic fatty liver disease” (TERS) RF-2021-12373889, Italian Ministry of Health Ricerca Finalizzata PNRR 2022 “RATIONAL: Risk stratification Of Nonalcoholic fAtty Liver” PNRR-MAD-2022-12375656; Italian Ministry of Health (Ministero della Salute), Rete Cardiologica “CV-PREVITAL” (Project Code RCR-2019-23669116_001); Italian Ministry of Health (Ministero della Salute), Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Ricerca Corrente; Fondazione Patrimonio Ca’ Granda, “Liver BIBLE” (PR-0361); Innovative Medicines Initiative 2 joint undertaking of European Union’s Horizon 2020 research and innovation programme and EFPIA European Union (EU) Programme Horizon 2020 (under Grant Agreement No. 777377) for the project LITMUS; The European Union, H2020-ICT-2018-20/H2020-ICT-2020-2 programme “Photonics” under grant Agreement No. 101016726—REVEAL; Gilead_IN-IT-989-5790; The European Union, HORIZON-MISS-2021-CANCER-02-03 programme “Genial” under Grant Agreement No. “101096312”; Italian Ministry of University and Research, PNRR—M4—C2 “di R&S su alcune Key Enabling Technologies” “National Center for Gene Therapy and Drugs based on RNA Technology” CN3 Spoke 4, group ASSET: A sex-specific approach to NAFLD targeting; MUR-bando PRIN 2022 project "DEFENDER" code PRIN202223LVALE_01 (G53D2300161000).
Funding
Open access funding provided by Università degli Studi di Milano within the CRUI-CARE Agreement. This work was supported by Ministero della Salute grant number (RF-2016-02364358, RF-2021-12373889, PNRR-MAD-2022-12375656).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of this article, VM wrote the first manuscript draft and LV supervised the process.
Corresponding author
Ethics declarations
Conflict of interest
Vittoria Moretti, Stefano Romeo, and Luca Valenti have no conflicts of interest to declare that are relevant to the content of this article.
Compliance with ethical requirements
This article does not contain any studies with human or animal subjects.
COI
LV has received speaking fees from MSD, Gilead, AlfaSigma, and AbbVie, served as a consultant for Gilead, Pfizer, AstraZeneca, Novo Nordisk, Intercept, Diatech Pharmacogenetics, Ionis Pharmaceuticals, Boehringer Ingelheim, and Resalis Therapeutics, and received unrestricted research grants from Gilead.
SR is consultant for Astra Zeneca, RIbocure Ab, Foresite Lab, Novartis, AMGEN, Sanofi, and Ultragenix. SR has received grants from Astra Zeneca for research in steatotic liver disease.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moretti, V., Romeo, S. & Valenti, L. The contribution of genetics and epigenetics to MAFLD susceptibility. Hepatol Int (2024). https://doi.org/10.1007/s12072-024-10667-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12072-024-10667-5