
Abstract
The functional role of the dopamine D4 receptor (D4R) and its main polymorphic variants has become more evident with the demonstration of heteromers of D4R that control the function of frontal cortico-striatal neurons. Those include heteromers with the α2A adrenoceptor (α2AR) and with the D2R, localized in their cortical somato-dendritic region and striatal nerve terminals, respectively. By using biophysical and cell-signaling methods and heteromer-disrupting peptides in mammalian transfected cells and rat brain slice preparations, here we provide evidence for a new functionally relevant D4R heteromer, the α1AR-D4R heteromer, which is also preferentially localized in cortico-striatal glutamatergic terminals. Significant differences in allosteric modulations between heteromers of α1AR with the D4.4R and D4.7R polymorphic variants could be evidenced with the analysis of G protein-dependent and independent signaling. Similar negative allosteric modulations between α1AR and D4R ligands could be demonstrated for both α1AR-D4.4R and α1AR-D4.7R heteromers on G protein-independent signaling, but only for α1AR-D4.4R on G protein-dependent signaling. From these functional differences, it is proposed that the D4.4R variant provides a gain of function of the α1AR-mediated noradrenergic stimulatory control of cortico-striatal glutamatergic neurotransmission, which could result in a decrease in the vulnerability for impulse control-related neuropsychiatric disorders and increase in the vulnerability for posttraumatic stress disorder.
Similar content being viewed by others


Introduction
The functional role of the dopamine D4 receptor (D4R) and its polymorphic variants in the brain is beginning to be understood with the realization that it can also be a target for norepinephrine and with the discovery of heteromers of D4Rs with other dopamine receptors and with several adrenoceptor subtypes (for review, see ref. [1]). This is exemplified in the pineal gland, which together with the retina and the frontal cortex are the main localizations of the D4R in the brain. In the pinealocytes, which are the melatonin-producing cells, D4Rs experience a significant circadian expression, with highest levels at the end of the dark period [2]. This is associated with an increased formation of heteromers of D4Rs with α1B and β1 adrenoceptors [3]. At darkness, norepinephrine, by also activating D4Rs promotes an allosteric inhibition of α1BR and β1R signaling in the respective heteromers, which results in a reduction in the synthesis and release of melatonin [3].
In the prefrontal cortex, D4Rs are expressed by GABAergic interneurons and by glutamatergic pyramidal neurons, both in their cortical somato-dendritic region and striatal nerve terminals [1]. Several studies have shown that D4Rs play a significant role in the modulation of the frontal-cortico-striatal neuronal function (for review, see ref. [1]). This role also depends on heteromerization, with α2A adrenoceptors (α2ARs) in the cortical perisomatic region [4], and with dopamine D2 receptors (D2Rs) in the cortico-striatal terminals [5, 6]. The understanding of the functional role of these D4R heteromers has been concomitant to the finding of different properties of the major human D4R gene (DRD4) polymorphic variants.
The human D4R gene (DRD4) displays a high number of polymorphisms in its coding sequence. The most extensive polymorphism is found in exon 3, a region that encodes the third intracellular loop (3IL) of the receptor [7,8,9]. This polymorphism includes a variable number of tandem repeats of a 48-base pair sequence. The most common polymorphisms contain 4 or 7 repeats (with allelic frequencies of about 60% and 20%, respectively), which encode a D4R with the respective number of repeats of a proline-rich sequence of 16 amino acids (D4.4R and D4.7R) [7,8,9]. Importantly, DRD4 polymorphisms have been associated with personality traits that constitute endophenotypes for impulse control-related neuropsychiatric disorders [1], with the most consistent associations found between the gene encoding D4.7R and attention-deficit hyperactivity disorder (ADHD) [8, 10,11,12] and substance use disorders (SUDs) [13].
Notably, clear qualitative differences in the functional and pharmacological properties of these polymorphic variants have only been observed when analyzing those properties upon heteromerization with α2ARs and D2Rs. Heteromerization with D4.7R, but not D4.4R, significantly increases the constitutive activity of the D2R and the signaling potency of dopamine, as compared with non-heteromerized D2R [14]. This provided a biochemical correlate of a gain of function of the D2R-D4.7R heteromers localized in glutamatergic terminals as mediators of an inhibition of cortico-striatal neurotransmission, which was demonstrated with immunohistochemical and in vivo optogenetic-microdialysis experiments in D4.7R knock-in mice expressing a humanized D4R with the 3IL of the human D4.7R [5].
Similarly, heteromerization of α2AR with D4.7R, but not D4.4R, significantly increases the signaling potency of norepinephrine for the α2AR [4]. On the other hand, D4.4R, but not D4.7R activation, allosterically inhibits α2AR-mediated signaling in their respective heteromers [4], comparatively to the negative allosteric inhibition of α1BR and β1R in the respective heteromers in the pineal gland [3]. We proposed that the main functional output of the activation of cortical α2AR-D4.7R heteromers is a decrease in the excitability of glutamatergic pyramidal neurons, which should provide an additional gain of function of D4R in its inhibitory control of frontal cortico-striatal neurotransmission [1].
Interestingly, another adrenoceptor, the α1A receptor (α1AR) is also expressed by frontal cortico-striatal pyramidal neurons and predominantly localized in cortico-striatal terminals, but also on its perisomatic region [15, 16]. Although, also in the frontal cortex α1ARs are preferentially localized presynaptically in glutamatergic terminals [15]. Activation of α1ARs in the prefrontal cortex and the striatum leads to an increased activity and glutamate release by the pyramidal cortico-striatal neuron [15, 16]. The α1AR has lower affinity than the α2AR and has been conceptualized as a receptor that mediates the effect of stress-induced norepinephrine release, with possible implications for the pathophysiology and treatment of posttraumatic stress disorder (PTSD) [17]. The ability of D4Rs to form heteromers with several adrenoceptor subtypes and their clear potential colocalization with α1ARs in the pyramidal cortico-striatal glutamatergic neuron, led us to investigate the possible existence of functional α1AR-D4R heteromers in vitro and in vivo, as well as the possible pharmacological differences that would depend on the D4.4R and D4.7R polymorphic variants.
Materials and Methods
Cell Culture and Transfection
Human Embryonic Kidney-293 T (HEK-293 T) cells and two previously characterized HEK‐293 T cell lines with tetracycline inducible expression of the D4R polymorphic variants D4.4R or D4.7R were used [4]. Cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 5% Fetal Bovine Serum (FBS) and kept in an incubator at 37 °C and 5% CO2. The inducible HEK‐293 T cells were obtained with the Flp‐In T‐Rex system and were maintained with hygromycin 50 μg/ml and blasticidin 15 μg/ml and the D4R variant expression was induced for 18‐24 h with administration of tetracycline (250 ng/ml). Cells were transiently transfected with cDNA corresponding to the specific fused or non-fused receptors, G protein subunits or β-arrestin-2 using polyethyleneimine (Sigma-Aldrich, Cerdanyola del Vallés, Spain) or Lipofectamine 2000 (in calcium release experiments). All experiments were performed 48 h after transfection.
DNA Constructs
The cDNAs of the human D4.4R, D4.7R, α1AR and α2AR expressed in the pcDNA3.1 vector were amplified without its stop codon using sense and antisense primers harbouring BamHI and EcoRI restriction sites and subcloned into pRluc-N1 (Rluc) or pEYFP-N1 (YFP) vectors. For BiLC assays, human α1AR, D4.4R and D4.7R expressing the amino acid residues 1–229 (nRluc) or 230–311 (cRluc) of the Rluc8 variant were subcloned into pcDNA3.1. For β-arrestin-2 recruitment assay, α1AR-YFP, non-fused D4.4R and D4.7R, and β-arrestin-2 fused to pRluc-N1 were used. For Gα protein activation assays the following human constructs were used: non-fused D4.4R, D4.7R and α1AR, Gαi1-Rluc (Rluc8 variant) with Rluc inserted at position 91, Gαs-Rluc with Rluc inserted at position 67, Gαq-Rluc with Rluc inserted at position 97, non-fused Gβ1, and Gγ2 fused to YFP (mVenus variant) at the N terminus.
TAT‐TM Peptides
Synthetic peptides with the amino acid sequences of human D4Rs and α1R transmembrane domains (TMs) (Peptide Synthesis Facility, University Pompeu Fabra, Barcelona) fused to a peptide derived from the HIV-transactivator of transcription (TAT: YGRKKRRQRRR) were used as receptor heteromer-disrupting molecules. The cell-penetrating TAT peptide binds to the phosphatidylinositol‐(4,5)-bisphosphate found on the inner surface of the membrane, allowing the right orientation of the peptide when inserted in the plasma membrane [18]. TM4, TM5, TM6 and TM7 peptides of the D4Rs and α1R were chosen since TM4, TM5 and/or TM6 are often involved in the interface of GPCR heteromers, and TM7 is often used as a negative control [4, 19,20,21,22]. HIV-TAT peptide was then fused to the N‐terminus of TM4 and TM6 and to the C-terminus of TM5 and TM7. The amino acid sequences were:
-
TAT‐TM4 de D4R: RRRQRRKKRGYGSRRQLLLIGATWLLSAAVAAPVLCGL.
-
TM5‐TAT de D4R: YVVYSSVCSFFLPCPLMLLLYWATFYGRKKRRQRRR.
-
TAT‐TM6 de D4R: RRRQRRKKRGYVLPVVVGAFLLCWTPFFVVHI.
-
TM7‐TAT de D4R: LVSAVTWLGYVNSALNPVIYTVFNAYGRKKRRQRRR.
-
TAT‐TM4 de α1R: RRRQRRKKRGYLMALLCVWALSLVISIGPLFGWRQ.
-
TM5‐TAT de α1R: PGYVLFSALGSFYLPLAIILVMYCYGRKKRRQRRR.
-
TAT‐TM6 de α1R: RRRQRRKKRGYLGIVVGCFVLCWLPFFLVMPIGSF.
-
TM7‐TAT de α1R: TVFKIVFWLGYLNSCINPIIYPCSYGRKKRRQRRR.
BRET and BiLC Assays
For the bioluminescence resonance energy transfer (BRET) assays, HEK-293 T cells were transiently co‐transfected with a constant amount of the cDNA encoding the Rluc-fused receptor and increasing amounts of cDNA encoding the YFP-fused receptor. The cell medium was removed and replaced with 0.1% glucose supplemented Hank’s Balanced Salt Solution (HBSS) buffer (140 mM NaCl, 5 mM KCl, 1.2 mM CaCl2, 0.4 mM MgSO4·7H2O, 0.5 mM MgCl2·6H2O, 0.3 mM Na2HPO4, 0.4 mM KH2PO4 and 4 mM NaHCO3) and cells were collected. The protein concentration in collected intact cell preparations was determined using the Bradford assay kit (Bio-rad; Munich, Germany), with bovine serum albumin dilutions as standards. For fluorescence quantification, 20 μg of protein were plated in 96-well black and transparent bottom microplates and fluorescence measured as the emission at 530 nm after 500 nm excitation in a Mithras LB 940 (Berthold Technologies, Bad Wildbad, Germany). Separately, for BRET measurements 20 μg of protein were plated in 96-well white microplates and 5 μM of Coelenterazine H (Invitrogen) was added one minute before BRET signal acquisition using the Mithras LB 940 reader. BRET signal was determined as the ratio of the light emitted by YFP (530 nm) over that emitted by coelenterazine H (485 nm). Rluc expression was also quantified by reading luminescence 10 min after the addition of coelenterazine H. Net BRET was defined as ((long-wavelength emission)/(short-wavelength emission)) – Cf, where Cf corresponds to ((long-wavelength emission)/(short-wavelength emission)) of the Rluc protein expressed individually. BRET is expressed as milliBRET units (mBU). Data were fitted to a nonlinear regression equation, assuming a single‐phase saturation curve, with GraphPad Prism 9 software. For bimolecular luminescent complementation (BiLC) assays, cells were co-transfected with the cDNA encoding the receptors of interest fused to Rluc hemiproteins (nRluc and cRluc). After 48 h, cells were treated or not with the indicated TAT-TM peptides (2 μM) for 4 h at 37 °C. The quantification of the receptor-reconstituted Rluc expression was measured at 485 nm after 10 min of adding coelenterazine H. Cells expressing the receptor fused to one hemiprotein showed similar luminescence levels to nontransfected cells.
G-Protein Activation and β-Arrestin-Recruitment BRET Assays
Variations of BRET assays were also performed to detect ligand-induced activation of distinct subtypes of Gα protein and β-arrestin recruitment. For G-protein activation assay, expression vectors coding different Rluc-fused Gα protein subunits and YFP-fused Gγ2 protein were co-transfected with the receptor or receptors of interest and non-fused Gβ1 constructs. For β-arrestin recruitment, Rluc (Rluc8 variant)-fused β-arrestin-2, α1AR-YFP and non-fused D4.4R and D4.7R constructs were co-transfected for BRET detection. As previously reported [14], cells were harvested, washed, and resuspended in 0.1% glucose supplemented Hank’s Balanced Salt Solution (HBSS) buffer. Approximately 200,000 intact cells/well were distributed in 96-well plates, and 5 μM coelenterazine H (substrate for luciferase) was added to each well. Two minutes after the addition of coelenterazine H, agonists were added to each well, whereas antagonists were added 10 min before the addition of the agonist. The acceptor fluorescence was quantified (excitation at 500 nm and emission at 540 nm for 1-s recordings) in Mithras LB940 (Berthold Technologies, Bad Wildbad, Germany) to confirm the constant expression levels across experiments. In parallel, the BRET signal from the same batch of cells was determined as the ratio of the light emitted by mVenus/YFP (530 nm) over that emitted by Rluc (485 nm) in PHERAstar Flagship microplate reader (BMG Lab technologies, Offenburg, Germany). The ligand induced events were calculated as the BRET change (BRET ratio for the corresponding drug minus the BRET ratio in the absence of the drug) observed after the addition of the ligands. BRET curves were analyzed by nonlinear regression using GraphPad software. All ligands tested are from Tocris Bioscience; Bristol, UK.
cAMP Accumulation Assay
The HEK-293 T cell lines with inducible expression of D4.4R or D4.7R were transfected with non-fused α1A receptor. Two hours before initiating the assay, cell culture medium was substituted by serum-free medium. Cells were then detached and resuspended in serum-starved medium containing 50 μM zardaverine, 0.1% BSA and 5 mM HEPES and were plated in 384-well microplates (1500 cells/well) and treated with the corresponding ligands. Cells were then pre-treated with the antagonists or vehicle for 15 min and then stimulated with agonists also for 15 min. In case of Gi-mediated inhibitory signaling, cells were stimulated for 15 min with forskolin after agonists treatment. Intracellular cAMP production was quantified by homogeneous time-resolved fluorescence (HTRF) energy transfer method using the Lance Ultra cAMP kit (PerkinElmer, Waltham, Massachusetts, US). Fluorescence readings at 665 nm were performed on a PHERastar Flagship Microplate Reader (BMG Labtech, Ortenberg, Germany) equipped with an HTRF optical module.
Intracellular Calcium Release
To determine intracellular calcium free concentration, HEK-293 T cells were co-transfected with cDNA encoding the receptor or receptors of interest and 3 μg of the GCaMP6 calcium sensor. Cells were harvested, washed, and resuspended in Mg2+-free Locke’s buffer (154 mM NaCl, 5.6 mM KCl, 3.6 mM NaHCO3, 2.3 mM CaCl2, 5.6 mM glucose, 5 mM HEPES, pH 7.4) supplemented with 10 μM glycine. The protein concentration in collected intact cell preparations was determined using the Bradford assay kit (Bio-rad; Munich, Germany), using bovine serum albumin dilutions as standards. Then, 40 µg of protein were plated in 96-well black, clear-bottom microplates and treated with the desired ligands. Fluorescence emission intensity of the GCaMP6 sensor was recorded for 150 s (30 flashes/well) at 515 nm upon excitation at 488 nm on an EnSpire® Multimode Plate Reader (Perkin Elmer; Wellesley, MA, United States).
Brain Slice Preparation
Male Sprague Dawley rats (2 months old; from the animal facility of the Faculty of Biology, University of Barcelona) were used. The animals were housed two per cage and kept on a 12 h dark/light cycle with food and water available ad libitum, and experiments were performed during the light cycle. All procedures were approved by the Ethical Committee for Animal Use of University of Barcelona (OB 408/18 and OB 409/18). Animals were killed by decapitation under 4% isoflurane anesthesia, and brains were rapidly removed, placed in ice-cold oxygenated (O2/CO2, 95%/5%) Krebs–HCO3− buffer (containing [in mM]: 124 NaCl, 4 KCl, 1.25 KH2PO4, 1.5 MgCl2, 1.5 CaCl2, 10 glucose, and 26 NaHCO3, pH 7.4), and sliced coronally at 4 °C using a brain matrix (Zivic Instruments). Slices of the prefrontal cortex or striatum (500 μm thick) were dissected at 4 °C in Krebs–HCO3− buffer; each slice was transferred into an incubation tube containing 1 ml of ice-cold Krebs–HCO3− buffer. The temperature was raised to 23 °C, and after 30 min the medium was replaced by 2 ml of fresh buffer or TM peptides prepared in Krebs–HCO3− buffer at 4 µM. Slices were incubated under constant oxygenation (O2/CO2, 95%/5%) at 30 °C for 4 h in an Eppendorf Thermomixer (5 Prime, Boulder, Colorado, USA). Then, the medium was replaced with 200 μl of fresh buffer and incubated for 30 min before the addition of any ligand. After incubation with the corresponding ligands, the solution was discarded, and slices were frozen on dry ice and stored at − 80 °C. The tissue was lysed by the addition of ice-sold lysis buffer and treated as described below for HEK‐293 T cells for ERK1/2 phosphorylation determination.
ERK1/2 Phosphorylation Assay
HEK‐293 T cells were co-transfected with α1AR and D4.4R or D4.7R. The day of the experiment, the culture medium was substituted by serum-starved medium 4 h before treatment with the ligands of interest for 10 min in case of antagonists and 7 min for agonists, at 37 °C and in humid atmosphere. Then, cells were placed in ice to stop the metabolism and cells were washed with ice‐cold PBS. Successively, ice-cold lysis buffer (50 mM Tris–HCl pH 7.4, 50 mM NaF, 150 mM NaCl, 45 mM-glycerophosphate, 1% Triton X-100, 20 μM phenylarsine oxide, 0.4 mM NaVO4 and protease inhibitor) was added. The cellular debris was removed by centrifugation at 13,000 g for 10 min at 4 °C, and the protein levels were quantified by the bicinchonic acid method, using bovine serum albumin dilutions as standard. The samples were stored and then processed for immunoblotting as described below.
Western Blotting
Determination of protein levels by immunoblotting was carried out in transfected cells or in brain slices to determine the level of ERK1/2 phosphorylation. Equivalent amounts of cell protein were separated by polyacrylamide gel electrophoresis on denaturing conditions (10% SDS). Proteins were transferred into polyvinylidene fluoride membranes and then treated with odyssey blocking buffer (LI‐COR Biosciences, Lincoln, Nebraska) for 1 h. Primary antibodies mixture of a mouse anti– phospho‐ERK1/2 antibody (1:2500; Sigma‐Aldrich) and rabbit anti‐ERK1/2 antibody (1:40,000; Sigma‐Aldrich) were added and kept over-night at 4 °C. After removal of the primary antibodies the 42‐ and 44‐kDa bands corresponding to ERK1 and ERK2 were visualized by the addition of a mixture of IRDye800 (anti‐mouse) antibody (1:10,000; Sigma‐Aldrich) and IRDye 680 (anti‐rabbit) antibody (1:10,000; Sigma‐Aldrich) for 2 h and scanned by the Odyssey infrared scanner (LICOR Biosciences). Band densities were quantified using the scanner software and exported to Excel (Microsoft, Redmond, WA). The level of phosphorylated ERK1/2 isoforms was normalized for differences in loading using the total ERK1/2 protein band intensities.
In situ Proximity Ligation Assay
Rat brain slices were fixed by immersion in 4% PFA solution for 1 h at 4 °C. Samples were then washed in 50 mM Tris‐HCl, 0.9% NaCl pH 7.8 buffer (TBS), cryopreserved in a 30% sucrose solution for 48 h at 4 °C, and stored at ‐20 °C until sectioning. 20 μm‐thick slices were cut coronally (frontal to bregma AP = 0) on a freezing cryostat (Leica Jung CM‐3000), mounted on slide glass and frozen at ‐20 °C until use. To perform the PLA, slices were thawed at 4 °C, washed in PBS, permeabilized with PBS containing 0.01% Triton X‐100 for 10 min, and successively washed with PBS. Heteromers were detected using the Duolink II in situ PLA detection Kit (Sigma-Aldrich) and following the instructions of the supplier. To detect α1AR‐D4R complexes, a mixture of equal amounts of mouse anti‐α1AR antibody (Thermo Scientific, Fremont, California) and goat anti‐D4R (sc‐1439) (Santa Cruz Biotechnology, Santa Cruz, California) antibody was used. Samples were further incubated with anti‐mouse plus and anti‐goat minus PLA probes. Slices were mounted using DAPI-containing mounting medium and observed in a Leica SP2 confocal microscope (Leica Microsystems, Mannheim, Germany) equipped with an apochromatic 63X oil‐immersion objective (N.A. 1.4), and a 405 nm and a 561 nm laser line. For each field of view, a stack of two channels (one per staining) and 9 to 15 Z stacks with a step size of 1 μm were acquired. Images were opened and processed with Image J software (National Institutes of Health, Bethesda, MD). Quantification of cells containing one or more red dots versus total cells (blue nucleus) was determined by using the Fiji package (https://fiji.sc/). Nuclei and red spots were counted on the maximum projections of each image stack. After getting the projection, each channel was processed individually. The blue nuclei were segmented by filtering with a median filter, subtracting the background, enhancing the contrast with the Contrast Limited Adaptive Histogram Equalization (CLAHE) plug‐in, and finally applying a threshold to obtain the binary image and the regions of interest (ROIs) around each nucleus. Red spot images were also filtered and thresholded to obtain the binary images. Red spots were counted in each of the ROIs obtained in the nuclei images.
Results
In vitro Identification of α1AR-D4.4R and α1AR-D4.7R Heteromers
The possible heteromerization of D4.4R and D4.7R human polymorphic variants with α1AR was first explored using the BRET biophysical approach. In this technique the bioluminescent donor (Rluc) and acceptor (YFP) are fused to the two putatively interacting receptors and BRET occurs when they are in very close proximity. A saturation curve indicates a specific interaction while a straight line indicates a random-collision non-specific interaction. The experiments were performed in HEK-293 T cells transiently co-transfected with the cDNA of one receptor fused to Rluc and increasing amounts of cDNA encoding the other receptor fused with YFP. Saturation BRET curves were obtained with D4.4R-Rluc and α1AR-YFP, and with α1AR-Rluc and D4.7R-YFP (Figs. 1A and B). The BRETmax obtained for the D4.4R-α1AR pair was 53 ± 5 mBU (in mean ± S.D. of milliBRET units, n = 4), and a significantly lower signal was obtained for the D4.7R-α1AR pair (39 ± 3 mBU, n = 6; non-paired t test: p < 0.001), while BRET50 values obtained for both pairs were not significantly different: 33 ± 7 and 25 ± 5 (in mean ± S.D.) for D4.4R-α1AR and D4.7R-α1AR, respectively. In contrast, linear plots were obtained in cells transfected with both combinations of fusion proteins of the α2AR-α1AR pair (Figs. 1A and B). These results could indicate a reduced ability of D4.7R to form heteromers with α1AR, as also suggested for D2R-D4.7R and α2AR-D4.7R heteromers, as compared with D4.4R [4, 6], although a reduced BRET between the intracellularly localized Rluc and YFP due to a hindrance effect related to the large 3IL of D4.7R could also be involved.

BRET experiments on heteromerization of α1AR with D4.4R and D4.7R. A HEK-293 T cells were transfected with a constant amount of D4.4 R-Rluc cDNA (0.11 μg) or α2AR-Rluc cDNA (0.008 μg), and with increasing amounts of α1AR-YFP cDNA (0.01 to 1.8 μg). B HEK-293 T cells were transfected with a constant amount of α1AR-Rluc cDNA (0.25 μg) and with increasing amounts of D4.7R-YFP cDNA (0.15 to 2.5 μg) or α2AR-YFP cDNA (0.2–2 μg). The relative amount of BRET is given as a function of 100 the ratio between the fluorescence of the acceptor (YFP) and the luciferase activity of the donor (Rluc). BRET is expressed as mili BRET units (mBU) of 3 to 6 different experiments. C, D Effect of TM peptides on α1AR-D4R heteromerization by BiLC. The figure shows the quantification of luminescence due to Rluc complementation in HEK-293 cells co-expressing α1AR-cRluc and D4.4R-nRluc in the absence (control) or presence of TM4, TM5, TM6 and TM7 peptides of D4R (C) and α1AR (D). Cells were treated for 4 h with vehicle or with the corresponding TM peptide (2 μM) before performing the complementation assay. Luminescence values (in percentage of control) represent means ± S.E.M. from 8 different experiments. The luminescence control values (without interfering peptides) were always between 120.000 and 150.000 relative luminescence units. Statistical differences in luminescence values were calculated by one-way ANOVA followed by Dunnett’s post-hoc test; ***: P < 0.001, versus control
BiLC experiments with α1AR-cRluc and D4.4R-nRluc also demonstrate a significant proximity of both receptors, compatible with α1AR-D4.4R heteromerization, which was significantly reduced with the incubation of TM4 and TM6 peptides but not with TM5 or TM7 peptides of either D4R and α1AR (Figs. 1C and D). These results indicate that TM4 and TM6 of both receptors form part of the heteromeric interface. The corresponding TM peptides can then be used as heteromer-disrupting tools to disclose the pharmacological properties of α1AR-D4R heteromers and their presence in native tissues.
Differences in D4.4R and D4.7R-Mediated G Protein Activation upon α1AR Co-Expression
HEK-293 T cells were co-transfected with D4.4R or D4.7R, the Gα subunit of the Gi protein (Gαi1) fused to Rluc, the Gγ2 subunit fused to YFP and non-fused Gβ1 subunit, without or with co-transfection with non-fused α1AR. Gi protein activation was analyzed as changes in the BRET signal induced by the endogenous agonists, dopamine and norepinephrine, in the absence and presence of the D4R antagonist L745870, or α1AR ligands, the α1AR agonist A61603 and the α1AR antagonist prazosin. The effect of A61603 alone was also analyzed. The antagonists were administered 10 min before the agonists. Data were fitted to sigmoidal concentration–response curves and EC50 and Emax values were deduced. As expected, in the absence of α1AR, D4.4R and D4.7R showed the same pharmacological profile, with similar EC50 and Emax values for dopamine and similar values for norepinephrine, which showed about 10 times less potency and the same efficacy than dopamine (Figs. 2A and B and Table 1), as previously described [14]. Also as expected, the selective α1AR agonist A61603 did not produce any effect and did not modify the response to dopamine, and the selective D4R antagonist L745870, but not the α1AR antagonist prazosin, antagonized the effect of dopamine (Figs. 2A and B and Table 1). Interestingly, co-transfection with α1AR did not modify this pharmacological profile for D4.7R, but it did for the D4.4R. In this case, the two α1AR ligands, A61603 and prazosin, promoted a significant shift to the right of the dopamine concentration–response curve, with a significant decrease in the EC50 values (of about 8 times), strongly suggestive of selective negative allosteric modulations by α1AR ligands, agonists or antagonists, of the Gi protein activating effect of dopamine in the α1AR-D4.4R heteromer (Figs. 2C and D and Table 1). Apart from the negative crosstalk shown with agonists of both receptors, the most demonstrative results of a dependence on α1AR-D4.4R heteromerization are those showing cross-antagonism, the ability of a selective antagonist of a GPCR to counteract the activation of another molecularly different GPCR.

BRET experiments on ligand-induced D4.4R and D4.7R-mediated Gi protein activation with and without α1AR co-expression. Concentration–response experiments of dopamine (DA), norepinephrine (NE) and the α1AR agonist A61603 in the presence of A61603 (1 µM), the D4R antagonist L745870 (1 µM) or the α1AR antagonist prazosin (1 µM) in HEK-293 T cells transiently transfected with Gαi1-Rluc, Gγ2-YFP, non-fused Gβ1 and D4.4R (A), D4.7R (B), D4.4R plus α1AR (C) or D4.7R plus α1AR (D). Ligand-induced changes in BRET values were measured as described in Material and Methods. BRET values in the absence of ligands were subtracted from the BRET values for each agonist concentration. Data from all the experiments per treatment were fitted to a sigmoidal dose–response function by nonlinear regression analysis per experiment and represent means ± S.E.M. (n = 3–8, performed in triplicate) (see Table 1 for EC50 and Emax values and statistical analysis)
Differences in α1AR-Mediated G Protein Activation Upon Co-Expression with D4.4R or D4.7R
Classically, α1AR is coupled to the Gαq/11 protein family, but it has also been shown to couple to the Gαs protein-cAMP signaling pathway [23, 24]. We therefore analyzed both G protein subtypes when studying α1AR-mediated G protein activation in BRET experiments. HEK-293 T cells were co-transfected with α1AR, the Gα subunit of the Gq or Gs proteins (Gαq or Gαs) fused to Rluc, the Gγ2 subunit fused to YFP and non-fused Gβ1 subunit, without or with co-transfection with D4.4R or D4.7R. Gq or Gs protein activation was analyzed as changes in the BRET signal induced by endogenous agonists norepinephrine and dopamine, the selective α1AR agonist A61603 and the selective D4R agonist A412997. A61603 was then used to analyze its possible interactions with D4R ligands, since, differently from norepinephrine, it did not promote significant D4R activation (see above). The effect of A61603 was then also analyzed in the presence of A412997, L745870 and prazosin. The antagonists were administered 10 min before the agonists.
The changes in BRET values can be positive or negative depending on the Gα subtype as well as on the position and orientation of the inserted Rluc [25]. In the present experiment, Gi1 and Gq activation produced a decrease in BRET values whereas Gs activation produced an increase in the BRET signal (Fig. 3). In the absence of D4Rs, norepinephrine promoted a significant Gq and Gs activation, while dopamine was mostly inefficient (less than 50% as compared to norepinephrine, at the highest 10 μM concentration), which did not allow reliable EC50 calculations, indicating that it should be at least two orders of magnitude higher than the EC50 values for norepinephrine (Figs. 3A and D). In both cases, A61603 was more potent and as effective as norepinephrine, and the effect of A61603 was counteracted by prazosin and not modified by A412997 or L745870 (Figs. 3A and D and Tables 2 and 3). The same pharmacological profile was observed with co-transfection with D4.7R (Figs. 3C and F), while upon co-transfection with D4.4R, both D4R ligands promoted a significant shift to the right of the A61603 concentration–response curves, with an increase of more than ten times in the EC50 values (Figs. 3B and E and Tables 2 and 3). This is also strongly suggestive of reciprocal selective negative allosteric modulations, by which D4R agonists or antagonists counteract the Gq and Gs protein activating effect of the α1AR agonist A61603 in the α1AR-D4.4R heteromer.

BRET experiments on ligand-induced α1AR-mediated Gq and Gs protein activation with and without D4.4R or D4.7R co-expression. Concentration–response experiments of dopamine (DA), norepinephrine (NE), the D4R agonist A412997 or the α1AR agonist A61603 alone or in the presence of A412997 (1 µM), the α1AR antagonist prazosin (1 µM) or the D4R antagonist L745870 (1 µM) in HEK-293 T cells transiently transfected with Gαq-Rluc (A-C) Gαs-Rluc (D-F), Gγ2-YFP, non-fused Gβ1 and α1AR (A, D), α1AR plus D4.4R (B, E) or α1AR plus D4.7R (C, F). Ligand-induced changes in BRET values were measured as described in Material and Methods. BRET values in the absence of ligands were subtracted from the BRET values for each agonist concentration. Data from all the experiments per treatment were fitted to a sigmoidal dose–response function by nonlinear regression analysis per experiment and represent means ± S.E.M. (n = 3–8, performed in triplicate) (see Tables 2 and 3 for EC50 and Emax values and statistical analysis)
Different Modulation of Adenylyl Cyclase and Calcium Signaling in α1AR-D4.4R and α1AR-D4.7R Cells
The consequence of the specific allosteric modulations between α1AR and D4R ligands on G protein activation demonstrated in cells co-expressing α1AR and D4.4R were analyzed at the level of G protein-dependent signaling. First, on adenylyl cyclase signaling, with cAMP accumulation experiments, where activation of Gs proteins increases cAMP formation, while activation of Gi proteins inhibits forskolin-induced cAMP accumulation. These experiments were performed in previously characterized inducible D4.4R and D4.7R cell lines [4] co-transfected with α1AR. The D4R agonist A412997 (10 nM) significantly inhibited forskolin-induced cAMP accumulation, and the α1AR agonist A61603 (10 nM) promoted a discrete but significant cAMP accumulation (Figs. 4A and B). In both α1AR-D4.4R and α1AR-D4.7R cells, prazosin (1 μM) did not modify the effect of forskolin and counteracted A61603-induced cAMP accumulation. In α1AR-D4.4R cells, but not in α1AR-D4.7R cells, prazosin also counteracted the effect of A412997, and A412997 counteracted A61603-induced cAMP accumulation (Figs. 4A and B).

Adenylyl cyclase and calcium signaling in α1AR-D4.4R and α1AR-D4.7R cells. A, B cAMP formation in tetracycline-inducible HEK-293 T cells expressing D4.4R (A) or D4.7R (B) and transiently co-transfected with α1AR. cAMP formation was induced by forskolin (FK, 200 nM; black bars) or with the α1R agonist A61603 (10 nM; white bars) in the presence or absence of the D4R agonist A412997 (10 nM) or the α1R antagonist prazosin (1 μM). Values are means ± S.E.M. of 3 to 5 experiments, expressed as % of cAMP induced by forskolin. Statistical differences between different treatments were analyzed by one-way ANOVA, followed by Dunnett’s post hoc test; *, **, *** and ****: P < 0.05, P < 0.01, P < 0.001 and < 0.0001, respectively, versus basal; ### and ####: P < 0.001 and P < 0.0001, respectively, versus forskolin. C-E Intracellular calcium mobilization in HEK-293 T cells transiently co-transfected with GCaMP6, D4.4R and α1AR (C, E) or D4.7R and α1AR (D, F). Cells were activated with 20 nM of A61603 (black), co-activated with A61603 and A412997 (blue) or pre-treated with 60 nM of prazosin (grey) or L745870 (red) before A61603 activation. C, D Representative experiments showing the intracellular calcium release curves over time. E, F Values are means ± S.E.M. of 4 experiments, expressed as % of maximal effect of A61603. Statistical differences between different treatments were analyzed by one-way ANOVA, followed by Dunnett’s post hoc test; ** and ***: P < 0.01 and P < 0.001, respectively
Intracellular calcium mobilization was then used as a correlative measure of Gq activation. Both in α1AR-D4.4R cells and α1AR-D4.7R cells, A61603 promoted a clear increase in the intracellular calcium signal, which was counteracted by prazosin. Only in α1AR-D4.4R cells, the effect of A61603 was significantly decreased by the D4R ligands, A412997 and L745870 (Figs. 4C-E). In summary, the G protein activation and the G protein-dependent signaling experiments demonstrate functional differences between D4.4R and D4.7R that depend on the co-expression with the α1AR. Specifically the D4.4R variant determines the appearance of reciprocal negative crosstalk and cross-antagonism between α1AR and D4.4R cells, which are pharmacological properties that are often simultaneously disclosed by GPCR heteromers [19, 22]. The negative results obtained in α1AR-D4.7R cells, so far would indicate the lack of allosteric interactions in the α1AR-D4.7R heteromer.
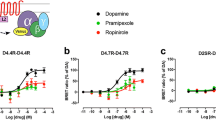
Similar Modulation of β-arrestin Recruitment and MAPK Signaling in α1AR-D4.4R and α1AR-D4.7R Cells
Before excluding the existence of allosteric interactions in the α1AR-D4.7R heteromer, we also studied G protein-independent signaling. Thus, in previous studies we found that allosteric modulations in GPCR heteromers can have functional selectivity, i.e., selectivity for a signaling pathway, such as for a G protein-dependent or G protein-independent pathway. For instance, in the dopamine D1 receptor (D1R)-D3R heteromer, there is a specific G protein-independent, β-arrestin-mediated synergistic interaction between D1R and D3R agonists [20]. The presence of these potentially independent allosteric interactions can also be modulated by different cellular mechanisms, such as intracellular calcium levels, as reported for the A2AR-D2R heteromer [26]. HEK-293 T cells were co-transfected with non-fused D4.4R or D4.7R, α1AR fused to YFP and β-arrestin-2 fused to Rluc. The β-arrestin recruitment is then quantified as changes in BRET signal induced by increasing concentrations of ligands. Importantly, this assay should constitute an additional method to reveal α1AR-D4.7R heteromers. Thus, the BRET detection of β-arrestin-2-Rluc recruitment by D4R agonists with α1AR fused to YFP implies a very significant proximity between both receptors, a β-arrestin recruitment by the GPCR heteromer. Similarly, interactions between D4R and α1AR ligands should imply allosteric interactions within the α1AR-D4.7R heteromer.
In fact, both in α1AR-D4.4R and α1AR-D4.7R cells, not only norepinephrine and A61603 promoted β-arrestin-2 recruitment, but also dopamine and the D4R agonist A412997 (Figs. 5A and B and Table 4), indicating the presence of functional α1AR-D4.4R and α1AR-D4.7R heteromers. Importantly, a qualitative different profile could be observed between both heteromers, with a lower relative efficacy of dopamine versus norepinephrine in the α1AR-D4.4R heteromer. In both heteromers, the potency of dopamine was higher than the one obtained in the G protein activation experiments and its relative potency versus norepinephrine was about two orders of magnitude. Importantly, negative crosstalk and cross-antagonism between α1AR and D4.R ligands could be observed in both α1AR-D4.4R and α1AR-D4.7R heteromers. In both cases, the concentration response-curve of A61603 was substantially shifted to the right, not only with prazosin, but also with dopamine or L745870 (Figs. 5C and D and Table 4). Also, for both α1AR-D4.4R and α1AR-D4.7R heteromers, the concentration response-curve of dopamine was substantially shifted to the right, not only with L745870, but also with prazosin (Figs. 5E and F and Table 4).

β-arrestin-2 recruitment-BRET experiments in HEK-293 T cells transiently transfected with D4.4R or D4.7R, α1AR-YFP and β-arrestin-2-Rluc. A, B Concentration–response curves induced by endogenous ligands dopamine (DA) or norepinephrine (NE), the α1AR agonist A61603 or the D4R agonist A412997 alone. C, D Concentration response-curve of A61603 in the presence of the α1AR antagonist prazosin (1 μM), the D4R antagonist L745870 (1 μM) or dopamine (10 nM). E, F Concentration response-curve of dopamine in the presence of the α1AR antagonist prazosin (1 μM) or the D4R antagonist L745870 (1 μM). After 7 min of drug exposure, BRET was measured as described in Materials and Methods. BRET values in the absence of ligands were subtracted from the BRET values for each condition. Data from all the experiments per treatment were fitted to a sigmoidal dose–response function by nonlinear regression analysis per experiment and represent means ± S.E.M. (n = 3–8, performed in triplicate) and are shown as a percentage of A61603 3 activation (see Table 4 for EC50 and Emax values and statistical analysis)
We also analyzed MAPK signaling (ERK1/2 phosphorylation), which is often a G protein-independent and β-arrestin-mediated signaling, and the results paralleled those of the β-arrestin-2 recruitment. In α1AR-D4.4R and α1AR-D4.7R cells, A412997 and A61603 promoted ERK1/2 phosphorylation which, in both cases, were counteracted by both prazosin and L745870 (Figs. 6A and B). Altogether, these experiments strongly support that α1ARs form functional heteromers with both D4.4R and D4.7R and that they show a differential profile in their allosteric interactions, with negative crosstalk and cross-antagonism that occur at the level of G protein-dependent and independent signaling for the α1AR-D4.4R heteromer and which are functionally selective, β-arrestin-dependent, for the α1AR-D4.7R heteromers.

MAPK signaling in α1AR-D4.4R and α1AR-D4.7R cells. A, B ERK1/2 phosphorylation in HEK-293 T cells transiently transfected with α1R and D4.4R (A) or D4.7R (B). ERK1/2 phosphorylation was induced by the α1R agonist A61603 (500 nM) or the D4R agonist A412997 (500 nM) in the presence or absence of the α1R antagonist prazosin (5 μM) or the D4R antagonist L745870 (5 μM). The values represent the mean ± S.E.M. of quantified immunoreactive bands corresponding to ERK1 and ERK2 of 4 to 8 experiments and expressed as percentage of values from non-treated cells. Statistical differences between different treatments were analyzed by one-way ANOVA, followed by Dunnett’s post hoc test; *** and ****: P < 0.001 and P < 0.0001, respectively, versus A412997; #, ## and ###: P < 0.05, P < 0.01 and P < 0.001, respectively, versus A61603
Identification of Functional α1AR-D4R Heteromers in the Rat Frontal Cortex and Striatum
The possible existence of α1AR-D4R heteromers in the rat brain, in the frontal cortex and striatum, was first analyzed by using the proximity ligation assay (PLA). PLA requires that both receptors be close enough (< 40 nm) to allow the two different antibody-based probes to ligate (see Materials and Methods). If the receptors are forming complexes, a punctate fluorescent signal can be detected by confocal microscopy. Red dots were detected surrounding DAPI-positive nuclei in striatal and cortical slices (Figs. 7C, D, E and F). The number of apparent cells with red dots was significantly higher than the number of dots from slices treated only with one primary antibody and both secondary antibodies (negative controls) (Figs. 7A, B, G and H). These results indicate the existence of complexes of α1AR and D4R in the rat brain, compatible with α1AR-D4R heteromers. Although the experiments did not allow to identify if the dots are preferentially expressed presynaptically in nerve terminals establishing contact with cell bodies, or postsynaptically in the somatodendritic area, they should be expected to mainly label cortico-striatal glutamatergic terminals, where both receptors can mostly be co-localized (see Introduction and Discussion).

Detection of α1aR-D4R complexes in rat brain by PLA. A, B Absence of complexes in the absence of α1aR antibody (negative controls) in striatal (A) and cortical slices (B). C, D α1AR-D4R complexes observed as red around blue-coloured DAPI-stained cell nuclei in striatal (C) and cortical slices (D) and (E, F) cropped figures of the main C and D images. G Number of cells containing one or more red spots, expressed as a percentage of the total number of cells. H r values (number of red spots/cells containing spots). Data are the mean ± S.E.M. of counts of 3 different experiments. Statistical differences versus negative control were analyzed by one‐way ANOVA followed by Dunnett’s post hoc test (***P < 0.001; ****P < 0.0001). Scale bar: 20 μm
We then used a more functional but also more demonstrative method to identify α1AR-D4R heteromers in rat cortical and striatal slices, based on the identification of a pharmacological property of the heteromer (biochemical fingerprint) and on its specific disruption by synthetic peptides that specifically disrupt α1AR-D4R heteromers (demonstrated by BiLC experiments, Figs. 1C and D). ERK1/2 phosphorylation induced by the D4R agonist A412997 and the α1AR agonist A61603 was analyzed in slices from rat frontal cortex or striatum, incubated in the absence or presence of the D4R antagonist L745870 or the α1AR antagonist prazosin, and incubated in the absence or presence of α1AR-D4R heteromer-disrupting peptides. In the absence of peptides, both in cortical and striatal slices, A412997 (1 μM) and A61603 (1 μM) promoted ERK1/2 phosphorylation, which, for both agonists, was significantly counteracted by L745870 and prazosin at a concentration (10 μM) that did not produce a significant effect versus basal values (Figs. 8A and B). Importantly, both in cortical and striatal slices, the L745870-mediated cross-antagonism of A61603 was significantly and selectively counteracted by the TM peptides of both α1AR and D4R that disrupted α1AR-D4R heteromerization (TM4 and TM6, but not TM5 and TM7) (Figs. 8C-F). Altogether, these experiments demonstrate that a significant proportion of frontal cortical and striatal α1AR and D4R form functional α1AR-D4R heteromers.

MAPK signaling in rat brain. A, B ERK1/2 phosphorylation in rat cortical (A) or striatal slices (B). ERK1/2 phosphorylation was induced by the α1R agonist A61603 (1 μM) or the D4R agonist A412997 (1 μM) in the presence or absence of the α1R antagonist prazosin (10 μM) or the D4R antagonist L745870 (10 μM). C-F Effect of TM peptides of α1AR and D4R. Slices were not pretreated (Ø) or were pre-treated for 4 h with 4 μM of TM4-TM7 peptides of α1AR (labelled in red) or D4R (labelled in green). Slices were not stimulated (vehicle), stimulated for 10 min with A61603 or pre-treated for 20 min with 10 µM of L745870 before A61603 treatment. The values represent the mean ± S.E.M. of quantified immunoreactive bands corresponding to ERK1 and ERK2 of 4 experiments and expressed as percentage of values from non-treated slices. Statistical differences between different treatments were analyzed by one-way ANOVA, followed by Dunnett’s post hoc test; * and ***: P < 0.05 and P < 0.001, respectively, versus A412997; #, ## and ###: P < 0.05, P < 0.01 and P < 0.001, respectively, versus A61603
Discussion
The present study demonstrates the ability of the D4R to form functional heteromers with α1AR in the brain, adding to the list of heteromers of D4R with other adrenoceptors, which includes α1BR and β1R in the pineal gland and α2AR in the frontal cortex [1]. In addition, it also adds to the studies that showed that the functional and pharmacological differences between the D4R polymorphic variants, more specifically between D4.4R and D4.7R, can be disclosed upon heteromerization with other GPCRs, such as α2AR and D2R [4,5,6, 14]. It has been suggested that α2AR-D4R and D2R-D4R, respectively localized in the cortical perisomatic region and in the striatal nerve terminals of pyramidal neurons, exert a key modulatory role of cortico-striatal glutamatergic neurotransmission [1]. This modulatory role is different depending on the D4R variant, which depends on the existence of different allosteric modulations in the respective heteromers, with the presence of D4.7R promoting a gain of function of the D2R-mediated dopaminergic and α2AR-mediated noradrenergic inhibitory control of cortico-striatal glutamatergic transmission [1].
Nevertheless, α1AR-D4R heteromers represent a significant population of functional interacting α1ARs and D4Rs localized in the rat frontal cortex and in the striatum, as demonstrated with PLA and MAPK activation experiments. In the striatum, the demonstration of α1AR-D4R heteromers implies their preferential role in the modulation of striatal glutamate release, in view of the preferential striatal localization of both receptors in striatal glutamatergic terminals [15, 16]. In this case, as discussed below, it is the presence of the D4.4R, and not the D4.7R, what might result on a gain of function of the α1AR-mediated noradrenergic stimulatory control of cortico-striatal glutamatergic neurotransmission.
The analysis of G protein activation and signaling in cells expressing α1ARs and D4.4Rs or D4.7Rs, also demonstrated pharmacological differences between both polymorphic variants that depend on heteromerization with α1ARs. Differently to the α1AR-D4.4R heteromer, the allosteric interactions in the α1AR-D4.7R heteromer were functionally selective and could only be observed in experiments of G protein-independent signaling (β-arrestin recruitment and MAPK activation). Thus, significant allosteric interactions could also be observed in the α1ARs-D4.4R heteromer in experiments of G protein activation and G protein-dependent signaling (adenylate cyclase activity or intracellular calcium mobilization). When present, the allosteric interactions between α1AR and D4.4R ligands were reciprocal and antagonistic, and agonists or antagonists of one of the receptors negatively modulated the result of the activation of the other molecularly different receptor (negative crosstalk or cross-antagonism).
The α1ARs-D4.4R heteromer is functionally like other GPCR heteromers constituted by two molecularly different GPCRs separately coupled to stimulatory and inhibitory G proteins, which promote neuronal activation and inhibition, respectively. For instance, the A2AR-D2R heteromer and D1R-D3R heteromers, with a tetrameric structure that allows the simultaneous coupling of Gs to an A2AR or D1R homodimer and Gi to a D2R or D3R homodimer [20,21,22, 26]. In these GPCR heteromers multiple and reciprocal G protein-dependent and independent allosteric interactions can be identified which can be subjected to differential control by different exogenous ligands or intracellular messengers. The output of these integrative devices will therefore depend on the final integrated signaling of the respective Gs and Gi targeted plasma membrane and intracellular effectors. In fact, some plasma membrane effectors, such as adenylyl cyclase and GIRKs oligomerize with GPCR heteromers, forming part of G protein-coupled-effector macromolecular membrane assemblies (GEMMA; [27]).
Irrespective of their coupling to stimulatory or inhibitory G proteins, activation of most GPCRs leads to MAPK signaling, which is often dependent on β-arrestin recruitment. There is still a significant lack of understanding of the functional neuronal and behavioral correlates of the isolated or combined activation or inhibition of the different GPCR-targeted cellular effectors. Nevertheless, it is generally accepted that the most immediate responses of plasma membrane effectors, which mediates early changes in neuronal excitability and neurotransmitter release, are mediated by their direct interaction with G protein subunits [27]. On the other hand, MAPK activation mediates more protracted gene-expression-mediated effects. It should therefore be expected that the lack of G protein-dependent allosteric interactions in the α1AR-D4.7R heteromer would determine significant functional neuronal and behavioural differences, as compared with the α1AR-D4.4R heteromer.
According to the results from G protein activation, norepinephrine can potentially bind to α1AR and, with higher concentrations, to the D4R, while dopamine seems to need exceedingly large concentrations to bind to α1AR. This would not support previous suggestions about α1AR being a target for endogenous dopamine [28, 29]. Although the striatum is classically a main target of the dopaminergic system, its more ventral component, the shell of the nucleus accumbens (NAc), also receives a substantial noradrenergic innervation, both in rodents and humans, and significant basal concentrations of noradrenaline can be detected in this striatal compartment by microdialysis and shown to significantly increase with amphetamine administration [30,31,32]. Also, a seminal study by Weinshenker and colleagues showed a significant role of α1AR in the modulation of glutamate and secondarily dopamine release and in the locomotor activating effects of cocaine and morphine [15]. So, the present study indicates that a significant population of these striatal α1ARs and D4Rs form functional heteromers.
The D4.4R and D4.7R polymorphic variants should determine significant differences in the integration of norepinephrine and dopamine in the ventral striatum operated by α1AR-D4.4R or α1AR-D4.7R heteromers. The separate activation of α1AR or D4R should lead to facilitation and inhibition of glutamate release, respectively, but upon norepinephrine release, the activation of α1AR should allosterically counteract D4.4R, but not D4.7R-mediated inhibition. We should then expect the D4.4R variant to provide a gain of function of the α1AR-mediated noradrenergic stimulatory control of frontal cortico-striatal glutamatergic neurotransmission. This could therefore imply a lower degree of cortico-striatal transmission during conditions of stress in the presence of α1AR-D4.7R as compared to α1AR-D4.4R heteromers, which would add to the lower degree of cortico-striatal transmission determined by the striatal D2R-D4.7R heteromers (maybe more prevalent in the dorsal striatum) and the cortical α2AR-D4.7R heteromers [1]. Therefore, as suggested for D2R-D4.7R and α2AR-D4.7R, the α1AR-D4.7R heteromers could also increase the vulnerability of impulse control-related neuropsychiatric disorders while it could also decrease the vulnerability of PTSD (see Introduction).
D4.4R and D4.7R confer significantly different functional and pharmacological properties to α1AR-D4R heteromers, which mediate a dopamine- and norepinephrine-dependent fine-tune modulation of the frontal cortico-striatal glutamatergic neuronal function. α1AR-D4R heteromers may explain a differential vulnerability for PTSD and the differential effect of D4R polymorphisms in the moderation of the impulsivity traits and their role in impulse control-related neuropsychiatric disorders, including ADHD, and more specifically, the association of D4.7R with impulse-control disorders.
Data Availability
All data needed to evaluate the conclusions in this study are present in the paper.
References
Ferré S, Belcher AM, Bonaventura J et al (2022) Functional and pharmacological role of the dopamine D4 receptor and its polymorphic variants. Front Endocrinol (Lausanne) 13:1014678. https://doi.org/10.3389/fendo.2022.1014678
Kim J-S, Bailey MJ, Weller JL et al (2010) Thyroid hormone and adrenergic signaling interact to control pineal expression of the dopamine receptor D4 gene (Drd4). Mol Cell Endocrinol 314:128–135. https://doi.org/10.1016/j.mce.2009.05.013
González S, Moreno-Delgado D, Moreno E et al (2012) Circadian-Related Heteromerization of Adrenergic and Dopamine D4 Receptors Modulates Melatonin Synthesis and Release in the Pineal Gland. PLoS Biol 10:e1001347. https://doi.org/10.1371/journal.pbio.1001347
Casado-Anguera V, Moreno E, Sanchez-Soto M et al (2021) Heteromerization between alpha2A adrenoceptors and different polymorphic variants of the dopamine D4 receptor determines pharmacological and functional differences. Implications Impulsive-Control Dis. Pharmacol Res 170:105745. https://doi.org/10.1016/j.phrs.2021.105745
Bonaventura J, Quiroz C, Cai NS et al (2017) Key role of the dopamine D4 receptor in the modulation of corticostriatal glutamatergic neurotransmission. Sci Adv 3:e1601631. https://doi.org/10.1126/sciadv.1601631
Gonzalez S, Rangel-Barajas C, Peper M et al (2012) Dopamine D4 receptor, but not the ADHD-associated D4.7 variant, forms functional heteromers with the dopamine D2S receptor in the brain. Mol Psychiatry 17:650–662. https://doi.org/10.1038/mp.2011.93
Wang E, Ding Y-C, Flodman P et al (2004) The Genetic Architecture of Selection at the Human Dopamine Receptor D4 (DRD4) Gene Locus. The Am J Human Gene 74:931–944. https://doi.org/10.1086/420854
LaHoste GJ, Swanson JM, Wigal SB et al (1996) Dopamine D4 receptor gene polymorphism is associated with attention deficit hyperactivity disorder. Mol Psych 1:121–124
Chang F-M, Kidd JR, Livak KJ et al (1996) The world-wide distribution of allele frequencies at the human dopamine D4 receptor locus. Hum Genet 98:91–101. https://doi.org/10.1007/s004390050166
Gizer IR, Ficks C, Waldman ID (2009) Candidate gene studies of ADHD: a meta-analytic review. Hum Genet 126:51–90. https://doi.org/10.1007/s00439-009-0694-x
Faraone SV, Perlis RH, Doyle AE et al (2005) Molecular Genetics of Attention-Deficit/Hyperactivity Disorder. Biol Psychiat 57:1313–1323. https://doi.org/10.1016/j.biopsych.2004.11.024
Li D, Sham PC, Owen MJ, He L (2006) Meta-analysis shows significant association between dopamine system genes and attention deficit hyperactivity disorder (ADHD). Hum Mol Genet 15:2276–2284. https://doi.org/10.1093/hmg/ddl152
Belcher AM, Volkow ND, Moeller FG, Ferre S (2014) Personality traits and vulnerability or resilience to substance use disorders. Trends Cogn Sci 18:211–217. https://doi.org/10.1016/j.tics.2014.01.010
Sanchez-Soto M, Yano H, Cai NS et al (2019) Revisiting the Functional Role of Dopamine D4 Receptor Gene Polymorphisms: Heteromerization-Dependent Gain of Function of the D4.7 Receptor Variant. Mol Neurobiol 56:4778–4785. https://doi.org/10.1007/s12035-018-1413-1
Mitrano DA, Schroeder JP, Smith Y et al (2012) Alpha-1 Adrenergic Receptors are Localized on Presynaptic Elements in the Nucleus Accumbens and Regulate Mesolimbic Dopamine Transmission. Neuropsychopharmacol 37:2161–2172. https://doi.org/10.1038/npp.2012.68
Rommelfanger KS, Mitrano DA, Smith Y, Weinshenker D (2009) Light and electron microscopic localization of alpha-1 adrenergic receptor immunoreactivity in the rat striatum and ventral midbrain. Neuroscience 158:1530–1540. https://doi.org/10.1016/j.neuroscience.2008.11.019
Ramos BP, Arnsten AFT (2007) Adrenergic pharmacology and cognition: Focus on the prefrontal cortex. Pharmacol Ther 113:523–536. https://doi.org/10.1016/j.pharmthera.2006.11.006
He SQ, Zhang ZN, Guan JS et al (2011) Facilitation of mu-opioid receptor activity by preventing delta-opioid receptor-mediated codegradation. Neuron 69:120–131. https://doi.org/10.1016/j.neuron.2010.12.001
Ferre S, Casado V, Devi LA et al (2014) G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev 66:413–434. https://doi.org/10.1124/pr.113.008052
Guitart X, Navarro G, Moreno E et al (2014) Functional selectivity of allosteric interactions within G protein-coupled receptor oligomers: the dopamine D1–D3 receptor heterotetramer. Mol Pharmacol 86:417–429. https://doi.org/10.1124/mol.114.093096
Moreno E, Casajuana-Martin N, Coyle M et al (2022) Pharmacological targeting of G protein-coupled receptor heteromers. Pharmacol Res 185:106476. https://doi.org/10.1016/j.phrs.2022.106476
Navarro G, Cordomi A, Casado-Anguera V et al (2018) Evidence for functional pre-coupled complexes of receptor heteromers and adenylyl cyclase. Nat Commun 9:1242. https://doi.org/10.1038/s41467-018-03522-3
Shibata K, Katsuma S, Koshimizu T et al (2003) alpha 1-Adrenergic receptor subtypes differentially control the cell cycle of transfected CHO cells through a cAMP-dependent mechanism involving p27Kip1. J Biol Chem 278:672–678. https://doi.org/10.1074/jbc.M201375200
Shinoura H, Shibata K, Hirasawa A et al (2002) Key amino acids for differential coupling of alpha1-adrenergic receptor subtypes to Gs. Biochem Biophys Res Commun 299:142–147. https://doi.org/10.1016/s0006-291x(02)02589-5
Yano H, Provasi D, Cai NS et al (2017) Development of novel biosensors to study receptor-mediated activation of the G-protein alpha subunits Gs and Golf. J Biol Chem 292:19989–19998. https://doi.org/10.1074/jbc.M117.800698
Navarro G, Aguinaga D, Moreno E et al (2014) Intracellular calcium levels determine differential modulation of allosteric interactions within G protein-coupled receptor heteromers. Chem Biol 21:1546–1556. https://doi.org/10.1016/j.chembiol.2014.10.004
Ferre S, Ciruela F, Dessauer CW et al (2022) G protein-coupled receptor-effector macromolecular membrane assemblies (GEMMAs). Pharmacol Ther 231:107977. https://doi.org/10.1016/j.pharmthera.2021.107977
Cucchiaroni ML, Freestone PS, Berretta N et al (2011) Properties of dopaminergic neurons in organotypic mesencephalic-striatal co-cultures–evidence for a facilitatory effect of dopamine on the glutamatergic input mediated by α-1 adrenergic receptors. Eur J Neurosci 33:1622–1636. https://doi.org/10.1111/j.1460-9568.2011.07659.x
Lin Y, Quartermain D, Dunn AJ et al (2008) Possible dopaminergic stimulation of locus coeruleus alpha1-adrenoceptors involved in behavioral activation. Synapse 62:516–523. https://doi.org/10.1002/syn.20517
Tong J, Hornykiewicz O, Kish SJ (2006) Identification of a noradrenaline-rich subdivision of the human nucleus accumbens. J Neurochem 96:349–354. https://doi.org/10.1111/j.1471-4159.2005.03546.x
Berridge CW, Stratford TL, Foote SL, Kelley AE (1997) Distribution of dopamine beta-hydroxylase-like immunoreactive fibers within the shell subregion of the nucleus accumbens. Synapse 27:230–241. https://doi.org/10.1002/(SICI)1098-2396(199711)27:3%3c230::AID-SYN8%3e3.0.CO;2-E
Delfs JM, Zhu Y, Druhan JP, Aston-Jones GS (1998) Origin of noradrenergic afferents to the shell subregion of the nucleus accumbens: anterograde and retrograde tract-tracing studies in the rat. Brain Res 806:127–140. https://doi.org/10.1016/s0006-8993(98)00672-6
Funding
Open Access funding provided thanks to the CRUE-CSIC agreement with Springer Nature. This work was supported by the Spanish “Ministerio de Economía y Competitividad” and the European Regional Development Fund of the European Union [SAF2014-54840-R and SAF2017-87629-R to V.C. and E.I.C.]; Grant No. PID2020-113938RB-I00 (funded by MCIN/AEI/1013039/501100011033) to E.M. and V.C. and the “Generalitat de Catalunya” [2017- SGR-1497 and 2021-SGR-00230]. S.F. was funded by the intramural funds of the National Institute on Drug Abuse (grant ZIA DA000493).
Author information
Authors and Affiliations
Contributions
Conceptualization: Vicent Casadó, Sergi Ferré, Estefanía Moreno and Enric I. Canela; Methodology: Patricia Homar-Ruano, Ning-Sheng Cai, Verònica Casadó-Anguera and Estefanía Moreno; Software: Patricia Homar-Ruano and Estefanía Moreno; Validation: Patricia Homar-Ruano and Estefanía Moreno; Formal analysis: Patricia Homar-Ruano and Estefanía Moreno; Investigation: Patricia Homar-Ruano and Estefanía Moreno; Resources: Vicent Casadó, Estefanía Moreno and Enric I. Canela; Data curation: Patricia Homar-Ruano; Writing—original draft preparation: Patricia Homar-Ruano, Sergi Ferré, Estefanía Moreno and Enric I. Canela; Writing—review and editing: Patricia Homar-Ruano, Sergi Ferré, Estefanía Moreno and Enric I. Canela; Visualization: Patricia Homar-Ruano, Ning-Sheng Cai, Verònica Casadó-Anguera, Vicent Casadó, Sergi Ferré, Estefanía Moreno and Enric I. Canela; Supervision: Estefanía Moreno and Enric I. Canela; Project administration: Estefanía Moreno and Enric I. Canela; Funding acquisition: Vicent Casadó, Estefanía Moreno and Enric I. Canela. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
The Ethical Committee for Animal Use of the University of Barcelona (OB 408/18 and OB 409/18) approved all procedures.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no financial or non-financial interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Estefanía Moreno and Enric I. Canela are co-senior authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Homar-Ruano, P., Cai, NS., Casadó-Anguera, V. et al. Significant Functional Differences Between Dopamine D4 Receptor Polymorphic Variants Upon Heteromerization with α1A Adrenoreceptors. Mol Neurobiol 60, 6566–6583 (2023). https://doi.org/10.1007/s12035-023-03476-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-023-03476-8