
Abstract
The coronavirus disease that presumably began in 2019 (COVID-19) is a highly infectious disease caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and has resulted in a pandemic. Initially, COVID-19 was thought to only affect respiration. However, accumulating evidence shows a wide range of neurological symptoms are also associated with COVID-19, such as anosmia/ageusia, headaches, seizures, demyelination, mental confusion, delirium, and coma. Neurological symptoms in COVID-19 patients may arise due to a cytokine storm and a heighten state of inflammation. The nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) is a central pathway involved with inflammation and is shown to be elevated in a dose-dependent matter in response to coronaviruses. NF-κB has a role in cytokine storm syndrome, which is associated with greater severity in COVID-19-related symptoms. Therefore, therapeutics that reduce the NF-κB pathway should be considered in the treatment of COVID-19. Neuro-COVID-19 units have been established across the world to examine the neurological symptoms associated with COVID-19. Neuro-COVID-19 is increasingly becoming an accepted term among scientists and clinicians, and interdisciplinary teams should be created to implement strategies for treating the wide range of neurological symptoms observed in COVID-19 patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The coronavirus disease 2019 (COVID-19) is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The World Health Organization termed the disease COVID-19 and the virus causing the disease SARS-CoV-2. In December 2019, the Wuhan Municipal Health Commission, China, reported a cluster of cases of pneumonia in the Wuhan, Hubei Province. The novel coronavirus was identified from these patients in January 2020. Full genomic sequencing and phylogenetic analysis revealed the beta-coronavirus is in the same subgenus as the severe acute respiratory syndrome (SARS) virus that caused the SARS-CoV epidemic in 2003 [1]. The structure of the receptor is very similar to the SARS coronavirus. SARS-CoV-2 uses the same receptor as SARS for cell entry, the angiotensin-1-converting enzyme 2 (ACE2) [2]. Based on information from the Johns Hopkins University Coronavirus Resource Center, as of April 2021, there are over 129,000,000 global cases with over 2,800,000 deaths. Unfortunately, these numbers are continuing to grow.
Our knowledge of COVID-19 is limited, but new findings are quickly accumulating. Approximately 5% of patients suffer critical disease including, respiratory failure, septic shock, multi-organ failure, and death [3]. These critically ill patients tend to have higher levels of pro-inflammatory mediators and cytokines, which is indicative of a “cytokine storm syndrome” [4]. Over activation of the nuclear factor kappa-light-chain enhancer of activated B cells (NF-κB) pathway is implicated in the severe/critical pathogenesis of the COVID-19 phenotype [5]. The NF-κB complex is expressed in both neurons and glia, and NF-κB is implicated in neurodegenerative diseases [6, 7]. During SARS-CoV and Middle East Respiratory syndrome coronavirus (MERS-CoV) outbreaks, viral proteins caused excessive NF-κB activation, possibly resulting in the high levels of disease severity and high fatality rates [8,9,10]. COVID-19 was first thought strictly to be a respiratory disease. However, as more studies emerged, COVID-19 was seen to have an impact in the brain [11]. In this review article, we summarize the accumulating evidence on the effect of COVID-19 on NF-κB and neurological functions.
Cellular Mechanisms of COVID-19 Infection
The spike glycoproteins SARS-CoV-2 has a high affinity to ACE2 receptors, which are present in bronchial epithelial cells, endothelial cells, and neurons. ACE2 receptors are expressed in various anatomical locations, such as the nasal cavity, lungs, heart, kidneys, intestines, and brain. SARS-CoV-2 attaches itself to ACE2 via its spike glycoprotein, allowing RNA to enter the cell and replicate the virus [12, 13]. The wide range of ACE2 expression in multiple organs may be the reason for the heterogeneity of COVID-19 symptoms. SARS-CoV-2’s regional binding domain results in a strong affinity for ACE2 receptors [14, 15].
ACE2 is expressed in the nasal cavity epithelia, and has been hypothesized that the olfactory epithelium is a common early infection site of SARS-CoV-2. After infection of the olfactory epithelium, SARS-CoV-2 enters the brain via the olfactory nerve and olfactory bulb, according to this hypothesis [16]. ACE2 is expressed in multiple brain structures including brainstem, cortex, striatum, hypothalamus, and hippocampus [17,18,19,20]. Since ACE2 is expressed in neurons and glial cells throughout the brain, it makes both types of cells vulnerable to SARS-CoV-2 [19]. ACE2 may influence GABA (gamma-aminobutyric acid) neurotransmission in the amygdala and potentially other structures in the brain, suggesting that SARS-CoV-2 may alter the excitatory/inhibitory balance of networks in the brain [21]. The expression level of ACE2 in the brain is lower than in other organs, and other receptors may play a role in SARS-CoV-2 infection of the brain [22]. CD147 (basigin) is an extracellular matrix metalloproteinase inducer and is implicated as another receptor to which the SAR-CoV-2 spike protein is able to bind [23]. CD147 is highly expressed in mouse brain tissue compared to lung tissue, suggesting that SARS-CoV-2 may bind to CD147 in the brain [24].
NF-κB and Inflammation
NF-κB is a transcription factor that regulates multiple aspects of immune function and mediates inflammatory responses. NF-κB induces the expression of pro-inflammatory genes such as those encoding cytokines and chemokines. NF-κB regulates the survival, activation, and differentiation of innate immune cells and inflammatory T cells. The NF-κB transcription factor consists of five proteins, p65 (RelA), RelB, c-Rel, p105/p50 (NF-κB1), and p100/52 (NF-κB2). In the inactive form, NF-κB is in the cytosol and interacts with the IκB proteins. The phosphorylation of IκBs by IκB kinase (IKK) leads to nuclear translocation of NF-κB, binding to their cognate DNA and activates transcription of different genes involved with inflammation, cell proliferation, and apoptosis [9]. NF-κB activators are diverse and include lipopolysaccharides, ionizing radiation, reactive oxygen species (ROS), cytokines such as tumour necrosis factor alpha (TNF-α) and interleukin 1-beta (IL-1β), and viral DNA and RNA [25]. NF-κB transcription factors promote the gene expression of many cytokines including IL-1, IL-2, IL-6, IL-12, TNF-α, LT-α, LT-β, and GM-CSF; chemokines such as IL-8, MIP-1, MCP1, RANTES, and eotaxin; and adhesion molecules including ICAM, VCAM, and E-selectin, acute phase proteins serum amyloid A, and inducible effector enzymes, such as inducible nitric oxide synthase; iNOS and cyclooxygenase-2; COX-2 [26]. Therefore, NF-κB is the primary transcription factor that regulates many cellular responses including early innate immunity, chronic inflammatory states, viral infections, septic shock syndrome, and multi-organ failure [25, 27]. There is accumulating evidence suggesting that sustained activation of NF-κB pathways is observed in neurodegenerative disorders, such as multiple sclerosis (MS) and Alzheimer’s disease [28, 29].
NF-κB and Coronavirus
The NF-κB signaling pathway is considered a proinflammatory pathway, largely due to its effect of the transcription of many different genes involved with inflammation [30]. The severe acute respiratory syndrome-associated coronavirus (SARS-CoV) was responsible for the worldwide outbreak of SARS in 2003. The nucleocapsid protein of SARS-CoV activates NF-κB in a dose-dependent manner. SARS-CoV lacking the Envelope (E) gene had reduced expression of proinflammatory cytokines, diminished neutrophil infiltration, and reduced lung pathology that resulted in increased survival of mice [8, 31]. Inhibition of the NF-κB pathway increased survival rates in mice infected with SARS-CoV suggesting that the activation of the NF-κB signaling pathway represents a major contribution to the inflammation induced after SARS-CoV infection [8]. The SARS-CoV spike (S) protein induces a cytokine response in infected mononuclear cells in the NF-κB pathway. The cytokine response initiated via Toll-like receptor (TLR) activation through a protein kinase C-dependent pathway of NF-κB was inhibited by NF-κB blockade (see Fig. 1) [32]. Therefore, understanding how the NF-κB pathway modulates inflammatory responses may aid in the development of novel therapeutics that block the NF-κB pathway to mitigate the cytokine storm and reduce the severity of disease and/or the mortality associated with COVID-19.

COVID infection and NF-κB signaling. This putative pathway suggests that coronaviruses are able to cause inflammation in human brain through nuclear factor kappa B (NF-κB)-dependent signaling. Binding of viral SARS-CoV to its receptors, such as CD147, angiotensin-converting enzyme 2 (ACE2), and the help of the transmembrane serine protease 2 (TMPRSS2), allows SARS-CoV to enter into host cells through cleaving/activating of viral envelope glycoproteins. Within the endosomes, viral single-stranded RNA virus activates the Toll-like receptors (TLRs), such as TLR3, TLR4, and TLR7/8. These receptors activate IKK which results in phosphorylation of the cytoplasmic inhibitor factor IκBα, which in turn leads to phosphorylation of IκBα, and subsequent degradation. As a result, NF-κB p50 and p65 are released from IκBα and translocate from the cytoplasm into the nucleus to induce transcription of various genes coding for pro-inflammatory proteins such as cytokines and chemokines. Activated NF-κB is associated with a variety of cytokine receptor- and TLR-mediated signal cascades, including binding of TNFα or IL-1 to their receptors. Excessive NF-κB activation triggers production of pro-inflammatory cytokines and a chemokine storm. T cells such as CD4 and CD8 are then activated at the site of infection by cytokines and a chemokine storm and promote further inflammation. Additionally, dendritic cells trigger adaptive immunity. This figure was developed using the BioRender online software tool
An in vitro human model that simulates the initial infection in alveolar epithelium type 2 cells using induced pluripotent stem cells showed inflammatory signaling that responded to SARS-CoV-2 infection within 24 h and the NF-κB signaling pathway predominated this response [33]. When damage occurs to alveolar epithelial type 2 cells, NF-κB pathway increased activation [34]. Additionally, macrophage pro-inflammatory polarization and cytokines release contribute to the enhanced inflammatory state [34]. This dysregulated inflammatory state in the lungs may lead to acute respiratory distress syndrome and may cause multiple organ failure, one of the most frequent causes of death in patients with COVID-19. Therefore, future therapeutic efforts may be directed at targeting alveolar epithelial type 2 cells to prevent acute respiratory distress syndrome.
NF-κB Activation
NF-κB transcription factors translocate into the nucleus and bind to κB-sites, which initiates transcription and production of proinflammatory mediators [35]. This is the “canonical pathway” of NF-κB since the essential modulator (NEMO), a regulatory subunit of the IKK complex is involved. The “non-canonical pathway” of NF-κB involves protein kinase R (PKR), which also mediates TNF-α. Activation of a subset of the tumour necrosis factor superfamily receptors occurs through B-cell activating factor receptor lymphotoxin β [26]. Activation of NF-κB via the “non-canonical pathway” is slow but long lasting with proinflammatory mediator production [36]. Beta coronavirus can cause hyperactivation of the NF-κB pathway via the myeloid differentiation primary response 88 (MyD88) pathway through pattern-recognition receptors (PPRs) [35, 37]. This results in the induction of many different cytokines and chemokines [5].
ACE2
ACE2 was identified on the binding protein for SARS-CoV-2. ACE2 converts angiotensin I (AngI) and angiotensin II (AngII) into the lung-protective Ang-(1–9) and Ang-(1–7), respectively. Increased levels of AngII are associated with decreased levels of ACE2 and are implicated in the propagation of severe inflammation from renin-angiotensin pathway dysregulation [38]. AngII can act as a proinflammatory cytokine via angiotensin-1-receptor (AT1R), which activates the NF-κB pathway [39]. This leads to the production of epidermal growth factor receptor (EGFR) ligands and TNF-α, which results in the further activation of NF-κB and the propagation of a “cytokine storm” [4, 5].
The Janus Kinase (JAK) signal transduction and activator of transcription factor 3 (STAT3) pathway can activate the NF-κB pathway. IL-6 activates the JAK-STAT pathway and phosphorylated STAT3, which translocates into the nucleus and results in IFN-γ reduction. This induces cytokine release syndrome. NF-κB activation produces IL-6. During COVID-19, the NF-κB and JAK-STAT pathways are activated and can hyperactivate the IL-6 amplifier response, which results in hyperactivation of NF-κB via STAT3 [5, 40, 41]. The MAPKs are serine-threonine kinases that regulate cellular responses during pathophysiological states. P38 mitogen-activated protein kinases (MAPKs) also increases NF-κB recruitment to chromatin targets [42]. MAPKs pathways are involved with the viral pathogenesis of coronaviruses. Inhibition of the P38 MAPK pathway ameliorated AngII organ damage [43]. The P38 MAPK pathway mediates TNF-α and IL-1β that can activate the NF-κB pathway [40, 44]. Therefore, hyperactivation of 38 MAPK and its cross-talk with NF-κB may result in inflammation, thrombosis, and vasoconstriction, which is associated with severe COVID-19 cases [45]. Stimulator of interferon genes (STING) is an adaptor molecule that links sensing of cytosolic DNA to the production of IFNs and NF-κB. COVID-19 can cause damage to self-DNA and hyperactive STING resulting in IFN-beta release and a cytokine storm following IRF-3 and NF-κB activation [46].
COVID-19 in the Brain
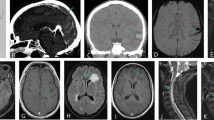
The effects of COVID-19 on the brain can be divided into two main forms, direct infection and secondary mechanisms, such as immune response or respiratory-induced hypoxia [11]. The SARS-CoV-2 RNA was detected in the cerebrospinal fluid of patients with COVID-19 [47]. The direct infection of the CNS by SARS-CoV-2 may occur via two routes, blood circulation into the brain and axonal transport through cranial nerves, such as the olfactory nerve [48]. The OC43 strain of coronavirus was shown to enter the CNS from the nasal cavity to the olfactory nerve. The OC43 strain was found in distant connections of the olfactory bulb, pyriform cortex, and brainstem [49, 50]. The brainstem has neural networks that are necessary for the generation of respiratory rhythms and disruption within the brainstem can result in lethality from respiration failure [51, 52]. SARS-CoV-2 may disrupt the brainstem, which may result in the respiratory impairments in COVID-19 patients. This may have significant implications for the management of COVID-19 patients with respiratory impairment [53]. SARS-CoV-2 can result in white matter demyelination in the brain and spinal cord [54]. In COVID-19-infected patients with neurological symptoms, 44% had abnormal MRI results with a cortical FLAIR signal abnormality present in 37% of the patients [55]. Accumulating evidence suggests that neurological symptoms in patients with COVID-19 are associated with higher disease severity and potential mortality [56].
Emerging data suggests that SARS-CoV-2 has various unexpected effects on neurological function. Reports from Wuhan, China, the suspected center of the COVID outbreak, described the occurrence of neurological symptoms in patients with SARS-CoV-2 [57,58,59]. For example, reports from China included symptoms such as headache, impaired consciousness, anosmia, dysgeusia, stroke, encephalopathy, myelitis, neuritic pain, myalgia, and rhabdomyolysis [60]. As the infection spread to many countries, evidence for the neurological symptoms accumulated across multiple countries. For instance, encephalopathy, agitation with confusion, and corticospinal signs were associated with severe COVID-19 in a case study from France [61]. A report from the UK observed impaired consciousness, acute cerebrovascular events, and muscle disease in COVID-19-infected patients [62]. These neurological impairments were seen in up to 50% of the most severe cases of COVID-19. Other reports include seizures, anosmia, ageusia, encephalitis, and Guillain-Barré Syndrome (GBS) associated with COVID-19 [59, 63, 64]. Also, reports have indicated that increased time in the ICU is associated with long-term cognitive deficits. It is unclear if the neurological symptoms in patients with COVID-19 are simply concurrent or if they are induced by the infection. However, given the accumulating reports of neurological effects in patients with COVID-19, we believe that it is critical to examine the impact of SARS-CoV-2 on the CNS. Similar to other viral infections such as MERS, herpes, varicella and cytomegalovirus that can activate the NF-κB pathway, COVID-19 has a similar neuro-pathogenesis profile.
Coronaviruses and Demyelination
Coronaviruses are known to cause demyelination, and to this end, coronaviruses serve as a rodent model for MS [65]. There have been many reports of GBS in patients with COVID-19 [56, 64, 66]. GBS is an immune-mediated peripheral neuropathy disorder characterized by muscle weakness and paresthesia, and is associated with auto immune system attacks on the myelin sheath of the peripheral nerves. The demyelination observed in patients with COVID-19 may be caused by multiple mechanisms. This includes direct invasion with the SARS-CoV-2 virus, hypoxia, pathological coagulation, and a pathological immune response. Whether COVID-19-related demyelination is a consequence of direct neuronal infection or an indirect route or a combination is not always clear, and a combination of many factors is possible [56].
The NF-κB pathway mediates inflammation in MS. MS is an inflammatory demyelinating disease characterized by repeated demyelination, which can result in disabling outcomes for patients. Studies of patients with MS have found increased levels of NF-κB in total peripheral blood mononuclear cells [67, 68]. The therapeutic mechanisms of many approved MS treatments block the NF-κB pathway immune response in the peripheral nervous system and CNS [69]. Patients with MS treated with dimethyl fumarate, which blocks the NF-κB pathway, had a mild form of COVID-19 [70]. The dimethyl fumarate prescribed to treat MS may have resulted in mild symptoms associated with COVID-19. Therefore, further research should evaluate the efficacy of MS therapeutics to be repurposed for the treatment of COVID-19.
Neuro-COVID-19
The term Neuro-COVID-19 is becoming increasingly used and increasingly accepted in scientific and clinical circles [11]. A Neuro-COVID-19 unit at the University of Brescia Hospital in Italy treated COVID-19 patients for stroke, delirium, seizures, encephalitis, and other neurological complications [71]. The CoroNerve Studies Group was created to study the neurological features of COVID-19 in the UK [72]. Other study groups have formed too. The Neuro-Covid-19 clinic of Northwestern Memorial Hospital in Chicago, IL, found that non-hospitalized COVID-19 “long haulers” also known as people with chronic COVID-19 syndrome had prominent brain fog and fatigue that affected their cognition and quality of life. People with chronic COVID-19 syndrome had clinical manifestations of COVID-19 and did not require hospitalization, but had neurological symptoms persisting at least 6 weeks from symptom onset [73]. Three months after COVID-19 onset and despite recovery from acute infection, neurological symptoms were prevalent including hyposmia/anosmia and cognitive deficits [74]. Cytokine activation appears to progress to a prolonged but less lethal and more incapacitating clinical outcome [75]. One of the most insidious, but least understood effects of chronic COVID-19 syndrome, is chronic fatigue, which is associated with shortness of breath, exhaustion, and general malaise [76]. These symptoms resemble chronic fatigue syndrome or myalgic encephalomyelitis with no known biomarker. COVID-19 does not have a well-defined treatment plan associated with the neurological symptoms observed in patients. Neuro-COVID-19 units should create interdisciplinary teams to implement strategies for treating COVID-19 patients. NF‑κB modulation may provide a scientific and clinical strategy for treating the neurological dysfunction observed in COVID-19 patients.
Potential Therapeutics of NF-κB Modulators in COVID‑19 Infection
The immunomodulation of NF-κB activation along with TNF-α inhibition may result in the reduction of a cytokine storm and decrease the severity of COVID-19. In mice infected with SARS-CoV, inhibition of NF-κB reduced inflammation and increased survival [8]. Cromolyn, an inhibitor of NF-κB, was shown to reduce inflammation and the cytokine storm in patients with COVID-19 [77]. Cromolyn may also reduce the activity of other cell types that produce inflammation [78]. The clinical effects of cromolyn have not been examined in detail for patients with COVID-19. Kaletra is a combination of two antiviral drugs, lopinavir and ritonavir, which are HIV protease inhibitors that have been repurposed for the treatment of COVID-19 and induces suppression of the NF-κB pathway [79, 80]. Treatment with kaletra did not affect the time to clinical improvement in hospitalized patients with severe COVID-19 [81]. Others have suggested that kaletra has the potential to be effective as a clinical treatment of COVID-19 at specific reduced doses such as 400/100 mg daily to improve drug tolerability [82]. Tocilizumab is used for the treatment of rheumatoid arthritis and has been repurposed for the treatment of COVID-19. Tocilizumab was shown to suppress the NF-κB pathway [83]. The survival of patients with severe COVID-19 is higher with tocilizumab treatment than with standard treatment [84]. However, chloroquine which is another repurposed drug for the treatment of COVID-19 is an NF-κB activator that increases expression of pro-inflammatory cytokines [85]. Furthermore, hydroxychloroquine reduced levels of TNF-α, TNF-1β, IgG, and IFN-γ, which in turn reduces the NF-κB pathway [86]. Many studies have shown positive effects of chloroquine and hydroxychloroquine such as decreased time to viral negativity, reduction in death, and shorten time to clinical recovery [87,88,89]. However, other clinical trials have not provided consistent evidence to support the therapeutic effects of chloroquine and hydroxychloroquine in the treatment of COVID-19 [90]. Factors such as the dose, age of patients, and the severity of COVID-19 should all be taken into account when treating patients with chloroquine and hydroxychloroquine. Interferon-β 1a has been repurposed for the treatment of COVID-19 and does not affect the NF-κB pathway [91]. The time to clinical improvement was decreased in patients receiving Interferon-β 1a treatment as compared to standard treatment [92]. Remdesivir and favipiravir inhibit viral replication and were repurposed for the treatment of COVID-19 and there is not any data on whether remdesivir and/or favipiravir affects the NF-κB pathway. Intravenous injection of remdesivir improved the clinical outcome of patients with COVID-19 [93]. Another study showed that COVID-19 patients treated with remdesivir had clinical improvement in 36 (68%) out of 53 patients [94]. Favipiravir treatment in patients with COVID-19 was associated with improved clinical outcomes and decreased time of viral clearance [95].
Dexamethasone is a glucocorticoid that inhibits the NF-κB pathway via increased expression of IκB [35, 96]. The increased expression of IκB inhibits NF-κB translocation into the nucleus, and as a result, NF-κB remains in the cytoplasm. Glucocorticoids are also immunomodulators that reduce IL-6 production and activity, which results in decreased cytokine feedback on the NF-κB pathway [97]. In patients with COVID-19, dexamethasone reduced the incidence of death compared to patients receiving invasive mechanical ventilation, and patients receiving oxygen without invasive mechanical ventilation [98]. Dexamethasone increased gene transcription of anti-inflammatory cytokines and decreased pro-inflammatory mediators [99]. Dexamethasone via its inhibition of the NF-κB pathway is an effective therapeutic in severe-critically ill COVID-19 patients.
Rescue therapies including inhaled nitric oxide in severe-critically ill COVID-19 patients resulted in different effects on systemic and cerebral oxygenation. The use of inhaled nitric oxide increased systemic oxygenation and cerebral oxygenation [100]. There is accumulating evidence that inhaled nitric oxide decreases the inflammatory cell-mediated lung injury by inhibiting neutrophil activation and subsequent pro-inflammatory cytokines [26]. Nitric oxide inhibits the NF-κB pathway by terminating the transcription process [101]. Given these results, the choice of rescue therapies that are adopted should take into account the lung and brain of patients. There are several ongoing clinical trials in progress measuring the effects of inhaled nitric oxide on clinical outcomes in patients with COVID-19 [102]. These clinical trials should provide information about the effectivity of inhaled nitric oxide for the treatment of COVID-19.
Camostat mesylate is a protease inhibitor used to treat pancreatitis and inflammatory diseases. Camostat mesylate inhibits TMPRSS2 to prevent SARS-CoV-2 fusion on cell surface and suppress SARS-CoV-2 replication in hamsters and SARS-CoV in mice [103]. There is one ongoing clinical trial examining the effect of camostat mesylate on the outcomes of patients with COVID-19 [104]. Once results of this clinical trial are available, it may inform clinicians about the effectiveness of camostat mesylate. Camostat mesylate could be used to suppress the NF-κB signaling pathway [26]. The NF-κB pathway is a redox-sensitive pathway, which becomes activated by oxidative stress [105]. Blocking the NF-κB pathway with antioxidants including vitamin A, vitamin C, glutathione, vitamin E, and zinc could have both prophylactic protection and prevent the progression of illness [106].
Vitamin D exerts neuroprotective effects via the NF-κB pathway and can prevent memory impairment [107]. The neuroprotective action of vitamin D is also associated with modulation of neurotrophins, which are important for survival, differentiation, and maintenance of nerve cells in both the peripheral nervous system (PNS) and CNS [108]. Vitamin D has immunomodulating properties via its activity as an immunosuppressant to protect neurons [109]. Vitamin D may prevent neurological symptoms associated with COVID-19 including loss of taste and smell, and headaches by stimulating the expression of neurotrophins such as nerve growth factor (NGF) [110]. The effect of COVID-19 was of a higher severity in people with vitamin D deficiency, and an increased risk for mortality in patients with vitamin D deficiency [111]. Therefore, COVID-19 patients should have vitamin D levels monitored and regulated to meet the targeted levels in medical guidelines.
N-acetylcysteine (NAC) is used to treat critically ill septic patients, and more recently for COVID-19 patients [112]. NAC is a precursor of the antioxidant glutathione, and boosts the immune system, suppresses viral replication, and reduces inflammation. NAC is an NF-κB inhibitor and works by downregulating the phosphorylation of IκB; it has an inhibitory effect against TNF-α mediated activation of the NF-κB pathway [26, 113]. As previously mentioned, NAC is a precursor of glutathione and the imbalance of the glutathione redox system is shown to be involved with many neurological disorders. Glutathione can regulate glutamate receptors including the N-methyl-D-aspartate (NMDA) receptor, which can contribute to glutamate excitotoxicity. Interactions between glutamate and glutathione can result in neuronal dysfunction and involve the NF-κB pathway [114]. Treatment with NAC was shown to have clinical improvement in critically ill COVID-19 patients [115].
Conclusions
The NF-κB pathway may play an important role in the progression of COVID-19 and the severe form of the illness. NF-κB inhibition may be a possible mechanism of action that the current drugs used to treat COVID-19 exert therapeutic effects. Although there is accumulating evidence that COVID-19 affects the brain, we are only beginning to understand the mechanisms by, and the extent to which, COVID-19 affects overall neurological functions. Neuro-COVID-19 is increasingly becoming recognized as a useful clinical/scientific construct in the evolving discussion about the development of approaches to the diagnosis, management, and treatment of neurological and neurocognitive effects of COVID-19. Given the large number of COVID-19 cases with neurological symptoms and the complexity of such cases, an effort involving the development of an international registry/database that can be shared and utilized by multispecialty collaborative teams of health care professionals and scientists all over the world should be launched [60]. The heterogeneity of neurological symptoms may stem from the various routes that SARS-CoV-2 can enter and affect the brain. The time scale of neurological symptoms should be further investigated classifying early neurological symptoms when the virus is detectable in the patient’s body compared to people with chronic COVID-19 syndrome who do not have virus detectable in their system, but experience neurological symptoms related to COVID-19 infection. Novel or repurposed therapeutics should evaluate both the acute phase of COVID-19 and the chronic COVID-19 syndrome. NF-κB inhibition in the context of neuro-COVID-19 should be further examined to develop novel strategies for targeting the NF-κB pathway in the brain of patients with COVID-19.
References
Wu F et al (2020) A new coronavirus associated with human respiratory disease in China. Nature 579(7798):265–269
Zhou P et al (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579(7798):270–273
Williamson EJ et al (2020) Factors associated with COVID-19-related death using OpenSAFELY. Nature 584(7821):430–436
Gao YM et al (2020) Cytokine storm syndrome in coronavirus disease 2019: A narrative review. J Intern Med
Hirano T, Murakami M (2020) COVID-19: a new virus, but a familiar receptor and cytokine release syndrome. Immunity 52(5):731–733
Adlimoghaddam A et al (2021) Nilotinib improves bioenergetic profiling in brain astroglia in the 3xTg mouse model of Alzheimer’s disease. Aging Dis 12(2):441–465
Adlimoghaddam A, Albensi BC (2021) The nuclear factor kappa B (NF-kappaB) signaling pathway is involved in ammonia-induced mitochondrial dysfunction. Mitochondrion 57:63–75
DeDiego ML et al (2014) Inhibition of NF-kappaB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J Virol 88(2):913–924
Oeckinghaus A, Ghosh S (2009) The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 1(4):a000034
Liao QJ et al (2005) Activation of NF-kappaB by the full-length nucleocapsid protein of the SARS coronavirus. Acta Biochim Biophys Sin (Shanghai) 37(9):607–612
Bougakov D, Podell K, and Goldberg E (2020) Multiple neuroinvasive pathways in COVID-19. Mol Neurobiol
Gupta A et al (2020) Extrapulmonary manifestations of COVID-19. Nat Med 26(7):1017–1032
Shang J et al (2020) Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci U S A 117(21):11727–11734
Shang J et al (2020) Structural basis of receptor recognition by SARS-CoV-2. Nature 581(7807):221–224
Yan R et al (2020) Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367(6485):1444–1448
Butowt R, Bilinska K (2020) SARS-CoV-2: olfaction, brain infection, and the urgent need for clinical samples allowing earlier virus detection. ACS Chem Neurosci 11(9):1200–1203
Kabbani N, Olds JL (2020) Does COVID19 infect the brain? If so, smokers might be at a higher risk. Mol Pharmacol 97(5):351–353
Xia H, Lazartigues E (2008) Angiotensin-converting enzyme 2 in the brain: properties and future directions. J Neurochem 107(6):1482–1494
Baig AM et al (2020) Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci 11(7):995–998
Xie W et al (2020) OIP5-AS1 attenuates microangiopathy in diabetic mouse by regulating miR-200b/ACE2. World Neurosurg 139:e52–e60
Wang L et al (2016) Increasing brain angiotensin converting enzyme 2 activity decreases anxiety-like behavior in male mice by activating central Mas receptors. Neuropharmacology 105:114–123
Lukiw WJ, Pogue A, and Hill JM (2020) SARS-CoV-2 infectivity and neurological targets in the brain. Cell Mol Neurobiol
Ulrich H, Pillat MM (2020) CD147 as a target for COVID-19 treatment: suggested effects of azithromycin and stem cell engagement. Stem Cell Rev Rep 16(3):434–440
Qiao J et al (2020) The expression of SARS-CoV-2 receptor ACE2 and CD147, and protease TMPRSS2 in human and mouse brain cells and mouse brain tissues. Biochem Biophys Res Commun 533(4):867–871
Zhang Q, Lenardo MJ, Baltimore D (2017) 30 years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell 168(1–2):37–57
Hariharan A et al (2020) The role and therapeutic potential of NF-kappa-B pathway in severe COVID-19 patients. Inflammopharmacology
Li Q, Verma IM (2002) NF-kappaB regulation in the immune system. Nat Rev Immunol 2(10):725–734
Liu T et al (2017) NF-kappaB signaling in inflammation. Signal Transduct Target Ther 2
Terai K, Matsuo A, McGeer PL (1996) Enhancement of immunoreactivity for NF-kappa B in the hippocampal formation and cerebral cortex of Alzheimer’s disease. Brain Res 735(1):159–168
Lawrence T (2009) The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 1(6):a001651
Day CW et al (2009) A new mouse-adapted strain of SARS-CoV as a lethal model for evaluating antiviral agents in vitro and in vivo. Virology 395(2):210–222
Dosch SF, Mahajan SD, Collins AR (2009) SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-kappaB pathway in human monocyte macrophages in vitro. Virus Res 142(1–2):19–27
Huang J et al (2020) SARS-CoV-2 infection of pluripotent stem cell-derived human lung alveolar type 2 cells elicits a rapid epithelial-intrinsic inflammatory response. Cell Stem Cell 27(6):962-973 e7
Carcaterra M, Caruso C (2021) Alveolar epithelial cell type II as main target of SARS-CoV-2 virus and COVID-19 development via NF-Kb pathway deregulation: a physio-pathological theory. Med Hypotheses 146:110412
D’Acquisto F, May MJ, Ghosh S (2002) Inhibition of nuclear factor kappa B (NF-B): an emerging theme in anti-inflammatory therapies. Mol Interv 2(1):22–35
Dorrington MG, Fraser IDC (2019) NF-kappaB signaling in macrophages: dynamics, crosstalk, and signal integration. Front Immunol 10:705
Birra D et al (2020) COVID 19: a clue from innate immunity. Immunol Res 68(3):161–168
Ingraham NE et al (2020) Understanding the renin-angiotensin-aldosterone-SARS-CoV axis: a comprehensive review. Eur Respir J 56(1)
Devaux CA, Rolain JM, Raoult D (2020) ACE2 receptor polymorphism: susceptibility to SARS-CoV-2, hypertension, multi-organ failure, and COVID-19 disease outcome. J Microbiol Immunol Infect 53(3):425–435
Battagello DS et al (2020) Unpuzzling COVID-19: tissue-related signaling pathways associated with SARS-CoV-2 infection and transmission. Clin Sci (Lond) 134(16):2137–2160
Brasier AR (2010) The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc Res 86(2):211–218
Saccani S, Pantano S, Natoli G (2002) p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat Immunol 3(1):69–75
Park JK et al (2007) p38 mitogen-activated protein kinase inhibition ameliorates angiotensin II-induced target organ damage. Hypertension 49(3):481–489
Sun SC (2017) The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol 17(9):545–558
Grimes JM, Grimes KV (2020) p38 MAPK inhibition: a promising therapeutic approach for COVID-19. J Mol Cell Cardiol 144:63–65
Berthelot JM, Liote F (2020) COVID-19 as a STING disorder with delayed over-secretion of interferon-beta. EBioMedicine 56:102801
Wang HY et al (2020) Potential neurological symptoms of COVID-19. Ther Adv Neurol Disord 13:1756286420917830
Politi LS, Salsano E, Grimaldi M (2020) Magnetic resonance imaging alteration of the brain in a patient with coronavirus disease 2019 (COVID-19) and Anosmia. JAMA Neurol 77(8):1028–1029
De Santis G (2020) SARS-CoV-2: A new virus but a familiar inflammation brain pattern. Brain Behav Immun 87:95–96
Dube M et al (2018) Axonal transport enables neuron-to-neuron propagation of human coronavirus OC43. J Virol 92(17)
Smith JC et al (1991) Pre-Botzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science 254(5032):726–729
Burgold T et al (2012) The H3K27 demethylase JMJD3 is required for maintenance of the embryonic respiratory neuronal network, neonatal breathing, and survival. Cell Rep 2(5):1244–1258
Hsieh YH et al (2020) Brainstem inflammation modulates the ventilatory pattern and its variability after acute lung injury in rodents. J Physiol 598(13):2791–2811
Zanin L et al (2020) SARS-CoV-2 can induce brain and spine demyelinating lesions. Acta Neurochir (Wien) 162(7):1491–1494
Kandemirli SG et al (2020) Brain MRI findings in patients in the intensive care Unit with COVID-19 infection. Radiology 297(1):E232–E235
Whittaker A, Anson M, Harky A (2020) Neurological manifestations of COVID-19: a systematic review and current update. Acta Neurol Scand 142(1):14–22
Asadi-Pooya AA, Simani L (2020) Central nervous system manifestations of COVID-19: a systematic review. J Neurol Sci 413:116832
Pleasure SJ, Green AJ, Josephson SA (2020) The spectrum of neurologic disease in the severe acute respiratory syndrome coronavirus 2 pandemic infection: neurologists move to the frontlines. JAMA Neurol 77(6):679–680
Mao L et al (2020) Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol 77(6):683–690
Roman GC et al (2020) The neurology of COVID-19 revisited: a proposal from the Environmental Neurology Specialty Group of the World Federation of Neurology to implement international neurological registries. J Neurol Sci 414:116884
Helms J et al (2020) Neurologic features in severe SARS-CoV-2 infection. N Engl J Med 382(23):2268–2270
Roberts CM et al (2020) COVID-19: a complex multisystem disorder. Br J Anaesth 125(3):238–242
Poyiadji N et al (2020) COVID-19-associated acute hemorrhagic necrotizing encephalopathy: imaging features. Radiology 296(2):E119–E120
Zhao H et al (2020) Guillain-Barre syndrome associated with SARS-CoV-2 infection: causality or coincidence? Lancet Neurol 19(5):383–384
Houtman JJ, Fleming JO (1996) Dissociation of demyelination and viral clearance in congenitally immunodeficient mice infected with murine coronavirus JHM. J Neurovirol 2(2):101–110
Alberti P et al (2020) Guillain-Barre syndrome related to COVID-19 infection. Neurol Neuroimmunol Neuroinflamm 7(4)
Eggert M et al (2008) Changes in the activation level of NF-kappa B in lymphocytes of MS patients during glucocorticoid pulse therapy. J Neurol Sci 264(1–2):145–150
Yan J et al (2018) Increased constitutive activation of NF-kappaB p65 (RelA) in peripheral blood cells of patients with progressive multiple sclerosis. J Neuroimmunol 320:111–116
Zhou Y et al (2020) Nuclear factor kappaB (NF-kappaB)-mediated inflammation in multiple sclerosis. Front Immunol 11:391
Capone F et al (2021) COVID-19 in multiple sclerosis patients treated with dimethyl fumarate. J Neurol
Talan J (2020) COVID-19: neurologists in Italy to colleagues in US: look for poorly-defined neurologic conditions in patients with the coronavirus. Neurology Today
Varatharaj A et al (2020) Neurological and neuropsychiatric complications of COVID-19 in 153 patients: a UK-wide surveillance study. Lancet Psychiatry 7(10):875–882
Graham EL et al (2021) Persistent neurologic symptoms and cognitive dysfunction in non-hospitalized Covid-19 “long haulers”. Ann Clin Transl Neurol
Rass V et al (2021) Neurological outcome and quality of life three months after COVID-19: a prospective observational cohort study. Eur J Neurol
Baig AM (2020) Deleterious outcomes in long-hauler COVID-19: the effects of SARS-CoV-2 on the CNS in chronic COVID syndrome. ACS Chem Neurosci 11(24):4017–4020
Marshall M (2020) The lasting misery of coronavirus long-haulers. Nature 585(7825):339–341
Mahase E (2020) Covid-19: what treatments are being investigated? BMJ 368:m1252
Yousefi H et al (2021) Repurposing existing drugs for the treatment of COVID-19/SARS-CoV-2 infection: a review describing drug mechanisms of action. Biochem Pharmacol 183:114296
Kariya R et al (2014) HIV protease inhibitor Lopinavir induces apoptosis of primary effusion lymphoma cells via suppression of NF-kappaB pathway. Cancer Lett 342(1):52–59
Dewan MZ et al (2009) An HIV protease inhibitor, ritonavir targets the nuclear factor-kappaB and inhibits the tumor growth and infiltration of EBV-positive lymphoblastoid B cells. Int J Cancer 124(3):622–629
Cao B et al (2020) A trial of lopinavir-ritonavir in adults hospitalized with severe Covid-19. N Engl J Med 382(19):1787–1799
Baldelli S et al (2020) Lopinavir/ritonavir in COVID-19 patients: maybe yes, but at what dose? J Antimicrob Chemother 75(9):2704–2706
Alraouji NN et al (2020) Tocilizumab potentiates cisplatin cytotoxicity and targets cancer stem cells in triple-negative breast cancer. Mol Carcinog 59(9):1041–1051
Ruiz-Antoran B et al (2021) Combination of tocilizumab and steroids to improve mortality in patients with severe COVID-19 infection: a Spanish, multicenter, cohort study. Infect Dis Ther 10(1):347–362
Park J et al (2003) Chloroquine induces activation of nuclear factor-kappaB and subsequent expression of pro-inflammatory cytokines by human astroglial cells. J Neurochem 84(6):1266–1274
Liang N et al (2018) Immunosuppressive effects of hydroxychloroquine and artemisinin combination therapy via the nuclear factor-kappaB signaling pathway in lupus nephritis mice. Exp Ther Med 15(3):2436–2442
Chen L, Z Z-Y, Fu J-G, Feng Z-P, Zhang S-Z, Han Q-Y (2020) Efficacy and safety of chloroquine or hydroxychloroquine in moderate type of COVID-19: a prospective open-label randomized controlled study
Yu B et al (2020) Low dose of hydroxychloroquine reduces fatality of critically ill patients with COVID-19. Sci China Life Sci 63(10):1515–1521
Chen Z, Hu J, Zhang Z, Jiang S, Han S, Yan D (2020) Efficacy of hydroxychloroquine in patients with COVID-19: results of a randomized clinical trial
Chen Y et al (2021) Hydroxychloroquine/chloroquine as therapeutics for COVID-19: truth under the mystery. Int J Biol Sci 17(6):1538–1546
Asadikaram G et al (2016) Interferon-beta 1a modulates expression of RAGE but not S100A12 and nuclear factor-kappaB in multiple sclerosis patients. NeuroImmunoModulation 23(5–6):345–351
Alavi Darazam I et al (2021) Role of interferon therapy in severe COVID-19: the COVIFERON randomized controlled trial. Sci Rep 11(1):8059
Holshue ML et al (2020) First case of 2019 novel coronavirus in the United States. N Engl J Med 382(10):929–936
Grein J et al (2020) Compassionate use of remdesivir for patients with severe Covid-19. N Engl J Med 382(24):2327–2336
Cai Q et al (2020) Experimental treatment with favipiravir for COVID-19: an open-label control study. Engineering (Beijing) 6(10):1192–1198
Ye Z et al (2020) Efficacy and safety of corticosteroids in COVID-19 based on evidence for COVID-19, other coronavirus infections, influenza, community-acquired pneumonia and acute respiratory distress syndrome: a systematic review and meta-analysis. CMAJ 192(27):E756–E767
Auphan N et al (1995) Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science 270(5234):286–290
Group RC et al (2021) Dexamethasone in hospitalized patients with Covid-19. N Engl J Med 384(8):693–704
Patel SK et al (2020) Dexamethasone: a boon for critically ill COVID-19 patients? Travel Med Infect Dis 37:101844
Robba C et al (2021) Early effects of ventilatory rescue therapies on systemic and cerebral oxygenation in mechanically ventilated COVID-19 patients with acute respiratory distress syndrome: a prospective observational study. Crit Care 25(1):111
Matthews JR et al (1996) Inhibition of NF-kappaB DNA binding by nitric oxide. Nucleic Acids Res 24(12):2236–2242
Ito K et al (2020) Major ongoing clinical trials for COVID-19 treatment and studies currently being conducted or scheduled in Japan. Glob Health Med 2(2):96–101
Zhao H et al (2021) Cross-linking peptide and repurposed drugs inhibit both entry pathways of SARS-CoV-2. Nat Commun 12(1):1517
Oroojalian F et al (2020) Novel insights into the treatment of SARS-CoV-2 infection: an overview of current clinical trials. Int J Biol Macromol 165(Pt A):18–43
Das KC (2001) c-Jun NH2-terminal kinase-mediated redox-dependent degradation of IkappaB: role of thioredoxin in NF-kappaB activation. J Biol Chem 276(7):4662–4670
Bauer SR et al (2020) What is the role of supplementation with ascorbic acid, zinc, vitamin D, or N-acetylcysteine for prevention or treatment of COVID-19? Cleve Clin J Med
Ali A et al (2021) Vitamin D exerts neuroprotection via SIRT1/nrf-2/ NF-kB signaling pathways against D-galactose-induced memory impairment in adult mice. Neurochem Int 142:104893
Di Somma C et al (2017) Vitamin D and neurological diseases: an endocrine view. Int J Mol Sci 18(11)
Koduah P, Paul F, Dorr JM (2017) Vitamin D in the prevention, prediction and treatment of neurodegenerative and neuroinflammatory diseases. EPMA J 8(4):313–325
Xu Y et al (2020) The importance of vitamin d metabolism as a potential prophylactic, immunoregulatory and neuroprotective treatment for COVID-19. J Transl Med 18(1):322
Kazemi A et al (2021) Association of vitamin D status with SARS-CoV-2 infection or COVID-19 severity: a systematic review and meta-analysis. Adv Nutr
Shi Z, Puyo CA (2020) N-Acetylcysteine to combat COVID-19: an evidence review. Ther Clin Risk Manag 16:1047–1055
Oka S et al (2000) N-acetylcysteine suppresses TNF-induced NF-kappaB activation through inhibition of IkappaB kinases. FEBS Lett 472(2–3):196–202
Bjorklund G et al (2021) The impact of glutathione metabolism in autism spectrum disorder. Pharmacol Res 166:105437
Assimakopoulos SF, Marangos M (2020) N-acetyl-cysteine may prevent COVID-19-associated cytokine storm and acute respiratory distress syndrome. Med Hypotheses 140:109778
Funding
We gratefully acknowledge the Research Manitoba Fellowship to Dr. Adlimoghaddam (Grant No: 1913), and support from the St. Boniface Hospital Research Foundation (Grant Nos.1406–3216 and 1410–3216), the Canadian Institute of Health Research (CIHR; Grant No. PJT-162144), the Alzheimer’s Society of Manitoba, the Honourable Douglas and Patricia Everett, Royal Canadian Properties Limited Endowment Fund (Grant No. 1403–3131) to Dr. Albensi.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethics Approval
Not Applicable.
Consent to Participate
Not Applicable.
Consent for Publication
All authors gave their consent for publication.
Conflict of Interest
Not applicable.
Disclaimer
Dr. Albensi previously held the Manitoba Dementia Research Chair and currently holds the Honorable Douglas and Patricia Everett, and Royal Canadian Properties Limited Endowment Fund Chair. The research was funded by Canadian Institutes of Health Research (CIHR), grant No. PJT-162144, and the St. Boniface Hospital Research Foundation, grant Nos. 1406–3216 and 1410–3216. Dr. Albensi is a Research Affiliate of the Centre on Aging at the University of Manitoba.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Davies, D.A., Adlimoghaddam, A. & Albensi, B.C. The Effect of COVID-19 on NF-κB and Neurological Manifestations of Disease. Mol Neurobiol 58, 4178–4187 (2021). https://doi.org/10.1007/s12035-021-02438-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02438-2