
Abstract
The enhancer of zeste homolog 2 (EZH2) is encoded by the Enhancer of zeste 2 polycomb repressive complex 2 subunit gene. EZH2 is involved in the cell cycle, DNA damage repair, cell differentiation, autophagy, apoptosis, and immunological modulation. The main function of EZH2 is to catalyze the methylation of H3 histone of H3K27Me3, which inhibits the transcription of target genes, such as tumor suppressor genes. EZH2 also forms complexes with transcriptions factors or directly binds to the promoters of target genes, leading to regulate gene transcriptions. EZH2 has been as a prominent target for cancer therapy and a growing number of potential targeting medicines have been developed. This review summarized the mechanisms that EZH2 regulates gene transcription and the interactions between EZH2 and important intracellular signaling molecules (Wnt, Notch, MEK, Akt) and as well the clinical applications of EZH2-targeted drugs.
Similar content being viewed by others


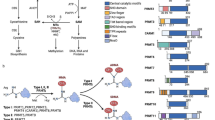
Function and structure of EZH2
Polycomb group (PcG) protein complexes inhibit transcription initiation of target gene by methylating histones on chromatin. PcG protein forms a stable multi-protein complex with some protein factors and acts collectively to keep their target genes in a transcriptional repression state. PRC1 (Polycomb repressive complex 1) and PRC2 (Polycomb repressive complex 2) are the two primary core protein complexes in mammalian cells [1]. Embryonic Ectoderm Development (EED), Zeste 12 inhibitor (Suppressor of Zeste 12, SUZ12), and EZH1 or EZH2 are the components of PRC2. The catalytic subunit of PRC2, Enhancer of zeste homolog 2 (EZH2), plays a crucial role in the H3 methylation pathway [2].
The EZH2 gene is located on chromosome 7q35 and comprises 20 exons that can code for 746 amino acids. There are EED interaction Domain (EID), Domain 1, Domain 2, Cysteine-rich domain (CXC Domain), and Enhancer domain of zeste and trithorax (SET Domain) in the EZH2 protein [3]. The SET domain can offer the active site of methyltransferase. Due to the solo EZH2 subunit lacks histone methyltransferase activity, it must be combined with the EED/ESC and SUZ12 domains to function [4].
Histone methylation modifications can specifically activate or inhibit gene transcriptional activity, which is linked to many human diseases. Histone methyltransferase EZH2 of the PRC2 complex promotes trimethylation of H3 histone 27 lysine, changing the structure of chromatin and limiting gene transcription.
EZH2 has the ability to directly methylate a variety of target molecules, including GATA4, STAT3, β-catenin, and the lysines at positions 510, 514, and 515 of PRC2 [5,6,7,8,9,10]. EZH2 may also bind to particular molecules directly, creating a ternary complex with Rel A and Rel B in the NF-κB component, EZH2 interacts to TCF, β-catenin, and ER to activate the c-Myc and cyclin D1 genes, which are located downstream [11, 12]. Another mode of action of EZH2 is to bind to the promoter region of target genes and affect gene transcription, commonly found in c-Myc and Notch1 [13, 14] (Fig. 1).

Domain composition and function of EZH2. Hierarchical analysis expression of EZH2 protein included five individual conservative structure area: EID structure area, Domain I, Domain II, CXC structure area, and SET Domain
EZH2-related diseases and signal pathways
EZH2/Wnt/β-Catenin signaling pathway
By binding to some long non-coding RNAs, such as NEAT1, EZH2 could control the enrichment of H3K27Me3 in various gene promoters and activate or inhibit the Wnt/β-catenin signaling pathway [15,16,17]. Additionally, EZH2 could assemble into a complex with β-catenin directly, control the expression of downstream FTO genes, and then increase the expression of c-Myc to promote the growth of tumor cells [18] (Fig. 2).

EZH2/Wnt/β-Catenin signal transduction and related diseases. Interrelationships between EZH2 and Wnt/β-Catenin and the resulting illnesses
MEK/ERK/EZH2 signal pathway
Elk-1 can bind to the EZH2 promoter after being phosphorylated, which increases EZH2 expression. The expression of EZH2 is decreased when the MEK-ERK-Elk-1 signaling pathway is compromised [19]. E2F4 can bind to EZH2 and activate MAPK signaling in AML (acute myeloid leukemia) patients. Additionally, EZH2 can block the MAPK signaling pathway’s ability to inhibit E2F4 [20]. Patients with diffuse large B-cell lymphoma develop resistance to EZH2 inhibitors as a result of MAPK signaling reduction of TNFSF10 and BAD expression through a FOXO3-dependent mechanism [21]. In non-small cell lung cancer cells with the KRASG12C mutation, MEK1 controls EZH2 expression [22]. EZH2 inhibits MEK-ERK1/2 signaling in vascular smooth muscle cells (VSMC), prevents aortic dissection (AD), inhibits ATG5 and ATG7 via H3K27Me3, inhibits autophagic cell death (ACD), and regulates the MEK-ERK1/2 signaling pathway [23]. CXXC4 is directly controlled by EZH2; it could inhibit MAPK signaling by severing the MEK1/2-ERK 1/2 connection [24] (Fig. 3).

MEK/ERK/EZH2 signal transduction and related diseases. Interrelationships between EZH2 and MEK/ERK and the resulting illnesses
EZH2/Notch signal pathway
Breast cancer and melanoma tumor cells proliferate more readily when EZH2 levels are elevated, which is related to the interaction of the histone methyltransferase NSD3 with EZH2 and RNA polymerase II to enhance H3K36me2/3 [25, 26]. PVT1, a long-chain non-coding RNA, can interact with EZH2 in non-small cell lung cancer (NSCLC) to mediate miR-497 promoter methylation and prevent miR-497 and YAP1 upregulation [27]. Notch1, Notch2, and Jagged 1 are inhibited by miR-34a, and HOTAIR, a long-chain non-coding RNA, could block miR-34a through H3K27me3 by EZH2 [28, 29]. Mutation or translocation of Notch1 or Notch2 in breast cancer can lead to the upregulation of PRC2/EZH2, promote the binding of EZH2 to the HES-1 transcription factor in the PTEN promoter region, and inhibit PTEN expression [30]. EZH2 inhibits Notch function by blocking the Notch ligand DLL4 and under the induction of TNF and promoting the methylation of the Notch1 promoter region [31, 32] (Fig. 4).

EZH2/Notch signal transduction and related diseases. Interrelationships between EZH2 and Notch and the resulting illnesses
EZH2/PI3K/Akt signal pathway
When JNK/STAT3 is activated, it results in the upregulation of IKBEK and miR-21, increases the phosphorylation of STAT3 and Akt, decreases the expression of Spry2, phosphorylates the 21st serine of EZH2 through p-Akt, inhibits the expression of H3K27Me3, PI3K, Id3, and increases the expression of Id2, Prdm1, and Eomes [33,34,35], and EZH2 can also interact with STAT3 and lead to increased STAT3 activity [36]. A significant amount of E2F1 is released by Akt, which then hyperphosphorylates Rb, activates cyclins and cyclin-dependent kinases (CDKs), and binds to the EZH2 promoter to increase EZH2 transcription [37]. When IGF1 binds to its receptor IGF1R, and VCAM-1 binds to VLA-4, it will activate the PI3K/Akt signaling pathway, promote the phosphorylation of EZH2, and lead to the increase of HIF-1, IGF1, and Bcl2, resulting in drug resistance [38]. By attaching to the EZH2 promoter, the transcription factor E2F7 increases H3K27Me3, decreases PTEN expression, and activates the Akt/mTOR signaling pathway [39, 40]. By activating TNFSF13B, EZH2 increases Akt phosphorylation, but when the level of the demethylase KDM2B is lowered, EZH2 inhibits the phosphorylation of PI3K and Akt [41, 42]. EZH2 has the ability to control the amount of VEGF-A through the PI3K/Akt pathway [43]. EZH2 control on PI3K can be accomplished by PIK3IP; however, EZH2 regulation requires ARID1A. Although ARID1A and EZH2 have a mutual inhibitory effect, EZH2 has the ability to inhibit PIK3IP1 and activate the PI3K/Akt pathway, while ARID1A has the ability to stimulate PIK3IP1 [44]. Cell Cycle-Related Kinase (CCRK) in immortalized human hepatocytes increases the amount of EZH2, promotes H3K27me3, and speeds tumor growth. At the same time, CCRK’s phosphorylation of Akt and EZH2 may stimulate CCRK transcription, forming a self-reinforcing regulatory circuit [45]. PTEN transcription will be significantly increased by MYC gene activation, while EZH2 phosphorylation will be decreased. Additionally, EZH2 can direct MYC to the IGF1R promoter region, where it can then activate IGF1R and Akt to aid in the development of tumors [46, 47]. The oncoprotein binding domain of Yin Yang 1 (YY1) could attract EZH2 in breast cancer, causing PTEN and PTENP1 to be down-regulated. The combination of YY1 and EZH2 inhibited the transcription of PTEN and PTENP1, resulting in increased phosphorylation of S473 and T308 of Akt and increased Akt activation [48]. FRA1/C-Jun can be activated by the ERK/AKT signaling pathway, which causes AP-1 to occupy the EZH2 promoter and cause primary EZH2 expression. Finally, trimethylates H3K27 to repress the ITG2 promoter and inhibit transcription [49] (Fig. 5).
Long non-coding RNA can also play a role in the EZH2 and Akt regulatory mechanism. Urothelial cancer associated 1 (UCA1) interacted directly with EZH2, increasing EZH2 expression and activating the Akt/GSK-3/cyclin D1 pathway. EZH2 may also interact directly with the cyclin D1 promoter, promoting cyclin D1 expression. Furthermore, Akt phosphorylation might indirectly affect EZH2 expression, generating a positive feedback loop with EZH2. Long-chain non-coding RNA PART1 (Prostate Androgen Regulated Transcript 1) inhibited downstream PI3K / Akt by boosting the production of PLZF (Promyelocytic Leukemia Zinc Finger) and subsequently recruiting EZH2 [50]. lncRNA-SNHG1 could bind to EZH2 and activated the PI3K/Akt/mTOR and Wnt/β-Catenin signaling pathways [51]. LINC01559 and CASC11 can bind EZH2 to the PTEN promoter, increase PTEN promoter methylation, inhibit PTEN, and activate PI3K/Akt [52, 53]. LINC00665 and lncRNA-AFAP1-AS1 can interact with EZH2 to activate the Akt signaling pathway, resulting in drug resistance in patients [54, 55] (Fig. 5).

EZH2 /Akt signal transduction and related diseases. Interrelationships between EZH2 and AKT and the resulting illnesses
EZH2 other related signal pathway
CHK1 is involved in the regulation of cell apoptosis. Inhibiting CHK1 has been shown to greatly increase the production of Caspase3 and Caspase9, as well as promote cell apoptosis [56]. By interacting with the promoter of CHK1, EZH2 could enhance the expression of CHK1, blocking the downstream apoptotic pathway. High expression of EZH2 and CHK1 has been linked to poor prognosis and treatment resistance in ovarian cancer patients [57], implying that EZH2 is also linked to other apoptotic pathways. EZH2 could enhance cell death and increase intestinal epithelial cell permeability by inactivating JAK2/STAT signaling through H3K27Me3 in inflammatory bowel illness [58]. Not only does EZH2 play a crucial part in cell death, but it is also a significant target in other disorders. The epithelial–mesenchymal transition (EMT) has been identified as a significant driver of tumor cell invasion and migration in cancer, and EZH2 is a crucial driver of EMT. The upstream pathway of EMT is driven by EZH2, according to studies. It could be linked to the signal axis TGF-MTA1-SOX4 [59]. The role of EZH2 in the progression of Myelodysplastic Syndromes (MDS) to Acute Myeloid Leukemia (AML) cannot be overlooked. In MDS, EZH2 is a downstream gene of the pRB-E2F pathway that plays a role in oncogene activation. The activation of the pRB-E2F pathway in high-risk or extremely high-risk MDS cells results in the overexpression of EZH2, which suppresses p53 and p15 and finally leads to AML. p53 can counteract the action of pRB-E2F in normal circumstances. The balance between p53 and pRB-E2F is essential for normal cell growth, but HO-1 disrupts this balance by over-activating the pRB-E2F pathway, which increases the inhibition of p53 and p15 from EZH2, allowing tumor cells to evade cell cycle-level checks and making chemotherapy drugs difficult to eliminate tumor cells [60]. Excessive stimulation of the CDK4/6-EZH2 pathway in psoriasis causes STAT3, IB, and the inflammatory response in the body to be triggered. Inhibitors targeting EZH2 could be a new sort of psoriasis therapy method for this pathway [61]. MYC boosted EZH2 expression and accelerated tumor growth in lymphomas via blocking miR-26a [62]. By suppressing miR-494, EZH2 can also boost MYC expression in a positive feedback loop. MYC can also form a complex with HDAC3 and EZH2 to block miR-29 expression, resulting in increased expression of downstream CDK6 and IGF1R and lymphoma [63]. Furthermore, through its interaction with DNA methyltransferase 1 (DNA methyltransferases, DNMT1), EZH2 can not only regulate histone methylation but also participate in DNA methylation and affect the transcription of downstream targets [64]. Upstream of EZH2-DNMT1, there are regulatory mechanisms. In non-small cell lung cancer, activation of SAPK/JNK inhibits the interaction between EZH2 and DNMT1 by suppressing SP1 and NF-B/p65 [65, 66]. lncRNA HOTAIR can also regulate EZH2-DNMT1 activity, enhance EZH2 expression, and cancer cell production [67] (Fig. 6).
Targeting EZH2 has become an important way to treat cancer
Melanoma, breast cancer, prostate cancer, lung and liver cancer, psoriasis, hematological malignancies, and other disorders have all been linked to EZH2 mutations and expression imbalances. At this time, EZH2 inhibitors can be split into two categories: those that inhibit EZH2 methyltransferase activity and those that damage the structure of PRC2. There are two types of inhibitors of EZH2 methyltransferase activity. S-adenosine-L-homocysteine (SAH) hydrolase inhibitors, for example, inhibit EZH2 indirectly by increasing SAH levels. The second is an S-adenosylmethionine (SAM) competitive inhibitor that inhibits EZH2 by occupying the SAM-binding site in the EZH2 binding pocket [68]. The use of EZH2 inhibitors in combination with other medications has also been shown to have a positive therapeutic impact [69]. Because EZH2 possesses both cancer suppressor and cancer promoter qualities, it is thought that drugs may be created to target EZH2’s cancer promoter domain without compromising its tumor suppressor activity (Table 2).
EZH2/EZH1 inhibitors
Tazemetostat was recently approved by the FDA for metastatic or advanced epithelioid sarcoma (ES) that is not suitable for surgical resection. Tazemetostat is a selective EZH2 inhibitor, in cell-free test, and Ki and IC50 are 2.5 nM and 11 nM, respectively, which are 35 times more selective than EZH1 [70]. In diffuse large B-cell lymphoma cells, EL1 can significantly inhibit the methyltransferase activity of EZH2, reduce H3K27Me3, and activate the expression of p16 to inhibit the proliferation of diffuse large B-cell lymphoma cells (DLBCL). EL1 can also inhibit the Y641F mutation of EZH2, the IC50 is 13 nM, and for WT EZH2, IC50 is 15 nM [71]. GSK126 is a potent and highly selective EZH2 methyltransferase inhibitor with IC50 is 9.9 nM in cell-free experiments. In vitro experiments, GSK126 could significantly inhibit EZH2 activity in DLBCL cells and induce tumor suppressor gene transcription. GSK126 also has inhibitory effect on EZH2 mutants [72]. CPI-169 is a potent and selective EZH2 inhibitor, the IC50 is 0.24 nM, 0.51 nM, and 6.1 nM for EZH2 WT, EZH2 Y641N, and EZH1. In non-Hodgkin’s lymphoma (NHL) cells, CPI-169 could significantly inhibit the activity of EZH2, reduce the trimethylation level of H3K27, and the safety of CPI-169 is reliable [73]. EPZ005687 could bind to the SAM pocket of the EZH2 SET domain. It is a SAM-competitive inhibitor of EZH2. The Ki is 24 nM. EPZ005687 could act on a variety of different lymphoma cells to reduce H3K27 methylation and prolong the G1 phase and shorten the S and G2/M phases. Interestingly, EPZ005687 has stronger inhibitory activity against EZH2 containing Tyr641 or Ala677 mutants, while WT EZH2 has a weaker effect [74]. EPZ011989 is an effective, selective, and orally active EZH2 inhibitor, the Ki < 3 nM. EPZ011989 has a significant tumor suppressor effect on mouse model of human B-cell lymphoma and EPZ011989 could reduce intracellular H3K27 methylation in human lymphoma cells with Y641F mutation [75]. ZLD10A is a new type of EZH2 inhibitor, which could inhibit the methyltransferase activity of EZH2 with high selectivity. The NHL cells treated with ZLD10A showed decreased H3K27 methylation and increased tumor cell apoptosis and also had a good inhibitory effect on EZH2 with Y641F and A677G mutants. ZLD10A’s IC50 for WT EZH2, Y641F, and A677G are 18.6 nM, 27.1 nM, and 0.9 nM [76]. GSK503 is a highly effective EZH2 inhibitor with potential anti-tumor potential. The Ki is about 3 ~ 27 nM. It has a significant inhibitory effect on the proliferation of DLBCL which carried GCB (Germinal Center B cells) cells and could also affect the lymphatic transformation [77]. JQEZ5 is an inhibitor of EZH2 lysine methyltransferase. IC50 is 11.1 nM, which inhibited the colony formation of human primary CD34+ chronic myeloid leukemia (CML) stem/progenitor cells [78]. GSK926 and GSK343 with the same maternal structure have EZH2 inhibitory activity and could significantly inhibit the trimethylation of H3K27, and the IC50 are 324 ± 174 nM and 174 ± 84 nM. GSK343 could effectively inhibit proliferation of breast cancer cells and prostate cancer cells [79]. PF-06726304 is a selective EZH2 inhibitor, Ki values are 0.7 nM and 3 nM for WT EZH2 and EZH2 (Y641N), and IC50 for H3K27me3 inhibition is 15 nM. In the subcutaneous Karpas-422 xenograft model, PF-06726304 could significantly inhibit tumor growth [80]. EZH2-IN-3 is an inhibitor of EZH2 and EZH1. The IC50 for EZH2 (WT) is 0.032 ± 0.019 nM. When the level of H3K27Me3 in the cell decreases, the effect of EZH2-IN-3 will change significantly. And it has a selective effect on the growth of diffuse large B-cell lymphoma cells [81]. Lirametostat (CPI-1205) is an inhibitor of selective histone lysine methyltransferase EZH2 with oral biological activity, and the IC50 is 2 nM and 52 nM for EZH2 and EZH1. It has potential anti-tumor activity. In multiple myeloma and plasmacytoma cell models, CPI-1205 could cause tumor cell apoptosis [82]. EBI-2511 is a potent and orally active EZH2 inhibitor, IC50 is 4 nM for EZH2 (A667G) and an IC50 value of 55 nM in the WSU-DLCL2 cell line. This compound is a scaffold based on Tazemetostat. In the mouse model of generalized xenotransplantation, the anti-tumor activity of EBI-2511 at the same dose is better than Tazemetostat, which provides a new compound skeleton to EZH2 inhibitors [83]. UNC1999 is a selective inhibitor of EZH2 and EZH1 with high oral bioavailability. In the cell-free test, the IC50 for EZH2 and EZH1 is 2 nM and 45 nM. UNC1999 is an effective autophagy inducer. It specifically inhibits H3K27me3/2 and selectively kills the diffuse large B-cell lymphoma cell line while carrying the EZH2 (Y641N) mutant [84]. Valemetostat (DS-3201, DS-3201b) is a selective EZH1/2 dual inhibitor (IC50 ≤ 10 nM), which could significantly inhibit the H3K27Me3 in adult T-cell leukemia lymphoma cells (ATL cells). At the same time, SLA and PAG1 genes are reactivated and normal immune function is restored by Valemetostat [85]. (R)-OR-S1 is a SAM-competitive, highly selective, oral EZH1/2 inhibitor. It has a good inhibitory effect on EZH2 (Y641F). The IC50 of EZH1 and EZH2 is 7.4 nM and 10 nM and the IC50 of H3K27Me3 is 0.47 nM. (R)-OR-S1 could significantly inhibit the growth of KARPAS-422 cells [86]. PF-06821497 is a lactam EZH2 inhibitor designed based on ligands and physicochemical properties. It has a good inhibitory effect on EZH2 (Y641N), and the Ki is 1.15 nM. It is suitable for Karpas-422 xenotransplantation. PF-06821497 also has a good inhibitory effect on tumor growth in mice [87]. DZNeP is an adenosine analogue and competitive S-adenosylhomocysteine hydrolase (SAH) hydrolase inhibitor. It inhibits the activity of EZH2 by increasing the content of SAH. DZNeP globally inhibits histone methylation. Strictly speaking, DZNeP is not a selective for the methylation process regulated by EZH2 [88]. Oxetinib (AZD9291) is a highly effective selective EGFR mutant inhibitor for the treatment of advanced non-small cell lung cancer. Interestingly, Oxetinib exhibits an affinity for EZH2 and can disrupt the interaction between EZH2 and EED, ultimately leading to impaired PRC2 function and reduced EZH2 expression [89] (Table 1).
PRC2 inhibitors
As the catalytic subunit of PRC2, EZH2 regulated epigenetic. It must be combined with EED and SUZ12. Therefore, destroying the structure of PRC2 or inhibiting the function of EED or SUZ12 can also indirectly inhibit the function of EZH2. However, if EZH2 does not play a regulatory role through PRC2, the effectiveness of inhibitors needs further research. Proteolysis targeting chimeras (PROTACs) have become a new technology in drug development. Researchers have synthesized two active compounds with the function of inhibiting PRC2 through this technology, PROTAC EED Degrader-1 and PROTAC EED Degrader- 2. The IC50 is 8.17 nM and 8.11 nM, both of them could inhibit the function of PRC2 by binding to EED (pKD1 is 9.02 ± 0.09 nM, pKD2 is 9.27 ± 0.05 nM) and could inhibit the proliferation of DLBCL cell lines with EZH2 mutations in vitro [90]. UNC6852 is a selective degradation agent of PRC2. The IC50 of this compound to EED is 247 nM. UNC6852 could block the histone methyl transfer of EZH2 and reduce the level of H3K27me3 in HeLa cells and diffuse large B-cell lymphoma (DLBCL) cells which contain mutations in EZH2 [91]. A-395 could interact with the H3K27me3 binding pocket in EED to prevent the allosteric activation of PRC2 catalytic activity. It can effectively inhibit the activity of PRC2 in vitro, and the IC50 is 18 ± 2 nM. A-395’s ability of regulation of H3K27Me3 is equivalent to GSK126 in Pfeiffer and Karpas-422 DLBCL cell lines [92]. MS1943 is an oral EZH2 selective degradation agent, which can effectively reduce EZH2 levels in cells. Its IC50 value of inhibiting EZH2 methyltransferase activity is 120 nM, which can effectively reduce the level of EZH2 in BT549, HCC70, MDA-MB-231, TNBC cells, KARPAS-422, SUDHL8 lymphoma cells, and PNT2 non-cancerous prostate cells [93]. Chemical resistance in prostate cancer can be overcome by inhibitors designed with EED as a target. LG1980 has been discovered to bind to EED, inhibiting the activity of EZH2 without affecting its catalytic function. LG1980 and EED have a Kd value of 2.71 μM, and LG1980 significantly inhibited the in vitro viability of ARCaPE-shEPLIN cells (IC50 = 0.26 μM) and C4-B-TaxR cells (IC50 = 6.87 μM) [94]. Astemizole, an FDA-approved drug, is a small-molecule inhibitor of the PRC2 EZH2-EED interaction. As tinidazole inhibits the activity of the EZH2-EED interaction, the stability of the PRC2 complex in cancer cells, and the activity of its methyltransferase. It can significantly inhibit the proliferation of DLBCLs and works well in combination with EPZ005687 [95]. The PRC2 cofactor Jarid2 can bind to EED with a Kd value of 3.0 mM following trimethylation of the lysine residue at position 116. This can result in allosteric stimulation of the PRC2 enzyme activity. Subsequent investigation revealed that Jarid2’s amino acid residues 114–118 were crucial for the protein’s ability to bind EED. Researchers created Jarid 2114–118-K116me3, which has a Kd value of 8.82 μM, as a result of these discoveries. Two more potent peptide mimicking ligands, UNC5114 and UNC5115, were eventually found through more thorough SAR investigation. The Kd values of UNC5114 and UNC5115 for EED in the ITC experiment were 0.68 μM and 1.14 μM, respectively. Unlike Jarid 2-K116me3, UNC5114 and UNC5115 prevent the binding of H3K27me3 to EED, which is how PRC2 is inhibited [96].
EED226 could change the conformation of PRC2 by binding to the H3K27Me3 pocket, therefore EED226 could inhibit the methyltransferase activity of PRC2 and reduce the level of H3K27Me3 in the cell. EED226 also exhibits inhibiting effects in Y641N EZH2 mutant cells. In the 14-day anti-proliferative activity experiment of KARPAS-422, the IC50 value of EED226 was 80 nM. By constructing the Karpas-422 cell xenograft model, EED226 has a good pharmacokinetic parameter [97]. Despite the fact that EED226 has demonstrated good tumor treatment, EZH2 selectivity, and bioavailability, there are still issues, such as modeling solubility and durability, as well as the presence of the mono-substantiated electron-rich furan ring. Researchers optimized the structure of EED226 and obtained MAK683. Researchers evaluated the performance of candidate compounds by assessing PRC2 activity, H3K27 methylation status, and Pfeiffer cell proliferation. The values of MAK683 are 9 ± 4 nM, 3 ± 2 nM, and 4 nM (GI50). MAK683 also showed good therapeutic effects on Karpas-422-derived xenograft mouse model [98]. Based on the structure–activity relationship of EED 226, researchers discovered a new EED inhibitor ZJH-16 by changing the substituent type. ZJH-16 directly binds to the H3K27me3 binding pocket of EED with high affinity (HTRF IC50 = 2.72 nM, BLI Kd = 4.4 nM), and ZJH-16 demonstrates volatile pharmaceutical (PK) properties and robust antagonist efficiency in a KARPAS-422 xenograft model [99]. The EED inhibitor BR001, which demonstrated significant EED targeting activity in competitive binding tests and had an IC50 value of 4.5 nM, was found based on the structure of EED226 and utilizing the scaffold picking approach. Inhibiting cell growth and dramatically lowering the expression level of H3K27me3 in KARPAS-422 cells are two effects of BR001. Without causing noticeable side effects, BR001 has strong anti-cancer efficacy in vivo in the Pfeiffer and KARPAS-422 xenograft tumor models. Nevertheless, the addition of anti-PD-1 therapy did not improve therapeutic impact [100]. In view of the achievements of the drug design idea of inhibiting the activity of PRC2 and reducing the level of methylation by binding to EED, the researchers continued to synthesize an active molecule with better inhibitory activity than EED226. EEDi5285 is an effective and oral EED inhibitor. EEDi5285 could bind to EED protein and the IC50 is 0.2 nM. It also shows amazing inhibitory activity in Pfeiffer and KARPAS-422 cell lines which carrying EZH2 mutations, the IC50 is 20 pM and 0.5 nM. EEDi5285 is also possess an excellent pharmacokinetic data and achieve complete tumor treatment effects on the KARPAS-422 xenograft model in mice [101]. Furthermore, EEDi-5273, a class of highly effective EED inhibitors with oral bioavailability greater than EEDi5285, was discovered through conformal restriction and system structure–activity relationship studies. With an IC50 value of 0.2 nM, EEDi-5273 has a high affinity for EED. With an IC50 value of 1.2 nM, EEDi-5233 effectively inhibits the growth of KARPAS-422 cells and achieves good tumor treatment effects in xenotransplantation animal models without side effects [102] (Table 1).
The impact of natural products on EZH2 and PRC2
Natural products derived from plants have been proven that has good anti-cancer activity, such as paclitaxel and vincristine. There are also a large number of natural products that can target EZH2. Gambogic acid (GNA) is derived from Garcinia hanburyi (Garcinia hanburyi Hook.f). It could bind to Cys668 in the EZH2 SET domain specifically and trigger ubiquitination of EZH2. According to the structure of GNA, the researchers have designed GNA002, which has much better activity. Through a xenograft tumor model confirm that when the anti-tumor activity is close, GNA002 has better safety than Cisplatin [103]. 16-Hydroxycleroda-3,13-dien-15,16-olide (PL3) is a diterpene compound isolated from Polyalthia longifolia, which could significantly inhibit EZH2 and SUZ12 in the PRC2 complex in dose-dependent manner, reduce the inhibition of Msk1, Set7, and Src1 genes by PRC2, and finally induce apoptosis of K562 cells [104]. Tanshinone I, a diterpene compound derived from Salvia miltiorrhiza. Tanshinone I could directly bind to EZH2 (Kd = 94.475 μM), thereby inhibiting the activity of PRC2 and the level of H3K27Me3. Tanshinone I could also increase the expression of MMP9 and ABCG2 and limit the growth of leukemia cells and function of malignant hematopoietic [105]. Curcumin is a natural polyphenol component, mainly derived from the rhizomes of turmeric (Curcuma longa Linn). Curcumin could inhibit the expression of EZH2, ASXL1, H3K27me3, and HOXA9 significantly. Curcumin could inhibit the proliferation and induce apoptosis in MDS cell lines from human and show anti-tumor activity in xenograft model mice [106]. Others like Triptolide, Ursolic acid, and Resveratrol are shown inhibitory to EZH2 [107].
Analysis of the current situation and prospects for EZH2 inhibitors
EZH2 encourages carcinogenesis and malignant transformation and is highly expressed in a variety of human cancers. Therefore, EZH2-targeted inhibition is a perfect drug design target. According to the aforementioned research, there is still a ton of flexibility in the structure and choice of targets for EZH2 inhibitors. By examining the structures of the “Deep Pocket,” “Aromatic Cage,” and “Tail,” researchers have continuously improved the main skeleton of EZH2 and EED inhibitors. They have also adopted strategies like changing substituents to improve the physicochemical characteristics and affinity of EZH2 inhibitors.
At this point, the main emphasis is on the EZH2 SET domain and disabling the interaction between EZH2-EED, which will allow for the correction of epigenetic flaws and the elimination of cancer. This approach has performed well in the treatment of some hematological tumors, but the effectiveness of EHZ2-related inhibitors in treating some solid tumors has not been satisfactory [108]. It is well known that EZH2 can be used as a single active molecule to extensively regulate physiological activities and that its role does not only depend on the activity of the PRC2-dependent methyltransferase. However, the non-classical function of EZH2 is largely ignored, and the design of existing inhibitors is only restricted to the pertinent domains of the methyltransferase function. Only by defining the non-classical function of EZH2’s mode of action will it be possible to develop EZH2 inhibitors with more thorough functionality. For instance, scientists have shown that EZH2’s N-terminal has a transcriptional activation region that mediates the binding of EZH2 to c-Myc, p300, and SMARCA4, thereby accelerating the growth of tumors. Based on this, scientists have developed a new class of EZH2 small-molecule degrading agent called MS177 that can significantly inhibit c-Myc-mediated gene upregulation in addition to targeting the catalytic and non-catalytic functions of EZH2. This agent can also degrade the components of the PRC2 complex. This scenario indicates that it is impossible to overlook EZH2’s non-classical catalytic function [109]. Existing inhibitors have functional flaws as a result of the diversity of EZH2 activities. At the same time, it is important to keep in mind that using EZH2 inhibitors might actually increase cell proliferation and improve DNA damage repair capabilities, which promotes the growth of tumors [110]. According to research, EZH2 inhibitors can activate H3K27ac, which makes cells more resistant to drugs. The therapeutic efficacy of tumors can be improved by giving BRD4 inhibitors to cells that have lost their sensitivity to EZH2 inhibitors [111]. In addition, CDK1, HSP90, or proteasome inhibitors prevent EZH2 from being degraded in AML cells, restoring the cells' sensitivity to medicines [112].
In summary, EZH2 plays a role in metabolic pathways, inflammation, and tumor growth [113, 114]. In addition, Tazemetostat or similar substance has brought EZH2 to the public notice as a crucial regulatory molecule of epigenetics, which gives us significant confidence that blocking EZH2 will eradicate the tumor. However, we should also be aware of the EZH2 inhibitors’ current drawbacks, such as their single target function and the risk of drug resistance and illness aggravation associated with overusing them. This necessitates a thorough assessment of EZH2’s structure and function, as well as the utilization of numerous pharmacological combination therapies to control the dangers associated with EZH2 inhibitors.

EZH2 and other signal transduction and related diseases. Interrelationships between EZH2 and other molecule and the resulting illnesses
References
Pasini D, Di Croce L. Emerging roles for polycomb proteins in cancer. Curr Opin Genet Dev. 2016;36:50–8.
Glancy E, Ciferri C, Bracken AP. Structural basis for PRC2 engagement with chromatin. Curr Opin Struct Biol. 2021;67:135–44.
Allis CD, Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406(6796):593–9.
Simon JA, Lange CA. Roles of the EZH2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647(1–2):21–9.
He A, Shen X, Ma Q, Cao J, von Gise A, Zhou P, et al. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev. 2012;26(1):37–42.
Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23(6):839–52.
Dasgupta M, Dermawan JK, Willard B, Stark GR. STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc Natl Acad Sci USA. 2015;112(13):3985–90.
Hoffmeyer K, Junghans D, Kanzler B, Kemler R. Trimethylation and acetylation of beta-catenin at lysine 49 represent key elements in ESC pluripotency. Cell Rep. 2017;18:2815–24.
Lee CH, Yu JR, Granat J, Saldana-Meyer R, Andrade J, LeRoy G, et al. Automethylation of PRC2 promotes H3K27 methylation and is impaired in H3K27M pediatric glioma. Genes Dev. 2019;33:1428–40.
Wang X, Long Y, Paucek RD, Gooding AR, Lee T, Burdorf RM, et al. Regulation of histone methylation by automethylation of PRC2. Genes Dev. 2019;33:1416–27.
Wu Z, Aau M, Guan P, Karuturi RK, Liou YC, Yu Q. Context-specific regulation of NF-κB target gene expression by EZH2 in breast cancers. Mol Cell. 2011;43(5):798–810.
Zhang Y, Hong M, Shang Y. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol. 2007;27(14):5105–19.
Böttcher M, Bruns H, Völkl S, Lu J, Chartomatsidou E, Papakonstantinou N, et al. Control of PD-L1 expression in CLL-cells by stromal triggering of the Notch-c-Myc-EZH2 oncogenic signaling axis. J Immunother Cancer. 2021;9(4):1889–98.
Zheng X, Pang B, Gu G, Gao T, Zhang R, Pang Q, et al. Melatonin inhibits glioblastoma stem-like cells through suppression of EZH2-notch1 signaling axis. Int J Biol Sci. 2017;13(2):245–53.
Li B, Yu F, Wu F, Hui T, A P, Liao X, et al. EZH2 impairs human dental pulp cell mineralization via the Wnt/β-catenin pathway. J Dent Res. 2018;97(5):571–9.
Chen Q, Cai J, Wang Q, Wang Y, Liu M, Yang J, et al. Long noncoding RNA NEAT1, regulated by the EGFR pathway, contributes to glioblastoma progression through the WNT/β-catenin pathway by scaffolding EZH2. Clin Cancer Res. 2018;24(3):684–95.
Khan H, Ni Z, Feng H, Xing Y, Wu X, Huang D, et al. Combination of curcumin with N-n-butyl haloperidol iodide inhibits hepatocellular carcinoma malignant proliferation by downregulating enhancer of zeste homolog 2 (EZH2) - lncRNA H19 to silence Wnt/β-catenin signaling. Phytomedicine. 2021;91:706–16.
Yang X, Shao F, Guo D, Wang W, Wang J, Zhu R, et al. WNT/β-catenin-suppressed FTO expression increases m6A of c-Myc mRNA to promote tumor cell glycolysis and tumorigenesis. Cell Death Dis. 2021;12(5):462–76.
Fujii S, Tokita K, Wada N, Ito K, Yamauchi C, Ito Y, et al. MEK-ERK pathway regulates EZH2 overexpression in association with aggressive breast cancer subtypes. Oncogene. 2011;30(39):4118–28.
Feng Y, Li L, Du Y, Peng X, Chen F. E2F4 functions as a tumour suppressor in acute myeloid leukaemia via inhibition of the MAPK signalling pathway by binding to EZH2. J Cell Mol Med. 2020;24(3):2157–68.
Bisserier M, Wajapeyee N. Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas. Blood. 2018;131(19):2125–37.
Riquelme E, Behrens C, Lin HY, Simon G, Papadimitrakopoulou V, Izzo J, et al. Modulation of EZH2 expression by MEK-ERK or PI3K-Akt signaling in lung cancer is dictated by different KRAS oncogene mutations. Cancer Res. 2016;76(3):675–85.
Li R, Yi X, Wei X, Huo B, Guo X, Cheng C, et al. EZH2 inhibits autophagic cell death of aortic vascular smooth muscle cells to affect aortic dissection. Cell Death Dis. 2018;9(2):180–95.
Lu H, Jin W, Sun J, Feng L, Lan H, Shen Q, et al. New tumor suppressor CXXC finger protein 4 inactivates mitogen activated protein kinase signaling. FEBS Lett. 2014;588(18):3322–6.
Manning CS, Hooper S, Sahai EA. Intravital imaging of SRF and notch signalling identifies a key role for EZH2 in invasive melanoma cells. Oncogene. 2015;34(33):4320–32.
Jeong GY, Park MK, Choi HJ, An HW, Park YU, Choi HJ, et al. NSD3-induced methylation of H3K36 activates notch signaling to drive breast tumor initiation and metastatic progression. Cancer Res. 2021;81(1):77–90.
Zeng SHG, Xie JH, Zeng QY, Dai SHH, Wang Y, Wan XM, et al. lncRNA PVT1 promotes metastasis of non-small cell lung cancer through EZH2-mediated activation of hippo/Notch1 signaling pathways. Cell J. 2021;23(1):21–31.
Kwon H, Song K, Han C, Zhang J, Lu L, Chen W, et al. Epigenetic silencing of miRNA-34a in human cholangiocarcinoma via EZH2 and DNA methylation: impact on regulation of notch pathway. Am J Pathol. 2017;187(10):2288–99.
Wasson CW, Abignano G, Hermes H, Malaab M, Ross RL, Jimenez SA, et al. Long non-coding RNA HOTAIR drives EZH2-dependent myofibroblast activation in systemic sclerosis through miRNA 34a-dependent activation of Notch. Ann Rheum Dis. 2020;79(4):507–17.
Pappas K, Martin TC, Wolfe AL, Nguyen CB, Su T, Jin J, et al. Notch and EZH2 collaborate to repress PTEN expression in breast cancer. Commun Biol. 2021;4(1):312–25.
Tsou PS, Campbell P, Amin MA, Coit P, Miller S, Fox DA, et al. Inhibition of EZH2 prevents fibrosis and restores normal angiogenesis in scleroderma. Proc Natl Acad Sci USA. 2019;116(9):3695–702.
Acharyya S, Sharma SM, Cheng AS, Ladner KJ, He W, Kline W, et al. TNF inhibits Notch-1 in skeletal muscle cells by EZH2 and DNA methylation mediated repression: implications in duchenne muscular dystrophy. PLoS ONE. 2010;5(8):479–88.
Zheng S, Xiao L, Liu Y, Wang Y, Cheng L, Zhang J, et al. DZNep inhibits H3K27me3 deposition and delays retinal degeneration in the rd1 mice. Cell Death Dis. 2018;9(3):310–24.
Chen B, Liu J, Chang Q, BeEZHold K, Lu Y, Chen F. JNK and STAT3 signaling pathways converge on Akt-mediated phosphorylation of EZH2 in bronchial epithelial cells induced by arsenic. Cell Cycle. 2013;12(1):112–21.
He S, Liu Y, Meng L, Sun H, Wang Y, Ji Y, et al. Ezh2 phosphorylation state determines its capacity to maintain CD8+ T memory precursors for antitumor immunity. Nat Commun. 2017;8(1):212521–36.
Chen X, Hao A, Li X, Du Z, Li H, Wang H, et al. Melatonin inhibits tumorigenicity of glioblastoma stem-like cells via the Akt-EZH2-STAT3 signaling axis. J Pineal Res. 2016;61(2):208–17.
Rizk M, Rizq O, Oshima M, Nakajima-Takagi Y, Koide S, Saraya A, et al. Akt inhibition synergizes with polycomb repressive complex 2 inhibition in the treatment of multiple myeloma. Cancer Sci. 2019;110(12):3695–707.
Kikuchi J, Koyama D, Wada T, Izumi T, Hofgaard PO, Bogen B, et al. Phosphorylation-mediated EZH2 inactivation promotes drug resistance in multiple myeloma. J Clin Invest. 2015;125(12):4375–90.
Zhang J, Ji F, Liu Y, Lei X, Li H, Ji G, et al. EZH2 regulates adult hippocampal neurogenesis and memory. J Neurosci. 2014;34(15):5184–99.
Yang R, Wang M, Zhang G, Bao Y, Wu Y, Li X, et al. E2F7-EZH2 axis regulates PTEN/Akt/mTOR signalling and glioblastoma progression. Br J Cancer. 2020;123(9):1445–55.
Sanches JGP, Song B, Zhang Q, Cui X, Yabasin IB, Ntim M, et al. The role of KDM2B and EZH2 in regulating the stemness in colorectal cancer through the PI3K/Akt pathway. Front Oncol. 2021;11:298–313.
Du X, Chen Y, Zhang Q, Lin J, Yu Y, Pan Z, et al. EZH2 Ameliorates Osteoarthritis by Activating TNFSF13B. J Bone Miner Res. 2020;35:956–65.
Geng J, Li X, Zhou Z, Wu CL, Dai M, Bai X. EZH2 promotes tumor progression via regulating VEGF-A/Akt signaling in non-small cell lung cancer. Cancer Lett. 2015;359(2):275–87.
Yamada L, Saito M, Thar Min AK, Saito K, Ashizawa M, Kase K, et al. Selective sensitivity of EZH2 inhibitors based on synthetic lethality in ARID1A-deficient gastric cancer. Gastric Cancer. 2021;24:60–71.
Feng H, Yu Z, Tian Y, Lee YY, Li MS, Go MY, et al. A CCRK-EZH2 epigenetic circuitry drives hepatocarcinogenesis and associates with tumor recurrence and poor survival of patients. J Hepatol. 2015;62(5):1100–11.
Kaur M, Cole MD. MYC acts via the PTEN tumor suppressor to elicit autoregulation and genome-wide gene repression by activation of the EZH2 methyltransferase. Cancer Res. 2013;73(2):695–705.
Kosalai ST, Morsy MHA, Papakonstantinou N, Mansouri L, Stavroyianni N, Kanduri C, et al. EZH2 upregulates the PI3K/Akt pathway through IGF1R and MYC in clinically aggressive chronic lymphocytic leukaemia. Epigenetics. 2019;14(11):1125–40.
Yi C, Li G, Wang W, Sun Y, Zhang Y, Zhong C, et al. Disruption of YY1-EZH2 interaction using synthetic peptides inhibits breast cancer development. Cancers (Basel). 2021;13(10):2402–25.
Ferraro A, Mourtzoukou D, Kosmidou V, Avlonitis S, Kontogeorgos G, Zografos G, et al. EZH2 is regulated by ERK/Akt and targets integrin alpha2 gene to control Epithelial-Mesenchymal Transition and anoikis in colon cancer cells. Int J Biochem Cell Biol. 2013;45(2):243–54.
Han H, Wang S, Meng J, Lyu G, Ding G, Hu Y, et al. Long noncoding RNA PART1 restrains aggressive gastric cancer through the epigenetic silencing of PDGFB via the PLZF-mediated recruitment of EZH2. Oncogene. 2020;39(42):6513–28.
Chen J, Wang F, Xu H, Xu L, Chen D, Wang J, et al. Long non-coding RNA SNHG1 regulates the Wnt/β-catenin and PI3K/Akt/mTOR signaling pathways via EZH2 to affect the proliferation, apoptosis, and autophagy of prostate cancer cell. Front Oncol. 2020;10:907–19.
Wang L, Bo X, Yi X, Xiao X, Zheng Q, Ma L, et al. Exosome-transferred LINC01559 promotes the progression of gastric cancer via PI3K/Akt signaling pathway. Cell Death Dis. 2020;11(9):723–35.
Han Y, Chen M, Wang A, Fan X. STAT3-induced upregulation of lncRNA CASC11 promotes the cell migration, invasion and epithelial-mesenchymal transition in hepatocellular carcinoma by epigenetically silencing PTEN and activating PI3K/Akt signaling pathway. Biochem Biophys Res Commun. 2019;508(2):472–9.
Liu X, Lu X, Zhen F, Jin S, Yu T, Zhu Q, et al. LINC00665 induces acquired resistance to gefitinib through recruiting EZH2 and activating PI3K/Akt pathway in NSCLC. Mol Ther Nucleic Acids. 2019;16:155–61.
Liu Y, Hu Q, Wang X. AFAP1-AS1 induces cisplatin resistance in non-small cell lung cancer through PI3K/Akt pathway. Oncol Lett. 2020;19(1):1024–30.
Myers K, Gagou ME, Zuazua-Villar P, Rodriguez R, Meuth M. ATR and Chk1 suppress a caspase-3-dependent apoptotic response following DNA replication stress. PLoS Genet. 2009;5(1):324–38.
Wen Y, Hou Y, Yi X, Sun S, Guo J, He X, et al. EZH2 activates CHK1 signaling to promote ovarian cancer chemoresistance by maintaining the properties of cancer stem cells. Theranostics. 2021;11(4):1795–813.
Zhou J, Yang Y, Wang YL, Zhao Y, Ye WJ, Deng SY, et al. Enhancer of zeste homolog 2 contributes to apoptosis by inactivating janus kinase 2/ signal transducer and activator of transcription signaling in inflammatory bowel disease. World J Gastroenterol. 2021;27(22):3073–84.
Li L, Liu J, Xue H, Li C, Liu Q, Zhou Y, et al. A TGF-β-MTA1-SOX4-EZH2 signaling axis drives epithelial-mesenchymal transition in tumor metastasis. Oncogene. 2020;39(10):2125–39.
He Z, Zhang S, Ma D, Fang Q, Yang L, Shen S, et al. HO-1 promotes resistance to an EZH2 inhibitor through the pRB-E2F pathway: correlation with the progression of myelodysplastic syndrome into acute myeloid leukemia. J Transl Med. 2019;17(1):366–84.
Müller A, Dickmanns A, Resch C, Schäkel K, Hailfinger S, Dobbelstein M, et al. The CDK4/6-EZH2 pathway is a potential therapeutic target for psoriasis. J Clin Invest. 2020;130(11):5765–81.
Sander S, Bullinger L, Klapproth K, Fiedler K, Kestler HA, Barth TF, et al. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood. 2008;112(10):4202–12.
Zhang X, Zhao X, Fiskus W, Lin J, Lwin T, Rao R, et al. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-Cell lymphomas. Cancer Cell. 2012;22(4):506–23.
Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439(7078):871–4.
Wu J, Zhao S, Tang Q, Zheng F, Chen Y, Yang L, et al. Activation of SAPK/JNK mediated the inhibition and reciprocal interaction of DNA methyltransferase 1 and EZH2 by ursolic acid in human lung cancer cells. J Exp Clin Cancer Res. 2015;34(1):99–110.
Li L, Wu J, Zheng F, Tang Q, Wu W, Hann SS. Inhibition of EZH2 via activation of SAPK/JNK and reduction of p65 and DNMT1 as a novel mechanism in inhibition of human lung cancer cells by polyphyllin I. J Exp Clin Cancer Res. 2016;35(1):112–25.
Xiang S, Zou P, Tang Q, Zheng F, Wu J, Chen Z, Hann SS. HOTAIR-mediated reciprocal regulation of EZH2 and DNMT1 contribute to polyphyllin I-inhibited growth of castration-resistant prostate cancer cells in vitro and in vivo. Biochim Biophys Acta Gen. 2018;1862(3):589–99.
Eich ML, Athar M, Ferguson JE 3rd, Varambally S. EZH2-targeted therapies in cancer: hype or a reality. Cancer Res. 2020;80(24):5449–58.
Kong Y, Zhang Y, Mao F, Zhang Z, Li Z, Wang R, et al. Inhibition of EZH2 enhances the antitumor efficacy of metformin in prostate cancer. Mol Cancer Ther. 2020;19(12):2490–501.
Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA. 2013;110(19):7922–7.
Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, et al. Selective inhibition of EZH2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci USA. 2012;109(52):21360–5.
McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492(7427):108–12.
Bradley WD, Arora S, Busby J, Balasubramanian S, Gehling VS, Nasveschuk CG, et al. EZH2 inhibitor efficacy in non-Hodgkin’s lymphoma does not require suppression of H3K27 monomethylation. Chem Biol. 2014;21(11):1463–75.
Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8(11):890–6.
Campbell JE, Kuntz KW, Knutson SK, Warholic NM, Keilhack H, Wigle TJ, et al. EPZ011989, a potent, orally-available EZH2 inhibitor with robust in vivo activity. ACS Med Chem Lett. 2015;6(5):491–5.
Song X, Zhang L, Gao T, Ye T, Zhu Y, Lei Q, et al. Selective inhibition of EZH2 by ZLD10A blocks H3K27 methylation and kills mutant lymphoma cells proliferation. Biomed Pharmacother. 2016;81:288–94.
Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013;23(5):677–92.
Xie H, Peng C, Huang J, Li BE, Kim W, Smith EC, et al. Chronic myelogenous leukemia- initiating cells require polycomb group protein EZH2. Cancer Discov. 2016;6(11):1237–47.
Verma SK, Tian X, LaFrance LV, Duquenne C, Suarez DP, Newlander KA, et al. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med Chem Lett. 2012;3(12):1091–6.
Kung PP, Rui E, Bergqvist S, Bingham P, Braganza J, Collins M, et al. Correction to design and synthesis of pyridone-containing 3,4-dihydroisoquinoline-1(2H)-ones as a novel class of enhancer of zeste homolog 2 (EZH2) inhibitors. J Med Chem. 2016;59(24):8306–25.
Garapaty-Rao S, Nasveschuk C, Gagnon A, Chan EY, Sandy P, Busby J, et al. Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse large B cell lymphoma cell growth. Chem Biol. 2013;20(11):1329–39.
Vaswani RG, Gehling VS, Dakin LA, Cook AS, Nasveschuk CG, Duplessis M, et al. Identification of (R)-N-((4-Methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a Potent and Selective Inhibitor of Histone Methyltransferase EZH2, Suitable for Phase I Clinical Trials for B-Cell Lymphomas. J Med Chem. 2016;59(21):9928–41.
Lu B, Shen X, Zhang L, Liu D, Zhang C, Cao J, et al. Discovery of EBI-2511: a highly potent and orally active EZH2 inhibitor for the treatment of non-Hodgkin’s lymphoma. ACS Med Chem Lett. 2018;9(2):98–102.
Konze KD, Ma A, Li F, Barsyte-Lovejoy D, Parton T, Macnevin CJ, et al. An orally bioavailable chemical probe of the Lysine Methyltransferases EZH2 and EZH1. ACS Chem Biol. 2013;8(6):1324–34.
Yamagishi M, Hori M, Fujikawa D, Ohsugi T, Honma D, Adachi N, et al. Targeting excessive EZH1 and EZH2 activities for abnormal histone methylation and transcription network in malignant lymphomas. Cell Rep. 2019;29(8):2321–37.
Honma D, Kanno O, Watanabe J, Kinoshita J, Hirasawa M, Nosaka E, et al. Novel orally bioavailable EZH1/2 dual inhibitors with greater antitumor efficacy than an EZH2 selective inhibitor. Cancer Sci. 2017;108(10):2069–78.
Kung PP, Bingham P, Brooun A, Collins M, Deng YL, Dinh D, et al. Optimization of orally bioavailable enhancer of zeste homolog 2 (EZH2) inhibitors using ligand and property-based design strategies: identification of development candidate(R)-5,8-dichloro-7-(methoxy(oxetan-3-yl)methyl)-2-((4-methoxy-6-methyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3,4-dihydroisoquinolin-1(2H)-one (PF-06821497). J Med Chem. 2018;61(3):650–65.
Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, et al. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8(6):1579–88.
Zhang KL, Shen QQ, Fang YF, Sun YM, Ding J, Chen Y. AZD9291 inactivates the PRC2 complex to mediate tumor growth inhibition. Acta Pharmacol Sin. 2019;40(12):1587–95.
Hsu JH, Rasmusson T, Robinson J, Pachl F, Read J, Kawatkar S, et al. EED-targeted PROTACs degrade EED, EZH2, and SUZ12 in the PRC2 complex. Cell Chem Biol. 2020;27(1):41–6.
Potjewyd F, Turner AW, Beri J, Rectenwald JM, Norris-Drouin JL, Cholensky SH, et al. Degradation of polycomb repressive complex 2 with an EED-targeted bivalent chemical degrader. Cell Chem Biol. 2020;27(1):47–56.
Huang Y, Zhang J, Yu Z, Zhang H, Wang Y, Lingel A, et al. Discovery of first-in-class, potent, and orally bioavailable embryonic ectoderm development (EED) inhibitor with robust anticancer efficacy. J Med Chem. 2017;60(6):2215–26.
Rej RK, Wang C, Lu J, Wang M, Petrunak E, Zawacki KP, et al. EEDi-5285: an exceptionally potent, efficacious, and orally active small-molecule inhibitor of embryonic ectoderm development. J Med Chem. 2020;63(13):7252–67.
Li X, Gera L, Zhang S, Chen Y, Lou L, Wilson LM, et al. Pharmacological inhibition of noncanonical EED-EZH2 signaling overcomes chemoresistance in prostate cancer. Theranostics. 2021;11(14):6873–90.
Kong X, Chen L, Jiao L, Jiang X, Lian F, Lu J, et al. Astemizole arrests the proliferation of cancer cells by disrupting the EZH2-EED interaction of polycomb repressive complex 2. J Med Chem. 2014;57(22):9512–21.
Barnash KD, The J, Norris-Drouin JL, Cholensky SH, Worley BM, Li F, et al. Discovery of peptidomimetic ligands of EED as allosteric inhibitors of PRC2. ACS Comb Sci. 2017;19(3):161–72.
He Y, Selvaraju S, Curtin ML, Jakob CG, Zhu H, Comess KM, et al. The EED protein-protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat Chem Biol. 2017;13(4):389–95.
Huang Y, Sendzik M, Zhang J, Gao Z, Sun Y, Wang L, et al. Discovery of the clinical candidate MAK683: an EED-directed, allosteric, and selective PRC2 inhibitor for the treatment of advanced malignancies. J Med Chem. 2022;65(7):5317–33.
Dong G, Zuo J, Yu J, Xu J, Gao G, Li GB, et al. Structure-based design of the indole-substituted triazolopyrimidines as new EED-H3K27me3 inhibitors for the treatment of lymphoma. J Med Chem. 2023;66(1):1063–81.
Dong H, Liu S, Zhang X, Chen S, Kang L, Chen Y, et al. An allosteric PRC2 inhibitor targeting EED suppresses tumor progression by modulating the immune response. Cancer Res. 2019;79(21):5587–96.
Ma A, Stratikopoulos E, Park KS, Wei J, Martin TC, Yang X, et al. Discovery of a first-in-class EZH2 selective degrader. Nat Chem Biol. 2020;16(2):214–22.
Rej RK, Wang C, Lu J, Wang M, Petrunak E, Zawacki KP, et al. Discovery of EEDi-5273 as an exceptionally potent and orally efficacious EED inhibitor capable of achieving complete and persistent tumor regression. J Med Chem. 2021;64(19):14540–56.
Wang X, Cao W, Zhang J, Yan M, Xu Q, Wu X, et al. A covalently bound inhibitor triggers EZH2 degradation through CHIP-mediated ubiquitination. EMBO J. 2017;36(9):1243–60.
Lin YH, Lee CC, Chang FR, Chang WH, Wu YC, Chang JG, et al. 16-hydroxycleroda-3,13-dien-15,16-olide regulates the expression of histone-modifying enzymes PRC2 complex and induces apoptosis in CML K562 cells. Life Sci. 2011;89(23–24):886–95.
Ma L, Zhang X, Wang Z, Huang L, Meng F, Hu L, et al. Anti-cancer effects of curcumin on myelodysplastic syndrome through the inhibition of enhancer of zeste homolog-2 (EZH2). Curr Cancer Drug Targets. 2019;19(9):729–41.
Huang Y, Yu SH, Zhen WX, Cheng T, Wang D, Lin JB, et al. Tanshinone I, a new EZH2 inhibitor restricts normal and malignant hematopoiesis through upregulation of MMP9 and ABCG2. Theranostics. 2021;11(14):6891–904.
Shahabipour F, Caraglia M, Majeed M, Derosa G, Maffioli P, Sahebkar A. Naturally occurring anti-cancer agents targeting EZH2. Cancer Lett. 2017;400:325–35.
Kim KH, Kim W, Howard TP, Vazquez F, Tsherniak A, Wu JN, et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat Med. 2015;21(12):1491–6.
Wang J, Yu X, Gong W, Liu X, Park KS, Ma A, Tsai YH, et al. EZH2 noncanonically binds cMyc and p300 through a cryptic transactivation domain to mediate gene activation and promote oncogenesis. Nat Cell Biol. 2022;24(3):384–99.
de Vries NA, Hulsman D, Akhtar W, de Jong J, Miles DC, Blom, et al. Prolonged Ezh2 depletion in glioblastoma causes a robust switch in cell fate resulting in tumor progression. Cell Rep. 2015;10(3):383–97.
Huang X, Yan J, Zhang M, Wang Y, Chen Y, Fu X, et al. Targeting epigenetic crosstalk as a therapeutic strategy for EZH2-aberrant solid tumors. Cell. 2018;175(1):186–99.
Göllner S, Oellerich T, Agrawal-Singh S, Schenk T, Klein HU, Rohde C, et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat Med. 2017;23(1):69–78.
Zhou J, Huang S, Wang Z, Huang J, Xu L, Tang X, et al. Targeting EZH2 histone methyltransferase activity alleviates experimental intestinal inflammation. Nat Commun. 2019;10(1):2427–38.
Lim HJ, Kim M. EZH2 as a potential target for NAFLD therapy. Int J Mol Sci. 2020;21(22):8617–32.
Acknowledgements
We are grateful to the Natural Science Foundation (217100030), no conflict of interest exists in the submission of this manuscript, and the manuscript is approved by all authors for publication.
Funding
We are grateful to Natural Science Foundation (217100030).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing Interests
No conflict of interest exists in the submission of this manuscript, and the manuscript is approved by all authors for publication.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, Y., Yang, Q. The roles of EZH2 in cancer and its inhibitors. Med Oncol 40, 167 (2023). https://doi.org/10.1007/s12032-023-02025-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12032-023-02025-6