
Abstract
IRF-1, a kind of transcription factor, is expressed in many cell types, except in early embryonal cells. IRF-1 has played an essential role in various physiological and pathological processes, including tumor immune surveillance, viral infection, development of immunity system and pro-inflammatory injury. However, the expression and function of IRF-1 in spinal cord injury (SCI) are still unknown. In this study, we have performed an acute SCI model in adult rats and investigated the dynamic changes of IRF-1 expression in the spinal cord. Western blot have shown that IRF-1 protein levels gradually increased, reaching a peak at day 3 and then gradually declined to a normal level at day 14 after SCI. Double immunofluorescence staining showed that IRF-1 immunoreactivity was found in neurons, but not in astrocytes and microglia. Additionally, colocalization of IRF-1/active caspase-3 was detected in neurons. In vitro, IRF-1 depletion, by short interfering RNA, obviously decreases neuronal apoptosis. In conclusion, this is the first description of IRF-1 expression in spinal cord injury. Our results suggested that IRF-1 might play crucial roles in CNS pathophysiology after SCI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Spinal cord injury is characteristically accompanied by a period of secondary cell degeneration, which occurs in injured tissue over a course of time after the initial damage and lead to neuronal death, microglial activation, inflammation, and reactive astrogliosis (Crowe et al. 1997; Dumont et al. 2001; Liu et al. 1997; McGraw et al. 2001). The primary injury is attributed to the initial mechanical insult due to the direct tissue damage and energy transformation (Tator 1996). The secondary injury may result from spinal cord edema, ischemia, free radical damage, electrolyte imbalance, excitotoxicity, inflammatory injury, or apoptosis. These factors cause neuronal cell death, astrocyte proliferation, and reactive gliosis, resulting in the formation of a dense astrocytic scar (McGraw et al. 2001; Raghupathi 2004). The apoptosis of post-mitotic cells, such as neurons occurring after the spinal cord injury, may lead to subsequent neurological dysfunction (Di Giovanni et al. 2003). Simultaneously, glial scar provides a physical and biochemical barrier to regeneration and plasticity, and it is a source of multiple inhibitory factors that affect functional recovery from spinal cord injury (SCI) (Davies et al. 1996; Silver and Miller 2004). However, the signaling events that contribute to neuronal apoptosis and glial scar after SCI remain to be further researched.
IRF-1, the primary member of the interferon (IFN) regulatory factor family, was initially identified as a key regulator of type I interferon (α/β). The IRF-1 messenger RNA is expressed in cell cycle-dependent accumulation at a low base level in many cell types except early embryonal cells (Watanabe et al. 1991). Recently, IRF-1 has been found participating in signal transduction of other ligands, including tumor necrosis factor and interleukin-12 (Galon et al. 1999), interleukin-1β (Geller et al. 1993), and CD40 ligand (Gupta et al. 1998). In addition, some studies have shown that IRF-1 may function as a potential tumor suppressor (Tanaka et al. 1994), such as that the loss of IRF-1 contributes to tumor development in conjunction with c-Ha-ras in vivo (Nozawa et al. 1999). IRF-1 is also connected with apoptosis induced by DNA damage or other stimuli. DNA damage causes apoptosis in mitogenically activated mature T lymphocytes which is dependent on IRF-1 instead of p53. In addition, IRF-1 induces apoptosis by the intrinsic pathway, instead of the extrinsic pathway, by increasing the expression of p53 unregulated modulator (PUMA) (Gao et al. 2010). However, in thymocytes, apoptosis is lived on p53 but not on IRF-1. Thus, the role of IRF-1 and p53 in regulating DNA damage-induced apoptosis is both cooperative and independent, depending on the type and different stage of the cell (Lallemand et al. 2002). Previous studies have demonstrated the important role of IRF-1 in cell apoptosis; whether IRF-1 could affect the apoptosis of neurons after SCI or not remains unclear.
In this study, we investigated temporal-spatial patterns of IRF-1at protein level and its colocalization with active caspase-3 using an acute SCI model on adult rats. The above data suggest that IRF-1 is involved in pathophysiological and biochemical progression after SCI. Our experiments were conducted to gain an insight into the functions of IRF-1 and its role in the cellular and molecular mechanisms underlying SCI and repair.
Materials and Methods
Spinal Cord Injury
Experiments were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals; all animal experiments were allowed by the Department of Animal Center, Medical College of Nantong University. Surgery male Sprague–Dawley (SD) rats (n = 48) with an average body weight of 250 g were subjected to SCI. The SD rats were deeply anesthetized by sodium pentobarbital (65 mg/kg, i.p.), and the surgery was operated under aseptic conditions. A laminectomy was performed at the level of the 9th thoracic vertebrae to expose a circle of dura mater. The contusion injuries (n = 40) were used by the NYU impactor; the exposed rat’s spinal cord was impacted by dropping a bar 10 g in weight and 2.0 mm in diameter from a height of 10 cm. The control group animals (n = 8) were anesthetized and surgically planned, but not undergo a spinal cord injury. The animals were permitted to recover on a 30 °C heating pad. Treatments after operative included saline (2.0 cm3, s.c.) for rehydration and Baytril (0.3 cm3, 22.7 mg/ml, s.c., twice daily) to prevent infection. The animals were sacrificed at 6 to 12 h and 1, 3, 5, 7 and 14 days after injury. Eight naive animals were used as non-injured controls. All efforts were made to decrease the number of animals used and their inflicting pain.
Western Blot Analysis
In order to get samples for Western blot analysis, the sham or injured spinal cords were taken out and cryopreserved at −80 °C for further use later. The portion of the spinal cord extending from 5 mm caudal to 5 mm rostral in the injury epicenter was immediately removed. To make the lysates, the frozen spinal cord samples were minced with eye scissors on ice. The samples were then well-distributed in lysis buffer (50 mmol/l Tris, 1 % NP-40, pH 7.5, 1 % SDS, 5 mmol/l EDTA, 1 % sodium deoxycholate, 1 mmol/l PMSF, 1 % Triton X-100, 1 mg/ml leupeptin, and 10 mg/ml aprotinin) and clarified by centrifugation for 20 min in a microcentrifuge at 4 °C. After the determination of its protein concentration with the Bradford assay (Bio-Rad), the resulting supernatant (50 μg of protein) was accepted to SDS–polyacrylamide gel electrophoresis (PAGE). Then, the separated proteins were transferred to a polyvinylidine difluoride membrane (Millipore) by a transfer apparatus at 350 mA for 2.5 h and blocked with 5 % nonfat milk. The membrane was incubated with primary antibodies against IRF-1 (anti-rabbit, 1:1000; Santa Cruz), active caspase-3 (anti-rabbit, 1:1000; Cell Signaling), and GAPDH (anti-rabbit, 1:1000; Santa Cruz). After incubating with an anti-mouse or anti-rabbit horseradish peroxidase-conjugated secondary antibody, the protein was visualized by an enhanced chemiluminescence system (ECL, Pierce Company, USA).
Immunohistochemistry and Immunofluorescent
After the different time decided, the sham and injured rats were terminally anesthetized and perfused through the ascending aorta with saline and next with 4 % paraformaldehyde. After perfusion, the sham and injured spinal cords were post-fixed in 4 % paraformaldehyde for 12 h and then dehydration in sucrose. After treatment with sucrose solutions, the spinal cords were embedded in O.T.C. compound. Next, they were cut into 4.5-μm frozen cross sections at two spinal cord levels, prepared, and examined. All of the sections were blocked with 10 % Donkey serum with 0.3 % Triton X-100 and 1 % (w/v) bovine serum albumin (BSA) for 2 h at RT and incubated overnight at 4 °C with anti-IRF-1 antibody (anti-rabbit, 1:100; Santa Cruz), followed by incubation in biotinylated secondary antibody (Vector Laboratories, Burlingame, CA, USA). Staining was visualized with DAB (Vector Laboratories). Cells with strong or moderate brown staining were counted as positive; cells with no staining were counted as negative; while cells with weak staining were scored separately.
For double immunofluorescent staining, sections were removed from the freezer and incubated in an oven at 37 °C for 40 min. The sections were incubated with rabbit polyclonal primary antibodies for IRF-1 (1:100), NeuN (neuron marker, 1:100), DAPI (nucleus marker 1:1000), GFAP (astrocytes marker, 1:100), and IBa1 (microglia marker, 1:100). Briefly, sections were incubated with both primary antibodies overnight at 4 °C, followed by a mixture of CY2- and CY3-conjugated secondary antibodies for 2 h at 25 °C. The stained sections were examined with a Leica fluorescence microscope (Germany).
Rat Primary Neurons
Both spinal cords were removed from the 1-day-old Sprague–Dawley rat pups, and the tissues were gently minced by forceps and then dissociated by incubating at 37 °C for 30 min, with occasional mixing, in 10 ml of phosphate-buffered saline. After centrifugation at 1200 rpm for 5 min, the tissues were removed in 5 ml DMEM containing 10 % FBS and gently aspirated several times to inactivate the trypsin. The procedures were carried out similarly to those previously described. After the medium had been decanted, the cells were resuspended in a Neurobasal medium. Cells were plated at a density of 4 × 104 cells/ml on poly-l-lysine-coated 6-well culture plates. Before the experimental treatments, cells were grown for 8 days to induce apoptosis by using glutamate.
ShRNA and Transfection
ShRNA IRF-1 corresponding to nucleotide sequences (5′- TGTGGATCTTGCCACATTT -3′, 5′- TCTGTCTATGGAGACTTTA -3′). PC12cells were transfected with specific shRNA IRF-1 or negative control, and the efficacy of IRF-1 inhibition was tested by Western blot analysis. Cells were transfected transiently with plasmid DNA using 10 μl Lipofectamine 2000 according to the manufacturer’s recommendations. Transfected cells were used for the subsequent experiments 48 h after transfection.
Annexin-V/PI Analysis of Primary Neurons Apoptosis
After the indicated treatment, primary neurons were digested using trypsin and suffered apoptotic analysis by Annexin-V-FLUOS staining kit (Roche Diagnostics, Basel, Switzerland) obeying the manufacturer’s instructions.
Quantitative Analysis
The number of IRF-1-positive cells in the spinal cord 2.5 mm rostral to the epicenter was counted in a 500 × 500-μm measuring frame. For each animal, a measure was taken from a section through the dorsal horn, the lateral funiculus, and the ventral horn. In order to avoid counting the same cell in more than one section, we counted every fifth section (50 μm apart). The cell counts were used to determine the total number of IRF-1-positive cells per square millimeter. We simultaneously quantified the percentage of cells with IRF-1 staining. Cells double-labeled for IRF-1 and other phenotypic markers were quantified too. To identify the proportion of each phenotype-specific marker-positive cells expressing IRF-1, a minimum of 200 phenotype specific marker-positive cells were counted in both the white matter and gray matter in each section, with the exception that NeuN is only conducted in the gray matter. We then recorded the number of cells double-labeled with IRF-1 and a cell-specific marker. Two or three adjacent sections per animal were sampled at 2.5 mm from the epicenter.
Statistical Analysis
All data was analyzed with the Stata 7.0 statistical software. All values were expressed as the mean ± SEM. One-way ANOVA followed by Tukey’s post hoc multiple comparison tests were used for the statistical analysis. P values less than 0.05 were considered statistically significant. Each experiment consisted of at least three replicates per condition.
Result
Expression Profiles of IRF-1 Following SCI
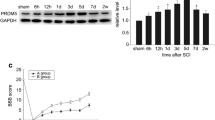
To investigate the expression pattern of IRF-1, a Western blot analysis was performed followed by the spinal cord injury. IRF-1 protein level was low in sham-operated spinal cords (sham) and gradually increased in the first 12 h after injury, reached a peak at day 3. After 14 days, IRF-1 expression returned to the level of sham-operated spinal cords (Fig. 1a, b). These results suggested that IRF-1 protein might be upregulated by an injury. In order to better understand the changes of IRF-1, we calibrate the densitometer by running a dilution series of protein added (by increasing dilutions of the protein extract with the 3d protein), a Western blot analysis was performed for the protein in different quantities, then measured the densitometric intensity, and set the densitometric intensity of 20 μ protein as 1.0. According to the measured values, we performed an actual calibration curve, and when the densitometric intensity of the measured values in Fig. 1a was brought into the curve, the result indicates an about 6-fold increase in IRF-1 protein expression after SCI. (Fig. 1c).

Change of IRF-1 expression at protein level following spinal cord injury. a Sample immunoblots detected for IRF-1 and GAPDH are shown. b Expression levels of IRF-1 were normalized against GAPDH, as estimated by optical density measurements. c A Western blot analysis was performed for the 3d protein in different quantities and then performed an actual calibration curve of the Western blot results, and when the densitometric intensity of the measured values in Fig. 1a was brought into the curve, the result indicate an about 6-fold increase in IRF-1 protein expression after SCI. The data are represented as means ± SEM (n = 3 rats per time point; *P < 0.05, significantly different from the sham-injured group)
The Expression and Distribution of IRF-1 in Sham and Injured Spinal Cord Tissue
To identify the expression and distribution changes of IRF-1 after SCI, immunohistochemistry with anti-IRF-1 antibody on transverse cryosections of the spinal cord was performed. IRF-1 was extensively expressed in the gray matter which usually indicated neurons, regardless of whether the spinal cord tissues were sham or injured. Notably, in sham-operated controls, immunostaining of IRF-1 was observed at low levels (Fig. 2a, c, e). However, the staining was increased after SCI (Fig. 2b, d, f). In addition, the number of IRF-1-positive cells in dorsal top and ventral bottom was counted and a significant difference was found between sham-operated spinal cords at day 3 after injury, which was paralleled with Western blot results (Fig. 2g).

Representative photographs of IRF-1 immunohistochemistry in adult rat spinal cord. Low-power views of transverse sections immunostained for IRF-1 in sham-operated spinal cord (a) and 3 days after injury (b). High-power views in the dorsal top (c, d) and in the ventral bottom (e, f). Immunostaining of IRF-1 shown that IRF-1 expression increases in the dorsal top (d) and ventral bottom (f) after injury. Bars 200 μm (a, b), 20 μm (c–f). Quantitative analysis of IRF-1-positive cells/mm2 in sham-operated rats and 3 days after SCI (g), indicating a obviously increase after SCI compare with sham-operated animals (n = 3 in sham-operated group, n = 3 in injured group; *P < 0.05 indicates significant difference compared with sham-operated animals; error bars SEM)
The Colocalization of IRF-1 and Different Phenotype-Specific Markers by Double Immunofluorescent Staining
To further confirm the cellular localization of IRF-1, double labeling immunofluorescent staining was shown with the following phenotype-specific markers: NeuN, GFAP, and IBa1 (Fig. 3a–r) in transverse cryosections of spinal cord within a 2-mm distance from the epicenter. Notably, IRF-1 expression was widely distributed in neurons and expression was significantly increased at 3 days after SCI, compared with the sham-operated group (P < 0.05). However, IRF-1 expression was not staining with astrocytes and microglia at sham-operated or 3 days after SCI (Fig. 3s).

Immunofluorescence staining for IRF-1 and different phenotype-specific markers in spinal cord of adult rats examined at 2.5 mm to the epicenter at day 3 after injury. a–r Tissue sections were immunolabeled with IRF-1 (red) and phenotype-specific markers (green), namely NeuN, GFAP, and IBa1. Merge of IRF-1 and phenotype-specific markers (yellow) is shown in the gray matter (a–f) and white matter (g–r). Bar 20 μm. s Quantitative analysis of cells co-expressing various phenotype-specific markers and IRF-1 (%) in gray matter and white matter. Changes of IRF-1 expression after SCI were increased in neurons (n = 3 in sham-injured group, n = 3 in injured group; *significant difference at P < 0.05 compared with sham-injured rats; error bars SEM)
Activated Caspase-3 and IRF-1 Immunoreactivity in Injured Spinal Cords
Reports have shown that neuronal apoptosis had played an important role in SCI. Then, Western blot analyses were prepared to examine the expression of activated caspase-3, which is a marker of apoptosis. Activated caspase-3 expression was significantly increased at 3 days after SCI (Fig. 4a, b). To probe the colocalization of IRF-1 with activated caspase-3 in injured spinal cords, double-immunofluorescent staining was performed. Colocalization of activated caspase-3 with IRF-1and NeuN was observed at 3rd day after SCI (Fig. 4h, n). In addition, glial cells might also cause apoptosis after SCI. We performed double-immunofluorescent staining of active caspase-3 with GFAP and IBa1. However, we could not find any colocalization in control or injured groups (Fig. 4o–z). These results indicate that IRF-1 might be associated with neuronal apoptosis after SCI.

Association of IRF-1 with apoptosis after SCI. a Western blot was analyzed for markers of apoptosis (active caspase-3) following SCI. b Bar chart showing the ratio of IRF-1 to GAPDH at each time point; the data are means ± SEM (n = 3 rats per time point, *P < 0.05, significantly different from the sham-injured group). c–z Double-immunofluorescence staining for IRF-1, NeuN, GFAP, and IBa1 with active caspase-3 in spinal cord at day 3 after SCI. Tissue sections labeled with active caspase-3 (red), NeuN or IRF-1 (green) are shown in the gray matter. Bar 20 μm. z1 Quantitative analysis of NeuN-, IRF-1-, GFAP-, and IBa1-positive cells expressing active caspase-3 in percent (n = 2 in sham-operated group, n = 3–5 in injured group; *P < 0.05, significantly different from the sham-injured group)
IRF-1 Knockdown Inhibits Neuronal Apoptosis
To investigate the effect of IRF-1 on neuronal apoptosis further, a neuronal apoptosis model was established by using glutamate to induce primary cultured neurons. Western blot showed that IRF-1, P53,PUMA, BAX, and active caspase-3 expression were increased (Fig. 5a). Then, we used IRF-1-specific siRNA to knockdown endogenous IRF-1 in primary neurons. The efficiency of the IRF-1-siRNA was tested by Western blot analysis. Western blot analysis showed IRF-1-siRNA #2 markedly reduced the level of IRF-1 expression compared with IRF-1-siRNA #1 (Fig. 5c). Furthermore, we use Western blot to investigate the active caspase-3 protein level and found that their expression were declined in IRF-1-specific siRNA (IRF-1-siRNA #2) cells. Our result displayed that siRNA reduced the protein levels of active caspase-3 compared with the control group (Fig. 5e). In order to further understand the relationship between IRF-1 and apoptosis, we use Annexin-V/PI to analyze apoptosis in primary neurons which was induced by glutamate. We found that apoptosis cells were obviously declined in IRF-1-siRNA#2 cells compared with control group (Fig. 5g).

Effects of IRF-1-specific siRNA on glutamate-induced primary neuron apoptosis. Cells were respectively transfected by nothing (normal control), control-siRNA, IRF-1-siRNA#1, and IRF-1-siRNA#2 for 48 h, then induced by glutamate (200 μM) for 12 h. a Western blot analysis for IRF-1, active caspase-3, in primary neurons after being induced by glutamate for different points. b Bar chart showing the ratio of IRF-1, active caspase-3, to GAPDH. c Efficiency of IRF-1-specific siRNA in primary neurons. Western blot analysis showing that IRF-1-siRNA#2 treatment markedly decreases IRF-1 levels at 48 h after siRNA transfection in neurons. d Bar chart showing the ratio of IRF-1 to GAPDH. e Western blot analysis showing that IRF-1-siRNA#2 treatment markedly decreases active caspase-3 levels at 48 h after siRNA transfection in neurons. f Bar chart showing the ratio of active caspase-3 to GAPDH. g Primary neurons were respectively transfected by nothing (normal control), control-siRNA, and IRF-1-siRNA#2 for 48 h, then on glutamate induction for 12 h, next subjected to flow cytometrical analysis, cells were transfected by IRF-1-siRNA#2 shown great role in reducing neuronal apoptosis. h Bar chart showing the ratio of the apoptosis of primary neurons (n = 9; *significant difference at P < 0.05 compared with sham-injured rats; error bars SEM)
Discussion
Traumatic spinal cord injury is a devastating disorder and a great financial burden on society. Recent studies suggest that apoptosis play important roles in the mediation of secondary SCI (Cheng et al. 2008). Thus, a further research of the apoptosis after SCI is necessary. In the current study, for the first time, we have revealed the expression profiles of IRF-1 after traumatic SCI. Western blot and immunohistochemistry analysis showed that IRF-1 was upregulated after injury and peaked at 3 days, and its expression was increased significantly in neurons, while no significant changes were detected in glial cells. In addition, expression patterns of P53,PUMA, BAX, and activate caspase-3 were parallel with that of IRF-1, which suggested the potential role of IRF-1 in regulating neuronal apoptosis. Finally, siRNA experiment showed that IRF-1 played an important role in regulating neuronal apoptosis in vitro. These data indicate that IRF-1 may participate in neuronal apoptosis after SCI.
SCI is often seriously disabling people, primarily the young, which brings a heavy burden in personal, financial, and societal terms (Johnson et al. 1997). SCI leads to tissue loss and associates with neurological dysfunction which causes mechanical damage and secondary biochemical and physiological responses (Dumont et al. 2001). Secondary injury mechanisms include neuronal apoptosis, glial cell activation, scar formation, and so on (Tator 1996). Neuronal apoptosis has an important role in secondary injury, diffusely apoptosis of neurons has been found following SCI (Lu et al. 2000) which are eliminated by the immune system and form a cavity (Willyard 2013). Furthermore, research has shown that the family of caspase played important roles in spinal cord nerve apoptosis (Liu et al. 1997). The upregulation of caspase family expression contributes to the spinal cord secondary injury, and selective inhibition of caspase effectively prevented spinal cord secondary injury (Wu et al. 2012). Thus, further understanding of the neuronal apoptosis in SCI can be useful for the treatment of SCI.
IRF-1 was the first member of IRFs family which was discovered to activate promoters in type I IFN genes. It has long been known that through IRF-1, many viruses can elicit the induction of type I IFN gene in many cell types by the activation of cytosolic PRRs (Yoneyama et al. 2004; Yoneyama et al. 2005). As is known, caspases are unique proteases that play important roles in an activation cascade downstream in the apoptosis mechanism. Some research has demonstrated that IRF-1 directly regulates the IFN-γ induced apoptosis by activation of caspase-1 gene expression in IFN-γ-sensitive ovarian cancer and other cancer cells (Kim et al. 2002). What is more, IRF-1 participated in induction of caspase-8 expression and apoptosis initiated by IFN-γ (Ruiz-Ruiz et al. 2004). Recently, further studies have indicated additional IRF-1 target genes. Activity of IRF-1 has been associated with downregulation of survivin (Pizzoferrato et al. 2004) and cyclin D1 (Kroger et al. 2007). Previous studies have also demonstrated that IRF-1 is involved in injury in lots of clinical setting, including ischemia–reperfusion injury (IR), shock, and transplantation. In liver warm IR model, IRF-1 knockdown mice are protected from organ injury after IR and that upregulation of IRF-1 results in liver damage even in the absence of injury (Tsung et al. 2006).
In conclusion, the present researches have demonstrated that IRF-1 expression changes after SCI suggesting that IRF-1 participates in the regulation of biochemical and physiological responses. Furthermore, knockdown of IRF-1 by siRNA can decrease glutamate-induced apoptosis in primary neurons. Our data may provide a novel insight for the treatment of SCI. However, the clear mechanism of IRF-1 in regulating SCI still needs further research.
Abbreviations
- IRF-1:
-
Interferon regulatory factor-1
- SCI:
-
Spinal cord injury
- CNS:
-
Central nervous system
- SD:
-
Sprague–Dawley
- NeuN:
-
Neuronal nuclear antigen
- GFAP:
-
Glial fibrillary acidic protein
- IBa1:
-
Ionized calcium-binding adapter molecule 1
- siRNA:
-
Short interfering RNA
References
Cheng C, Gao S, Zhao J, Niu S, Chen M, Li X, Qin J, Shi S, Guo Z, Shen A (2008) Spatiotemporal patterns of postsynaptic density (PSD)-95 expression after rat spinal cord injury. Neuropathol Appl Neurobiol 34:340–356
Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS (1997) Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med 3:73–76
Davies SJ, Field PM, Raisman G (1996) Regeneration of cut adult axons fails even in the presence of continuous aligned glial pathways. Exp Neurol 142:203–216
Di Giovanni S, Knoblach SM, Brandoli C, Aden SA, Hoffman EP, Faden AI (2003) Gene profiling in spinal cord injury shows role of cell cycle in neuronal death. Ann Neurol 53:454–468
Dumont RJ, Okonkwo DO, Verma S, Hurlbert RJ, Boulos PT, Ellegala DB, Dumont AS (2001) Acute spinal cord injury, part I: pathophysiologic mechanisms. Clin Neuropharmacol 24:254–264
Galon J, Sudarshan C, Ito S, Finbloom D, O’Shea JJ (1999) IL-12 induces IFN regulating factor-1 (IRF-1) gene expression in human NK and T cells. J Immunol 162:7256–7262
Gao J, Senthil M, Ren B, Yan J, Xing Q, Yu J, Zhang L, Yim JH (2010) IRF-1 transcriptionally upregulates PUMA, which mediates the mitochondrial apoptotic pathway in IRF-1-induced apoptosis in cancer cells. Cell Death Differ 17:699–709
Geller DA, Nguyen D, Shapiro RA, Nussler A, Di Silvio M, Freeswick P, Wang SC, Tweardy DJ, Simmons RL, Billiar TR (1993) Cytokine induction of interferon regulatory factor-1 in hepatocytes. Surgery 114:235–242
Gupta S, Xia D, Jiang M, Lee S, Pernis AB (1998) Signaling pathways mediated by the TNF- and cytokine-receptor families target a common cis-element of the IFN regulatory factor 1 promoter. J Immunol 161:5997–6004
Johnson RL, Gabella BA, Gerhart KA, McCray J, Menconi JC, Whiteneck GG (1997) Evaluating sources of traumatic spinal cord injury surveillance data in Colorado. Am J Epidemiol 146:266–272
Kim EJ, Lee JM, Namkoong SE, Um SJ, Park JS (2002) Interferon regulatory factor-1 mediates interferon-gamma-induced apoptosis in ovarian carcinoma cells. J Cell Biochem 85:369–380
Kroger A, Stirnweiss A, Pulverer JE, Klages K, Grashoff M, Reimann J, Hauser H (2007) Tumor suppression by IFN regulatory factor-1 is mediated by transcriptional down-regulation of cyclin D1. Cancer Res 67:2972–2981
Lallemand C, Palmieri M, Blanchard B, Meritet JF, Tovey MG (2002) GAAP-1: a transcriptional activator of p53 and IRF-1 possesses pro-apoptotic activity. EMBO Rep 3:153–158
Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, Dong HX, Wu YJ, Fan GS, Jacquin MF, Hsu CY, Choi DW (1997) Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci. 17:5395–5406
Lu J, Ashwell KW, Waite P (2000) Advances in secondary spinal cord injury: role of apoptosis. Spine (Phila Pa 1976) 25:1859–1866
McGraw J, Hiebert GW, Steeves JD (2001) Modulating astrogliosis after neurotrauma. J Neurosci Res 63:109–115
Nozawa H, Oda E, Nakao K, Ishihara M, Ueda S, Yokochi T, Ogasawara K, Nakatsuru Y, Shimizu S, Ohira Y, Hioki K, Aizawa S, Ishikawa T, Katsuki M, Muto T, Taniguchi T, Tanaka N (1999) Loss of transcription factor IRF-1 affects tumor susceptibility in mice carrying the Ha-ras transgene or nullizygosity for p53. Genes Dev 13:1240–1245
Pizzoferrato E, Liu Y, Gambotto A, Armstrong MJ, Stang MT, Gooding WE, Alber SM, Shand SH, Watkins SC, Storkus WJ, Yim JH (2004) Ectopic expression of interferon regulatory factor-1 promotes human breast cancer cell death and results in reduced expression of survivin. Cancer Res 64:8381–8388
Raghupathi R (2004) Cell death mechanisms following traumatic brain injury. Brain Pathol 14:215–222
Ruiz-Ruiz C, Ruiz de Almodovar C, Rodriguez A, Ortiz-Ferron G, Redondo JM, Lopez-Rivas A (2004) The up-regulation of human caspase-8 by interferon-gamma in breast tumor cells requires the induction and action of the transcription factor interferon regulatory factor-1. J Biol Chem. 279:19712–19720
Silver J, Miller JH (2004) Regeneration beyond the glial scar. Nat Rev Neurosci. 5:146–156
Tanaka N, Ishihara M, Taniguchi T (1994) Suppression of c-myc or fosB-induced cell transformation by the transcription factor IRF-1. Cancer Lett 83:191–196
Tator CH (1996) Experimental and clinical studies of the pathophysiology and management of acute spinal cord injury. J Spinal Cord Med 19:206–214
Tsung A, Stang MT, Ikeda A, Critchlow ND, Izuishi K, Nakao A, Chan MH, Jeyabalan G, Yim JH, Geller DA (2006) The transcription factor interferon regulatory factor-1 mediates liver damage during ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol 290:G1261–G1268
Watanabe N, Sakakibara J, Hovanessian AG, Taniguchi T, Fujita T (1991) Activation of IFN-beta element by IRF-1 requires a posttranslational event in addition to IRF-1 synthesis. Nucleic Acids Res 19:4421–4428
Willyard C (2013) Stem cells: a time to heal. Nature 503:S4–S6
Wu J, Kharebava G, Piao C, Stoica BA, Dinizo M, Sabirzhanov B, Hanscom M, Guanciale K, Faden AI (2012) Inhibition of E2F1/CDK1 pathway attenuates neuronal apoptosis in vitro and confers neuroprotection after spinal cord injury in vivo. PLoS ONE 7:e42129
Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T (2004) The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5:730–737
Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo YM, Gale Jr M, Akira S, Yonehara S, Kato A, Fujita T (2005) Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol 175:2851–2858
Conflict of Interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Jian Zhao and Chen Chen contribute equally to this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, J., Chen, C., Xiao, JR. et al. An Up-regulation of IRF-1 After a Spinal Cord Injury: Implications for Neuronal Apoptosis. J Mol Neurosci 57, 595–604 (2015). https://doi.org/10.1007/s12031-015-0642-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12031-015-0642-2