
Abstract
Gliostatin/thymidine phosphorylase (GLS/TP) is known to have angiogenic and arthritogenic activities in the pathogenesis of rheumatoid arthritis (RA). The novel oral Janus kinase (JAK) inhibitor baricitinib has demonstrated high efficacy in RA. However, the effect of baricitinib on fibroblast-like synoviocytes (FLSs), a key component of invasive synovitis, has not been still elucidated. This study investigated whether GLS/TP production could be regulated by JAK/signal transducers and activators of transcription (STAT) signaling in FLSs derived from patients with RA. FLSs were cultured and stimulated by interferon (IFN)γ in the presence of baricitinib. Expression levels of GLS/TP were determined using reverse transcription-polymerase chain reaction (RT-PCR), enzyme-linked immunosorbent assay (ELISA), and immunocytochemistry. Phosphorylation of STAT proteins was investigated by Western blot. In cultured FLSs, GLS/TP mRNA and protein levels were significantly induced by treatment with IFNγ and these inductions were suppressed by baricitinib treatment. Baricitinib inhibited IFNγ-induced STAT1 phosphorylation, while JAK/STAT activation played a pivotal role in IFNγ-mediated GLS/TP upregulation in RA. These results suggested that baricitinib suppressed IFNγ-induced GLS/TP expression by inhibiting JAK/STAT signaling, resulting in the attenuation of neovascularization, synovial inflammation, and cartilage destruction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease characterized by hyperplastic inflammatory synovia and neovascularization [1]. Fibroblast-like synoviocytes (FLSs) are a key component of this inflammation and cause joint destruction by the production of growth factors, proangiogenic factors, and various cytokines and chemokines [2]. Pathogenetic pathways in RA involve T cells and B cells, macrophages, and monocytes with inflammatory cytokines such as tumor necrosis factor (TNF) α, interleukin (IL)-1β and IL-6, and interferon (IFN) γ. The action of these cytokines on synovial fibroblasts, chondrocytes, and osteoclasts leads to joint destruction [3,4,5]. Interestingly, IFNγ promotes the invasive potential of FLSs through the Janus kinase (JAK) pathway [6].
Gliostatin (GLS) has thymidine phosphorylase (TP) activity and is known as platelet-derived endothelial cell growth factor (PD-ECGF) [7]. GLS/TP induces angiogenesis by stimulating the proliferation and chemotactic migration of endothelial cells [8], inhibits the growth of glial cells [9], and promotes tumor proliferation and metastasis [10]. GLS/TP was expressed in inflamed synovial tissues of patients with RA. We reported that GLS/TP levels in synovial fluids and sera were higher in patients with RA compared to patients with osteoarthritis (OA) and healthy controls [11]. In cultured FLSs, GLS/TP expression was found to be upregulated by inflammatory cytokines, such as TNFα [12], IL-1β [13], and GLS/TP itself [14]. In addition, the combination of TNFα and IFNγ synergistically augmented the expression of GLS/TP in FLSs [15]. We previously reported that direct injection of GLS/TP into rabbit knees led to pronounced RA-like synovitis [16], and that serum GLS/TP levels were decreased in RA patients treated with tacrolimus, one of the conventional synthetic disease-modifying antirheumatic drugs (csDMARDs) [17], and with surgeries such as synovectomy and total joint arthroplasty [18]. We reported that the GLS/TP promoter has seven Sp-1 binding sites and that the Sp-1 inhibitor mithramycin suppressed GLS/TP production in FLSs [19]. Further studies revealed that mithramycin, a Sp-1 inhibitor, suppressed the production of GLS/TP-induced matrix metalloproteases [20]. Therefore, GLS/TP has important roles in the pathogenesis of RA, and the suppression of GLS/TP production might be an effective therapy in RA. However, mithramycin as a therapeutic drug is clinically used for only certain malignant tumors due to its cytotoxicity. Several studies have also reported the production of mithramycin analogs that might mitigate the toxicity of the parent drug [21]. However, therapeutics that can be used in clinical practice have yet to be developed. Thus, it is necessary to identify a new strategy to control GLS/TP expression.
The JAK/signal transducer and activator of transcription (STAT) signaling pathways mediate the production of many cytokines and growth factors related to RA [22]. Novel oral JAK inhibitors, which are orally administered low molecular weight compounds, have demonstrated high efficacy in RA. Of the JAK inhibitors currently available, tofacitinib (JAK1 and JAK3 inhibitor), baricitinib (JAK1 and JAK2 inhibitor), upadacitinib (JAK1 inhibitor), filgocitinib (JAK 1 inhibitor), and peficitinib (pan-JAK inhibitor) have been widely used in many regions for RA treatment [23,24,25,26,27]. All current JAK inhibitors approved and in development have a significant effect on JAK1. JAK1 is involved in the signaling transduction of IL-6, IFN, IL-2, and IL-15 [28]
In our previous study, the JAK inhibitor tofacitinib inhibited TNFα-induced GLS/TP expression in rheumatoid FLSs [29]. Baricitinib is a selective inhibitor of JAK1 and JAK2 and exhibits moderate inhibitory activity against TYK2, while its inhibitory activity against JAK3 is limited [30]. The effect of baricitinib on GLS/TP production remains to be elucidated, as its pharmacological activity differs from that of tofacitinib. Therefore, we investigated the GLS/TP production effect of IFNγ and the inhibitory action of the novel JAK inhibitor baricitinib in FLSs derived from RA patients.
Materials and methods
Patients
Samples of synovial tissue were obtained from nine patients with RA at the time of undergoing surgeries of total joint arthroplasty or synovectomy. All patients fulfilled the 2010 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) criteria for the diagnosis of RA [31]. The clinical characteristics of these patients are shown in Table 1. This study was approved by the Nagoya City University Ethics Committee, and informed consent was obtained from all the patients upon their enrollment in the study, in accordance with the Declaration of Helsinki.
Preparation of FLSs
Human FLSs were obtained from the superficial layer of synovial tissues with synovitis. These synovial tissue specimens were minced and treated with 0.1% trypsin for 10 min. Dissociated cells were collected by centrifugation (600g for 10 min) and washed three times. The primary synoviocytes were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Wako Pure Chemical Industries, Osaka, Japan) supplemented with penicillin (100 units per ml), streptomycin (100 μg/ml), and 10% fetal bovine serum (FBS) (Gibco, Gaithersburg, MD, USA) at 37 °C in a 5% CO2 atmosphere. Cells between passages three through eight were used in the experiments as previously described [11,12,13]. The cell population was homogeneous and displayed typical FLS morphology. The cultures were completely free of lymphocytes, monocytes, and endothelial cells. FLSs were cultured to confluence in 6-well plates (1 × 105 cells/well) for reverse transcription-polymerase chain reaction (RT-PCR), enzyme immunoassay (EIA), and Western blotting.
Cell viability assay
FLSs (4 × 104/well) were incubated with baricitinib (0 to 1 μM) (Selleck Chemicals, Houston, TX, USA) and recombinant human IFNγ (0 to 1000 pg/ml) (R&D Systems, Minneapolis, MN, USA) in 96-well plates in 100 μl of medium for 24 h, after which 10 μl of WST-8 was added (Cell Counting Kit-8; Dojindo Laboratories, Kumamoto, Japan) to each well. The mixture was then incubated in the plate for 2 h at 37 °C, after which the absorbance was measured at 450 nm using a microplate reader (SpectraMax 340PC384; Molecular Devices, Sunnyvale, CA, USA).
RT-PCR
Confluent FLSs were incubated in the absence of baricitinib with IFNγ (0, 30, 100, or 1000 pg/ml) for 18 h, and confluent FLSs were incubated in the presence or absence of 0.3 μM baricitinib for 6 h and then with 100 pg/ml IFNγ for 0–24 h. Confluent FLSs were incubated in the presence or absence of 0.1 to 1 μM baricitinib for 6 h and then with 100 pg/ml IFNγ for 18 h. GLS/TP gene expression was assessed by RT-PCR. Total RNA was extracted from FLSs using an RNeasy® mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions, and cDNA was prepared with PrimeScript™ RT Master Mix (Takara Bio Inc., Otsu, Japan). The cDNA was then subjected to real-time PCR using a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) with GoTaq® qPCR Master Mix (Promega, Madison, WI, USA) and gene-specific primers. The PCR protocol was as follows. Initial denaturation was performed for 10 min at 95 °C, followed by 40 amplification cycles of 5 s at 95 °C and 1 min at 60 °C. After confirming amplification of GLS/TP and β-actin cDNA with the same efficiency, the relative expression levels of GLS/TP were normalized to the endogenous control β-actin. The GLS/TP and β-actin cDNA were amplified using the following primers: GLS/TP, 5′-GAGGCACCTTGGATAAGCTGGA-3′ and 5′-GCTGTCACATCTCTGGCTGCATA-3′; and β-actin, 5′-TGGCACCCAGCACAATGAA-3′ and 5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′.
Western blotting
Confluent FLSs were incubated with 100 pg/ml IFNγ in the presence of baricitinib (0.3 μM) in a 6-well plate. After treatment, the FLSs were recovered and gently homogenized in 10 mM Tris-HCl (pH 6.8) containing 0.5% SDS and a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA) on ice. The protein content was estimated using a BCA protein assay reagent kit (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of total protein were separated using 10% polyacrylamide gels containing sodium dodecyl sulfate (SDS) and then electro-transferred to polyvinylidene difluoride (PVDF) membranes (Immobilon-P; Millipore, Billerica, MA, USA). The blots were blocked with 5% skim milk in Tris-buffered saline with Tween 20 (TBS-T: 20 mM Tris-HCl, pH 7.6; and 137 mM NaCl; 0.1% Tween 20) overnight at 4 °C, and then incubated with the primary antibodies against GLS/TP (Cat# DM292, Acris Antibodies, San Diego, CA, USA), antibodies against phospho-STAT1 (Cat# 7649S, Cell Signaling Technology, Denver, MA, USA) or antibodies against the corresponding total STAT1 (Cat# 9172S, Cell Signaling Technology) diluted in TBS-T overnight at 4 °C. Actin was used as a gel loading control. After three washes with TBS-T, membranes were developed with the appropriate horseradish peroxidase-conjugated secondary antibodies (1,3000 dilution; Cat# NA934V, GE Healthcare, Little Chalfont, UK) for 1 h at room temperature and washed three times with TBS-T. The bands were visualized using enhanced chemiluminescence (Amersham Biosciences Corp, Piscataway, NJ, USA). ImageJ software was used to quantify the gel bands (http://rsbweb.nih.gov/ij/. Accessed 3 Nov. 2021; National Institutes of Health, Bethesda, MD, USA).
Immunocytochemistry
Confluent FLSs were incubated with 100 pg/ml IFNγ in the presence of baricitinib (0.3 μM) on chambered slides coated with BD Matrigel Matrix (BD Biosciences, Franklin Lakes, NJ, USA). Cells were fixed in 3% paraformaldehyde for 30 min, permeabilized with 0.2% Triton X-100 for 5 min, washed with PBS, and blocked for 60 min with blocking solution comprising 3% bovine serum albumin, 0.1% glycine, and 0.1% sodium azide in PBS at room temperature. After washing, the cells were labeled overnight with the appropriate primary antibody (1:100 anti-GLS/TP antibody; Acris Antibodies) at 4 °C. Cells were washed and labeled with an Alexa Fluor 594-labeled (red) goat anti-mouse IgG (1:1000; Cat# ab150116, Invitrogen, Carlsbad, CA, USA) secondary antibody. After washing, the sections were mounted on glass slides containing ProLong Gold Antifade with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). The stained cells were visualized using a confocal laser-scanning microscope (Nikon, Tokyo, Japan), and the total intensity of immunostaining in five random fields was quantified using ImageJ software. The numbers of cells were counted in each of the fields. The data were presented as the mean staining intensity per cell.
Statistical analysis
All data were entered into an electronic database and analyzed by using GraphPad Prism 7 (GraphPad Software, Inc., San Diego, CA, USA). Data are presented as the mean ± standard error of the mean (SEM) unless otherwise stated. The statistical significance of the differences between the two groups was analyzed using one-way analysis of variance (ANOVA) and Tukey’s post hoc test. In all cases, P values less than 0.05 were considered statistically significant.
Results
Effects of baricitinib on IFN𝛾-induced GLS/TP production in cultured FLSs
The effects of IFN𝛾 and baricitinib on FLSs were investigated by measuring GLS/TP expression. Initially, a cell viability assay was used to confirm that the concentrations of baricitinib and IFN𝛾 used in this study were nontoxic (Fig. 1A).

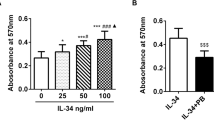
A Baricitinib and interferon (IFN)γ did not alter the viability of fibroblast-like synoviocytes (FLSs). No significant difference was found compared to the control. B IFNγ-induced gliostatin/thymidine phosphorylase (GLS/TP) mRNA expression in FLSs derived from patients with rheumatoid arthritis (RA). Confluent FLSs were incubated in the absence of baricitinib with IFNγ (0, 30, 100, or 1000 pg/ml) for 18 h. GLS/TP mRNA expression levels were normalized to those of β-actin. C Baricitinib suppressed IFNγ-induced GLS/TP expression in FLSs from RA patients. Confluent FLSs were incubated in the presence or absence of 0.3 μM baricitinib for 6 h and then with 100 pg/ml IFNγ for 0–24 h. D Baricitinib suppressed IFNγ-induced GLS/TP expression in FLSs. Confluent FLSs were incubated in the presence or absence of 0.1 to 1 μM baricitinib for 6 h and then with 100 pg/ml IFNγ for 18 h. GLS/TP mRNA expression levels were normalized to those of β-actin. Control FLSs were cultured without additional agents. The results are presented as the mean ± standard error of the mean (SEM) of experiments performed in triplicate. The statistical significance of differences between groups was calculated by one-way analysis of variance (ANOVA) and Tukey’s multiple comparisons test. Compared with controls: *P < 0.005. Compared with samples treated without baricitinib: †P < 0.05, ††P < 0.001. BAR, baricitinib
Confluent FLSs were cultured with IFN𝛾 (0, 30, 100, 1000 pg/ml) for 18 h, and IFN𝛾 was observed to induce GLS/TP mRNA expression in RA FLSs. GLS/TP mRNA expression increased in response to treatment with increasing concentrations of IFN𝛾. GLS/TP mRNA expression stimulated by 100 pg/ml IFN𝛾 was 16.3-fold higher than in the absence of IFN𝛾 stimulation (Fig. 1B).
To investigate the time course of GLS/TP-mediated induction of mRNA expression, confluent FLSs were cultured in 6-well plates with or without 0.3 μM baricitinib for 6 h and then further incubated with 100 pg/ml IFN𝛾 for the indicated times (Fig. 1C). GLS/TP mRNA expression increased in response to treatment with IFN𝛾 (100 pg/ml), and the peak was observed following 18 h of treatment. GLS/TP mRNA expression at 18 h was 8.6-fold higher than pretreatment levels. These increases in GLS/TP mRNA expression were significantly suppressed by treatment with 0.3 μM baricitinib compared with treatment with IFNγ alone (Fig. 1C).
We pretreated FLSs with 0 to 1 μM baricitinib for 6 h and subsequently incubated them with 100 pg/ml IFN𝛾 for 18 h. Treatment with IFNγ alone significantly increased GLS/TP mRNA expression levels. The GLS/TP levels were 11.3-fold higher than those in the control groups. Treatment with baricitinib suppressed these increases in a dose-dependent manner (Fig. 1D). Treatment with baricitinib alone did not affect GLS/TP mRNA expression. Induction of IFN𝛾-stimulated GLS/TP mRNA was significantly suppressed by treatment with 0.3 or 1 μM baricitinib.
Similarly, GLS/TP protein expression levels were significantly induced by treatment with IFN𝛾 alone (Fig. 2A). Western blotting showed that GLS/TP protein expression levels were 1.5-fold higher in the treatment group than in the control group (Fig. 2B). Induction of IFN𝛾-stimulated GLS/TP protein was significantly suppressed by treatment with 1 μM baricitinib (Fig. 2B).

Baricitinib suppressed interferon (IFN)γ-induced gliostatin/thymidine phosphorylase (GLS/TP) protein expression in fibroblast-like synoviocytes (FLSs) from RA patients (RA). A Confluent FLSs were incubated in the presence or absence of 0.1 to 1 μM baricitinib for 6 h and then with 100 pg/ml IFNγ for 18 h. GLS/TP protein levels were assessed by Western blotting. B Band density was measured using ImageJ. The statistical significance of differences between groups was calculated by Tukey’s multiple comparisons test. Compared with controls: *P < 0.001. Compared with samples treated without baricitinib: †P < 0.001. Immunocytochemical detection of GLS/TP in FLSs from RA patients. C FLSs were treated with or without 0.3 μM baricitinib for 6 h before being incubated with or without IFNγ (100 pg/ml) for 18 h, after which they were immunostained with a GLS/TP antibody (red). The cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). The scale bar represents 100 μm. Control FLSs were cultured without additional agents. D The total intensity of immunostaining in a random field was quantified by ImageJ, and the number of cells in the field was counted. The data (intensity/cell) are presented as the mean ± standard error of the mean (SEM) of five determinations. The statistical significance of differences between groups was calculated by one-way analysis of variance (ANOVA) and Tukey’s multiple comparisons test. Compared with controls: *P < 0.05. Compared with samples treated with IFNγ alone: †P < 0.05. BAR, baricitinib
FLSs were cultured to confluence and then treated with 0.3 μM baricitinib for 6 h, followed by incubation with 100 pg/ml IFN𝛾 for 18 h. The FLSs were subsequently immunostained for GLS/TP, which showed that untreated cells displayed weakly diffuse cytoplasmic staining. Treatment with baricitinib alone had no effect on GLS/TP staining. GLS/TP protein expression was significantly induced by treatment with IFNγ alone (Fig. 2C). GLS/TP protein expression was 1.7-fold higher in cells treated with IFNγ alone relative to control cells, and the induction was significantly suppressed by treatment with baricitinib. GLS/TP expression was 0.5-fold higher in cells treated with IFNγ and baricitinib than in cells treated with IFN𝛾 alone (Fig. 2D).
Baricitinib inhibits IFN𝛾-mediated induction of phospho-STAT1 expression
FLSs were pretreated with 0 or 0.3 μM baricitinib for 6 h and then incubated with 100 pg/ml IFNγ for the indicated time (Fig. 3). IFNγ significantly induced STAT1 phosphorylation following 15 min of treatment. STAT1 phosphorylation in the treated group was 3.2-fold higher than in the control group at 15 min. Pretreatment with baricitinib inhibited IFNγ-induced STAT1 phosphorylation at 15 min. STAT1 phosphorylation in the sample treated with baricitinib was 0.28-fold higher than that in the sample treated with IFNγ alone at 15 min of treatment (Fig. 3).

Baricitinib inhibits interferon (IFN)γ induction of phospho-signal transducer and activator of transcription (STAT)1. A Confluent fibroblast-like synoviocytes (FLSs) were treated with IFNγ (100 pg/ml) for the indicated duration. IFNγ induced phospho-STAT1 expression. These cells were incubated in the presence or absence of 0.3 μM baricitinib for 6 h before being incubated with IFNγ as indicated. Baricitinib (0.3 μM) inhibited IFNγ-induced STAT1 phosphorylation. B Band density was measured using ImageJ. The results are expressed as the mean ± standard error of the mean (SEM) of three experiments involving FLSs. The statistical significance of differences between groups was calculated by one-way analysis of variance (ANOVA) and Tukey’s multiple comparisons test. Compared with samples treated for 0 h: *P < 0.001. Compared with samples treated with IFNγ alone: †P < 0.001. BAR, baricitinib
Discussion
IFNγ is a cytokine that coordinates a diverse array of cellular programs through transcriptional regulation of immunologically relevant genes [32]. Aberrant IFNγ expression has been associated with a number of autoinflammatory and autoimmune diseases, including RA. Kokkonen and colleagues reported that the plasma levels of IFNγ are significantly increased in RA patients compared with healthy controls. The median IFNγ concentration in the sera of RA patients was 164.8 (71.2–793.2) pg/ml and that in health controls was 77.1 (48.4–127.6) pg/ml (P = 0.002) [33]. Similarly, the concentration of IFNγ in synovial fluid from patients with RA was reported to be higher than that in synovial fluid from patients with osteoarthritis (OA). Seventy-two percent of synovial fluid from RA patients, but just 7% of synovial fluid from OA patients contained > 10 pg/ml. The median concentration of IFNγ in synovial fluid from patients with RA was 17 pg/ml [34]. IFNγ is produced predominantly by natural killer (NK) and natural killer T (NKT) cells as part of the innate immune response, and by Th1 CD4 and CD8 cytotoxic T lymphocyte (CTL) effector T cells once antigen-specific immunity develops [35]. In RA patients, macrophages stimulated with IFNγ produce various cytokines, such as TNFα, IL-1β, and IL-6, and directly induce increases in antigen processing and immune pathways [3, 36]. Karonitsch and colleagues observed that IFNγ induces changes in synovial architecture and accelerates the migration of FLSs. They demonstrated that these effects are abolished by a small interfering RNA that knocks down JAK2 [6]. It remains unknown whether IFNγ acts directly on FLSs. GLS/TP has IFNγ binding sites in the promoter region [17]; therefore, we focused on the direct effect of IFNγ on GLS/TP production in RA FLSs.
First, we confirmed that IFNγ induced GLS/TP mRNA and protein production in a dose-dependent manner and that 100 pg/ml IFNγ significantly increased the expression of GLS/TP (p < 0.001). Induction of GLS/TP production by IFNγ was inhibited by baricitinib in a dose-dependent manner. Our immunocytochemical studies revealed that GLS/TP staining was weak and diffuse in the cytoplasm of FLSs in the absence of treatment. GLS/TP staining was increased following treatment with IFNγ and suppressed by combined treatment with baricitinib at a concentration of 0.3 μM. IFNγ binds a type II IFN receptor that is composed of two subunits, IFNGR1 and IFNGR2, which are associated with JAK1 and JAK2, respectively [37], and baricitinib selectively inhibits JAK1 and JAK2 [30]. Our results suggest that GLS/TP expression might be induced through the JAK1 or JAK2/STAT signaling pathway in FLSs stimulated with IFNγ.
Second, we confirmed that STAT1 was phosphorylated by IFNγ after 15 min and that STAT1 phosphorylation was inhibited by baricitinib. The GLS/TP gene promoter contains binding sites for STAT1, such as an interferon-stimulated response element (ISRE) and a γ-activated sequence (GAS) [38, 39]. Binding of IFNγ to its receptor results in tyrosine phosphorylation of the dormant cytoplasmic protein STAT1, which then translocates to the nucleus and binds to ISRE and GAS [40]. Baricitinib may suppress GLS/TP expression by inhibiting STAT1 phosphorylation. There have been several reports examining STAT1 phosphorylation, and stimulation of STAT1 phosphorylation by TNFα in FLSs requires 3 to 4 h [28, 41].
Similarly, the protein synthesis inhibitor cycloheximide significantly decreases GLS/TP mRNA production induced by TNFα in a dose-dependent manner [19]. Overall, these results suggest that IFNγ-induced GLS/TP gene transcription is directly regulated by STAT1 binding to the GLS/TP gene promoter in FLSs and that GLS/TP gene transcription is required for de novo protein synthesis in FLSs treated with TNFα.
In conclusion, ours is the first study to demonstrate that baricitinib downregulates GLS/TP mRNA and protein expression in RA FLSs. Our data suggest that JAK inhibitors, including baricitinib, could affect not only immune cells but also FLSs in RA. We revealed that IFNγ might directly affect GLS/TP production in FLSs via the JAK/STAT signaling pathway as well as the novel mechanism underlying the inhibitory effect of baricitinib on IFNγ-induced GLS/TP production in FLSs.
References
Firestein GS, McInnes IB. Immunopathogenesis of rheumatoid arthritis. Immunity. 2017;46:183–96. https://doi.org/10.1016/j.immuni.2017.02.006.
Nygaard G, Firestein GS. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat Rev Rheumatol. 2020;16:316–33. https://doi.org/10.1038/s41584-020-0413-5.
Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–38. https://doi.org/10.1016/S0140-6736(16)30173-8.
Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537–45. https://doi.org/10.1172/JCI36389.
Lee A, Qiao Y, Grigoriev G, Chen J, Park-Min KH, Park SH, Ivashkiv LB, Kalliolias GD. Tumor necrosis factor α induces sustained signaling and a prolonged and unremitting inflammatory response in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2013;65:928–38. https://doi.org/10.1002/art.37853.
Karonitsch T, Beckmann D, Dalwigk K, Niederreiter B, Studenic P, Byrne RA, Holinka J, Sevelda F, Korb-Pap A, Steiner G, Smolen JS, Pap T, Kiener HP. Targeted inhibition of Janus kinases abates interfon gamma-induced invasive behaviour of fibroblast-like synoviocytes. Rheumatology (Oxford). 2018;57:572–7. https://doi.org/10.1093/rheumatology/kex426.
Asai K, Nakanishi K, Isobe I, Eksioglu YZ, Hirano A, Hama K, Miyamoto T, Kato T. Neurotrophic action of gliostatin on cortical neurons. Identity of gliostatin and platelet-derived endothelial cell growth factor. J Biol Chem. 1992;267:20311–6.
Miyadera K, Sumizawa T, Haraguchi M, Yoshida H, Konstanty W, Yamada Y, Akiyama S. Role of thymidine phosphorylase activity in the angiogenic effect of platelet derived endothelial cell growth factor/thymidine phosphorylase. Cancer Res. 1995;55:1687–90.
Ueki T, Nakanishi K, Asai K, Okouchi Y, Isobe I, Eksioglu YZ, Kato T, Kohno K. Neurotrophic action of gliostatin on cocultured neurons with glial cells. Brain Res. 1993;622:299–302. https://doi.org/10.1016/0006-8993(93)90833-9.
Nakajima Y, Gotanda T, Uchimiya H, Furukawa T, Haraguchi M, Ikeda R, Sumizawa T, Yoshida H, Akiyama S. Inhibition of metastasis of tumor cells overexpressing thymidine phosphorylase by 2-deoxy-l-ribose. Cancer Res. 2004;64:1794–801. https://doi.org/10.1158/0008-5472.can-03-2597.
Takeuchi M, Otsuka T, Matsui N, Asai K, Hirano T, Moriyama A, Isobe I, Eksioglu YZ, Matsukawa K, Kato T, Tada T. Aberrant production of gliostatin/platelet-derived endothelial cell growth factor in rheumatoid arthritis. Arthritis Rheum. 1994;37:662–72. https://doi.org/10.1002/art.1780370509.
Waguri Y, Otsuka T, Sugimura I, Matsui N, Asai K, Moriyama A, Kato T. Gliostatin/platelet-derived endothelial cell growth factor as a clinical marker of rheumatoid arthritis and its regulation in fibroblast like synoviocytes. Br J Rheumatol. 1997;36:315–21. https://doi.org/10.1093/rheumatology/36.3.315.
Tanikawa T, Waguri-Nagaya Y, Kusabe T, Aoyama M, Asai K, Otsuka T. Gliostatin/thymidine phosphorylase-regulated vascular endothelial growth-factor production in human fibroblast-like synoviocytes. Rheumatol Int. 2007;27:553–9. https://doi.org/10.1007/s00296-006-0258-5.
Muro H, Waguri-Nagaya Y, Mukofujiwara Y, Iwahashi T, Otsuka T, Matsui N, Moriyama A, Asai K, Kato T. Autocrine induction of gliostatin/platelet- derived endothelial cell growth factor (GLS/PD-ECGF) and GLS-induced expression of matrix metalloproteinases in rheumatoid arthritis synoviocytes. Rheumatology (Oxford). 1999;38:1195–202. https://doi.org/10.1093/rheumatology/38.12.1195.
Toyoda Y, Tabata S, Kishi J, Kuramoto T, Mitsuhashi A, Saijo A, Kawano H, Goto H, Aono Y, Hanibuchi M, Horikawa H, Nakajima T, Furukawa T, Sone S, Akiyama S, Nishioka Y. Thymidine phosphorylase regulates the expression of CXCL10 in rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheum. 2014;66:560–8. https://doi.org/10.1002/art.38263.
Waguri-Nagaya Y, Otsuka T, Sugimura I, Matsui N, Asai K, Nakajima K, Tada T, Akiyama S, Kato T. Synovial inflammation and hyperplasia induced by gliostatin/platelet-derived endothelial cell growth factor in rabbit knees. Rheumatol Int. 2000;20:13–9. https://doi.org/10.1007/s002960000067.
Yamagami T, Waguri-Nagaya Y, Ikuta K, Aoyama M, Asai K, Otsuka T. FK506 inhibition of gliostatin/thymidine phosphorylase production induced by tumor necrosis factor-α in rheumatoid fibroblast-like synoviocytes. Rheumatol Int. 2011;31:903–9. https://doi.org/10.1007/s00296-010-1411-8.
Muro H, Waguri-Nagaya Y, Otsuka T, Matsui N, Asai K, Kato T. Serum gliostatin levels in patients with rheumatoid factor-negative and -positive rheumatoid arthritis and changes of these levels after surgical treatments. Clin Rheumatol. 2001;20:331–6. https://doi.org/10.1007/s100670170022.
Ikuta K, Waguri-Nagaya Y, Kikuchi K, Yamagami T, Nozaki M, Aoyama M, Asai K, Otsuka T. The Sp1 transcription factor is essential for the expression of gliostatin/thymidine phosphorylase in rheumatoid fibroblast-like synoviocytes. Arthritis Res Ther. 2012;14:R87. https://doi.org/10.1186/ar3811.
Tatematsu N, Waguri-Nagaya Y, Kawaguchi Y, Oguri Y, Ikuta K, Kobayashi M, Nozaki M, Asai K, Aoyama M, Otsuka T. Mithramycin has inhibitory effects on gliostatin and matrix metalloproteinase expression induced by gliostatin in rheumatoid fibroblast-like synoviocytes. Mod Rheumatol. 2018;28:495–505. https://doi.org/10.1080/14397595.2017.1350332.
Núñez LE, Nybo SE, González-Sabín J, Pérez M, Menéndez N, Braña AF, Shaaban KA, He M, Morís F, Salas JA, Rohr J, Méndez C. A novel mithramycin analogue with high antitumor activity and less toxicity generated by combinatorial biosynthesis. J Med Chem. 2012;55:5813–25. https://doi.org/10.1021/jm300234t.
Ivashkiv LB, Hu X. The JAK/STAT pathway in rheumatoid arthritis: pathogenic or protective? Arthritis Rheum. 2003;48:2092–6. https://doi.org/10.1002/art.11095.
Taylor PC. Clinical efficacy of launched JAK inhibitors in rheumatoid arthritis. Rheumatology (Oxford). 2019;58(Suppl 1):i17–26. https://doi.org/10.1093/rheumatology/key225.
Tanaka Y, Atsumi T, Amano K, Harigai M, Ishii T, Kawaguchi O, Rooney TP, Akashi N, Takeuchi T. Efficacy and safety of baricitinib in Japanese patients with rheumatoid arthritis: subgroup analyses of four multinational phase 3 randomized trials. Mod Rheumatol. 2018;28:583–91. https://doi.org/10.1080/14397595.2017.1392057.
Clark JD, Flanagan ME, Telliez J-B. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J Med Chem. 2014;57:5023–38. https://doi.org/10.1021/jm401490p.
Fleischmann R, Pangan AL, Song IH, Mysler E, Bessette L, Peterfy C, Durez P, Ostor AJ, Li Y, Zhou Y, Othman AA, Genovese MC. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III, double-blind, randomized controlled trial. Arthritis Rheum. 2019;71:1788–800. https://doi.org/10.1002/art.41032.
Takeuchi T, Tanaka Y, Tanaka S, Kawakami A, Iwasaki M, Katayama K, Rokuda M, Izutsu H, Ushijima S, Kaneko Y, Shiomi T, Yamada E, van der Heijde D. Efficacy and safety of peficitinib (ASP015K) in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III randomised, double-blind, placebo-controlled trial (RAJ4) in Japan. Ann Rheum Dis. 2019;78:1305–19. https://doi.org/10.1136/annrheumdis-2019-215164.
Choy EH. Clinical significance of janus kinase inhibitor selectivity. Rheumatology (Oxford). 2019;58:953–62. https://doi.org/10.1093/rheumatology/key339.
Kawaguchi Y, Waguri-Nagaya Y, Tatematsu N, Oguri Y, Kobayashi M, Nozaki M, Asai K, Aoyama M, Otsuka T. The Janus kinase inhibitor tofacitinib inhibits TNF-α-induced gliostatin expression in rheumatoid fibroblast-like synoviocytes. Clin Exp Rheumatol. 2018;36:559–67.
Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, Covington MB, Thomas B, Collier P, Favata MF, Wen X, Shi J, McGee R, Haley PJ, Shepard S, Rodgers JD, Yeleswaram S, Hollis G, Newton RC, et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol. 2010;184:5298–307. https://doi.org/10.4049/jimmunol.0902819.
Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, Combe B, Costenbader KH, Dougados M, Emery P, Ferraccioli G, Hazes JM, Hobbs K, Huizinga TW, Kavanaugh A, et al. Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010;62:2569–81. https://doi.org/10.1002/art.27584.
Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–89. https://doi.org/10.1189/jlb.0603252.
Kokkonen H, Soderstrom I, Rocklov J, Hallmans G, Lejon K, Dahlqvist SR. Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum. 2010;62:383–91. https://doi.org/10.1002/art.27186.
Steiner G, Tohidast-Akrad M, Witzmann G, Veseley M, Studnicka-Benke A, Gal A, Kunaver M, Zenz P, Smolen JS. Cytokine production by synovial T-cells in rheumatoid arthritis. Rheumatology (Oxford). 1999;38:202–13. https://doi.org/10.1093/rheumatology/38.3.202.
Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101. https://doi.org/10.1016/S0065-2776(07)96002-2.
Kato M. New insights into IFN-γ in rheumatoid arthritis: role in the era of JAK inhibitors. Immunol Med. 2020;43:72–8. https://doi.org/10.1080/25785826.2020.1751908.
Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–86. https://doi.org/10.1038/nri1604.
Song S, Ling-Hu H, Roebuck KA, Rabbi MF, Donnelly RP, Finnegan A. Interleukin-10 inhibits interferon-γ-induced intercellular adhesion molecule-1 gene transcription in human monocytes. Blood. 1997;89:4461–9.
Goto H, Kohno K, Sone S, Akiyama S, Kuwano M, Ono M. Interferon γ-dependent induction of thymidine phosphorylase/platelet-derived endothelial growth factor through γ-activated sequence-like element in human macrophages. Cancer Res. 2001;61:469–73.
Darnell JE Jr, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science (Washington DC). 1994;264:1415–21.
Rosengren S, Corr M, Firestein GS, Boyle DL. The JAK inhibitor CP-690,550 (tofacitinib) inhibits TNF-induced chemokine expression in fibroblast-like synoviocytes: autocrine role of type I interferon. Ann Rheum Dis. 2012;71:440–7. https://doi.org/10.1136/ard.2011.150284.
Acknowledgements
We thank Seiko Ukai for the excellent technical assistance.
Funding
This work was supported by JSPS KAKENHI Grant Numbers 19K18473 and 20K09465.
Author information
Authors and Affiliations
Contributions
YJ, AM, and YW-N conducted the study, performed experiments, and drafted the manuscript. YK and HY performed experiments. HS, SY, HS, KA, MN, and HM participated in the design and coordination of the study. MN and YW-N collected patient samples and provided critical comments on the interpretation of the data. All authors read and approved the final manuscript for submission.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval and consent to participate
Ethical approval for this study was granted by the Ethics Committee of Nagoya City University Graduate School of Medical Sciences (#621) and according to the Declaration of Helsinki. Informed consent was obtained from all patients prior to sample collection.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Joyo, Y., Kawaguchi, Y., Yonezu, H. et al. The Janus kinase inhibitor (baricitinib) suppresses the rheumatoid arthritis active marker gliostatin/thymidine phosphorylase in human fibroblast-like synoviocytes. Immunol Res 70, 208–215 (2022). https://doi.org/10.1007/s12026-022-09261-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12026-022-09261-4